Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup


Abstract
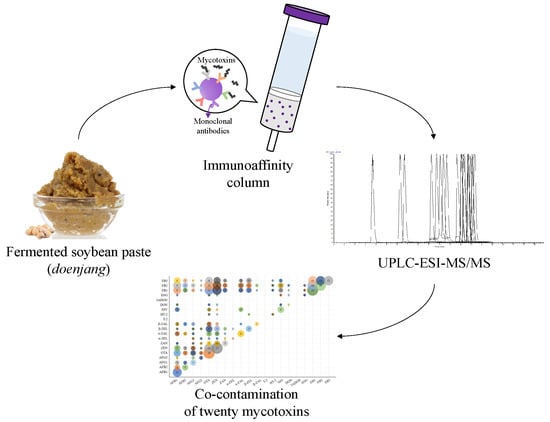
:
1. Introduction
2. Results and Discussion
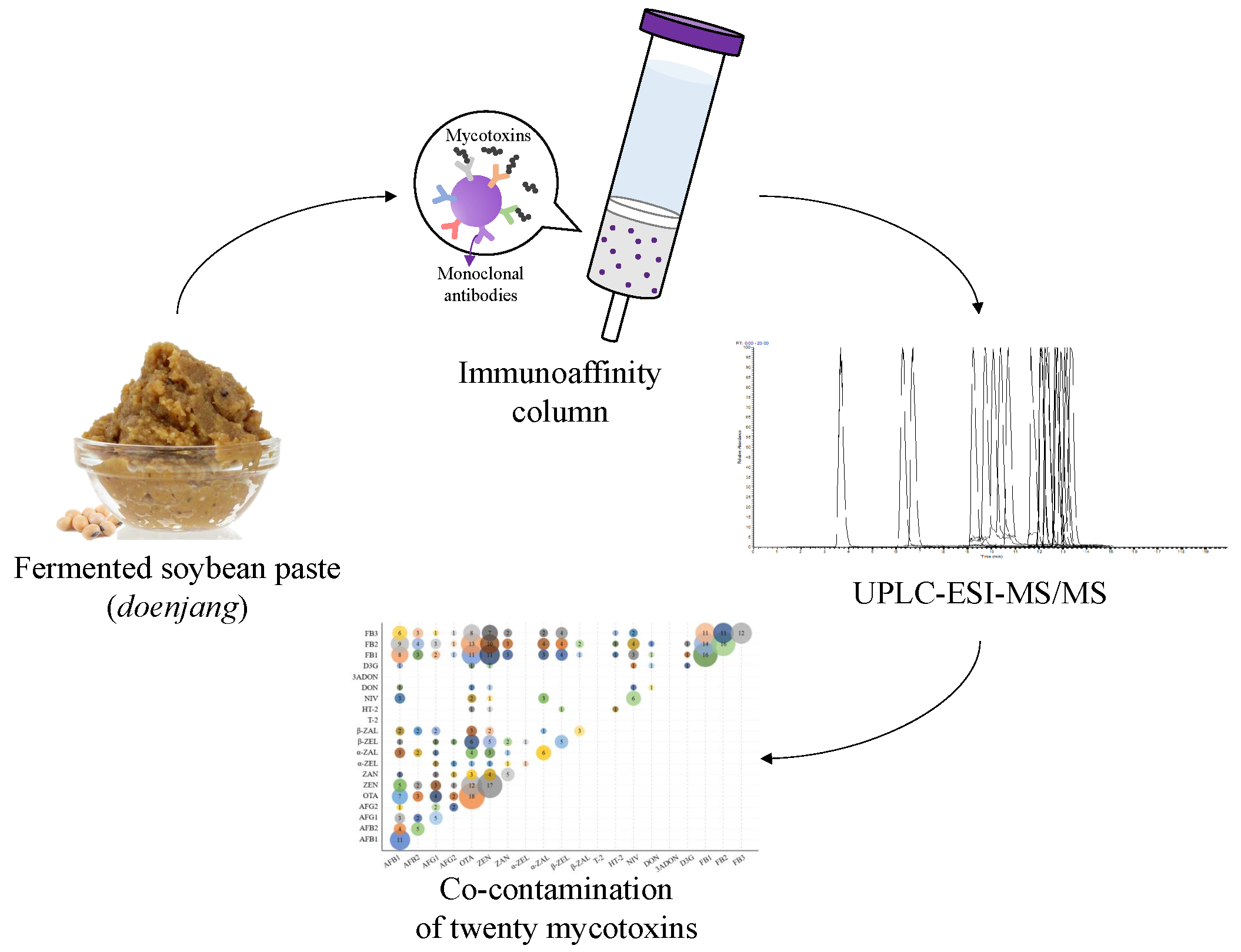
2.1. Optimization of LC-MS/MS Parameters
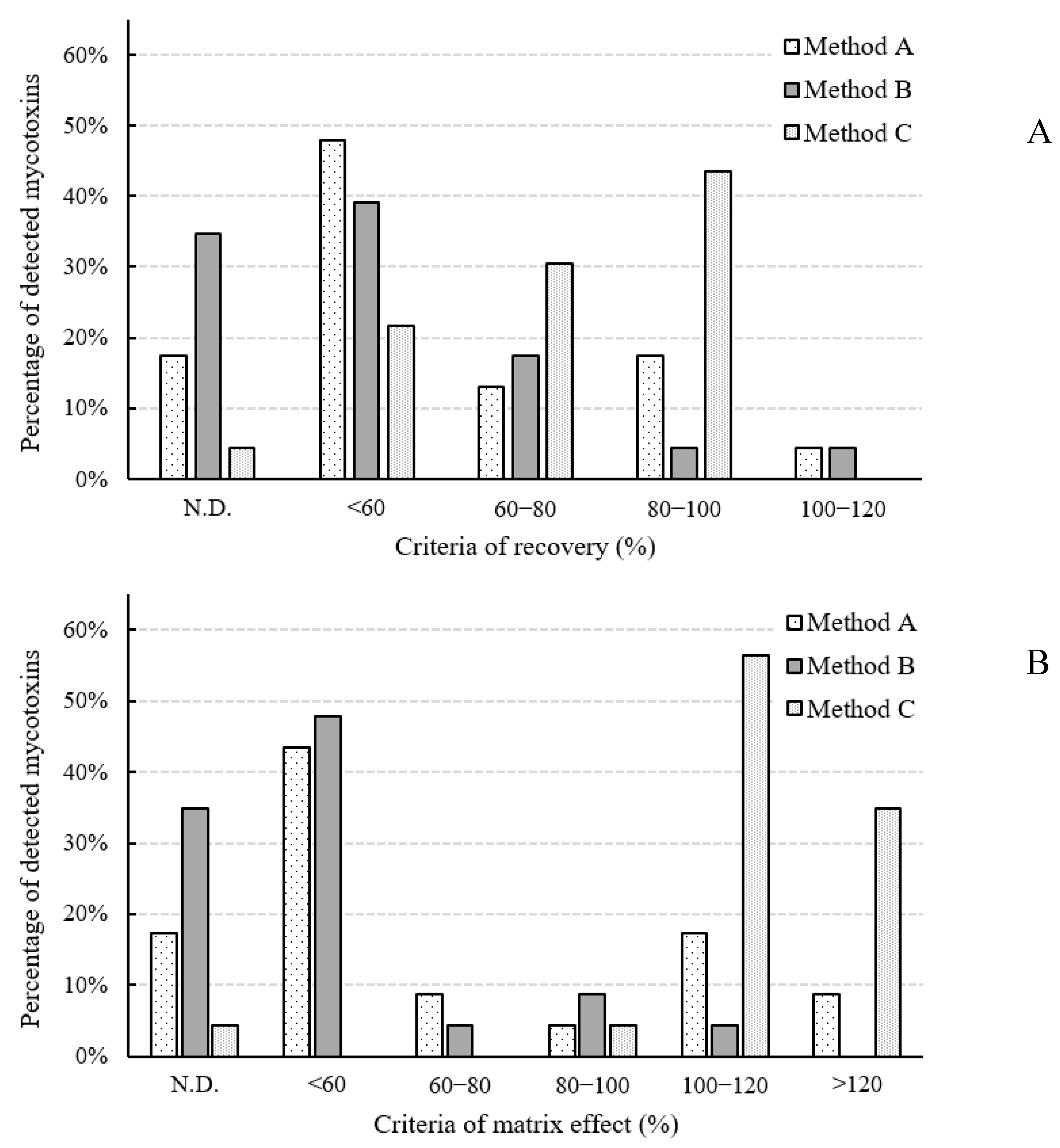
2.2. Optimization of Sample Preparation Method
2.2.1. SPE Method (Method A)
2.2.2. QuEChERS (Method B)
2.2.3. IAC Cleanup (Method C)
2.3. Method Validation
2.3.1. Linearity, LOD, LOQ, and Matrix Effect
2.3.2. Recovery, Precision, and Trueness
2.3.3. Measurement Uncertainty
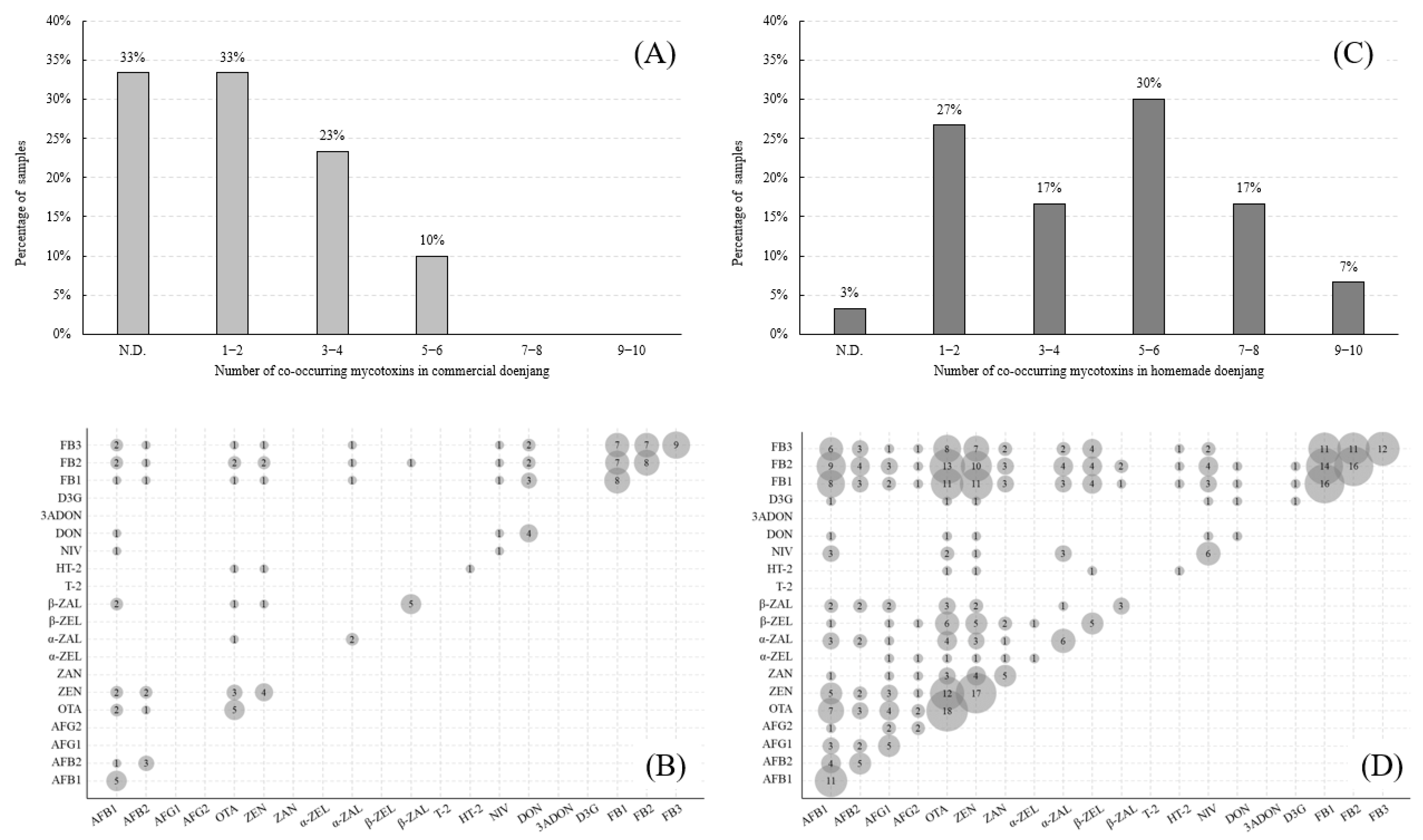
2.4. Application to Actual Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sampling
3.3. Sample Preparation
3.3.1. Method A
3.3.2. Method B
3.3.3. Method C
3.4. LC-MS/MS Conditions
3.5. Method Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sweeney, M.J.; Dobson, A.D. Molecular biology of mycotoxin biosynthesis. FEMS Microbiol. Lett. 1999, 175, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Kabak, B.; Dobson, A.D.; Var, I. Strategies to prevent mycotoxin contamination of food and animal feed: A review. Crit. Rev. Food Sci. Nutr. 2006, 46, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Lee, Y.W.; Bullerman, L.B. Changes of aflatoxins during the ripening and storage of Korean soy sauce and soybean paste and the characteristics of the changes. Part 3. Korean Public Health Res. 2000, 1, 21–28. [Google Scholar]
- Ahn, S.; Lee, S.; Lee, J.; Kim, B. Accurate determination of ochratoxin A in Korean fermented soybean paste by isotope dilution-liquid chromatography tandem mass spectrometry. Food Chem. 2016, 190, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kok, W.T. Derivatization reactions for the determination of aflatoxins by liquid chromatography with fluorescence detection. J. Chromatogr. B Biomed. Sci. Appl. 1994, 659, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Ok, H.E.; Kim, H.J.; Shim, W.B.; Lee, H.; Bae, D.H.; Chung, D.H.; Chun, H.S. Natural occurrence of aflatoxin B1 in marketed foods and risk estimates of dietary exposure in Koreans. J. Food. Protect. 2007, 70, 2824–2828. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Yoon, M.H.; Hong, H.G.; Joe, T.S.; Lee, I.S.; Park, J.H.; Ko, H.U. A survey of the presence of aflatoxins in food. J. Food Hyg. Saf. 2008, 23, 108–112. [Google Scholar]
- Kim, K.Y.; Nam, M.J.; Nam, B.R.; Ryu, H.J.; Song, J.E.; Shim, W.B.; Lee, S.H.; Jeong, D.H. Determination of total aflatoxins in foods by parallelism of ELISA and LC/MS/MS. JEHS 2010, 36, 52–60. [Google Scholar] [CrossRef]
- Kim, M.; Kim, Y.S. Detection of foodborne pathogens and analysis of aflatoxin levels in home-made doenjang samples. Prev. Nutr. Food Sci. 2012, 17, 172–176. [Google Scholar] [CrossRef]
- Park, J.W.; Yoo, M.S.; Kuk, J.H.; Ji, Y.A.; Lee, J.H. Simultaneous determination and monitoring of aflatoxin and ochratoxin A in food. J. Food Hyg. Saf. 2013, 28, 75–82. [Google Scholar] [CrossRef]
- Kim, D.H.; Hong, S.Y.; Kang, J.; Cho, S.; Lee, K.; An, T.; Lee, C.; Chung, S. Simultaneous determination of multi-mycotoxins in cereal grains collected from South Korea by LC/MS/MS. Toxins 2017, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Connolly, L.; Frizzell, C.; Elliott, C.T. Cytotoxic assessment of the regulated, co-existing mycotoxins aflatoxin B1, fumonisin B1 and ochratoxin, in single, binary and tertiary mixtures. Toxicon 2014, 90, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Vejdovszky, K.; Hahn, K.; Braun, D.; Warth, B.; Marko, D. Synergistic estrogenic effects of fusarium and alternaria mycotoxins in vitro. Arch. Toxicol. 2017, 91, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, S.; Bao, X.; Luo, C.; Yang, H.; Wang, J.; Zhao, S.; Zheng, N. Transcriptional and proteomic analysis revealed a synergistic effect of aflatoxin M1 and ochratoxin A mycotoxins on the intestinal epithelial integrity of differentiated human Caco-2 cells. J. Proteome Res. 2018, 17, 3128–3142. [Google Scholar] [CrossRef] [PubMed]
- Hird, S.J.; Lau, B.P.Y.; Schuhmacher, R.; Krska, R. Liquid chromatography-mass spectrometry for the determination of chemical contaminants in food. Trends Anal. Chem. 2014, 59, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Eshelli, M.; Qader, M.; Jambi, E.; Hursthouse, A.; Rateb, M. Current status and future opportunities of omics tools in mycotoxin research. Toxins 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Frenich, A.G.; Romero-González, R.; del Mar Aguilera-Luiz, M. Comprehensive analysis of toxics (pesticides, veterinary drugs and mycotoxins) in food by UHPLC-MS. Trends Anal. Chem. 2014, 63, 158–169. [Google Scholar] [CrossRef]
- Krska, R.; Schubert-Ullrich, P.; Molinelli, A.; Sulyok, M.; MacDonald, S.; Crews, C. Mycotoxin analysis: An update. Food Addit. Contam. 2008, 25, 152–163. [Google Scholar] [CrossRef]
- Pereira, V.; Fernandes, J.; Cunha, S. Mycotoxins in cereals and related foodstuffs: A review on occurrence and recent methods of analysis. Trends Food Sci. Technol. 2014, 36, 96–136. [Google Scholar] [CrossRef]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.; Powers, S.; Visconti, A. Simultaneous LC–MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef]
- Gambacorta, S.; Solfrizzo, H.; Visconti, A.; Powers, S.; Cossalter, A.; Pinton, P.; Oswald, I. Validation study on urinary biomarkers of exposure for aflatoxin B1, ochratoxin A, fumonisin B1, deoxynivalenol and zearalenone in piglets. World Mycotoxin J. 2013, 6, 299–308. [Google Scholar] [CrossRef]
- Wallin, S.; Gambacorta, L.; Kotova, N.; Lemming, E.W.; Nälsén, C.; Solfrizzo, M.; Olsen, M. Biomonitoring of concurrent mycotoxin exposure among adults in Sweden through urinary multi-biomarker analysis. Food Chem. Toxicol. 2015, 83, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Y.; Lin, H.Y.; Chen, Y.C.; Su, W.T.; Wang, S.C.; Chiueh, L.C.; Shin, Y.C. Development of a quantitative multi-mycotoxin method in rice, maize, wheat and peanut using UPLC-MS/MS. Food Anal. Methods 2013, 6, 727–736. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; Visconti, A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and fusarium toxins in cereals and derived products by liquid chromatography–tandem mass spectrometry after multi-toxin immunoaffinity clean up. J. Chromatogr. A 2014, 1354, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Choe, B.C.; Choi, E.J.; Jeong, H.J.; Hwang, Y.S.; Shin, G.Y.; Kim, J.H. Survey of mycotoxins in commonly consumed Korean grain products using an LC-MS/MS multimycotoxin method in combination with immunoaffinity clean-up. Food Sci. Biotechnol. 2015, 24, 1193–1199. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, D.H.; Moon, J.Y.; An, J.A.; Kim, Y.W.; Chung, S.H.; Lee, C. Distribution analysis of twelve mycotoxins in corn and corn-derived products by LC-MS/MS to evaluate the carry-over ratio during wet-milling. Toxins 2018, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef]
- Soleimany, F.; Jinap, S.; Faridah, A.; Khatib, A. A UPLC–MS/MS for simultaneous determination of aflatoxins, ochratoxin A, zearalenone, DON, fumonisins, T-2 toxin and HT-2 toxin, in cereals. Food Control. 2012, 25, 647–653. [Google Scholar] [CrossRef]
- Frenich, A.G.; Romero-González, R.; Gómez-Pérez, M.L.; Vidal, J.L. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar] [CrossRef]
- Dong, H.; Xian, Y.; Xiao, K.; Wu, Y.; Zhu, L.; He, J. Development and comparison of single-step solid phase extraction and QuEChERS clean-up for the analysis of 7 mycotoxins in fruits and vegetables during storage by UHPLC-MS/MS. Food Chem. 2019, 274, 471–479. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, C.; O’Kiely, P.; Elliott, C.; Danaher, M. Development and validation of an UHPLC-MS/MS method for the determination of mycotoxins in grass silages. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2015, 32, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, N.; Xian, H.; Wei, D.; Shi, L.; Feng, X. A single-step solid phase extraction for the simultaneous determination of 8 mycotoxins in fruits by ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1429, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gómez, J.V.; Gimeno-Adelantado, J.V.; Jiménez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Union. 2014, 147, 29–43.
- Commission Decision (EC) No 2002/657 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Commun. 2002, 8, 1–29.
- Tolosa, J.; Graziani, G.; Gaspari, A.; Chianese, D.; Ferrer, E.; Mañes, J.; Ritieni, A. Multi-mycotoxin analysis in durum wheat pasta by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Toxins 2017, 9, 59. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; González-Peñas, E. An LC–MS/MS method for multi-mycotoxin quantification in cow milk. Food Chem. 2017, 218, 378–385. [Google Scholar] [CrossRef]
- European Commission. Report on the Relationship between Analytical Results, Measurement Uncertainty, Recovery Factors and the Provisions of EU Food and Feed Legislation, with Particular Reference to Community Legislation Concerning. 2004, p. 8. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_sampling_analysis-eport_2004_en.pdf (accessed on 10 October 2019).
- Park, K.Y.; Hwang, K.M.; Jung, K.O.; Lee, K.B. Studies on the standardization of doenjang (Korean soybean paste) 1. Standardization of manufacturing method of doenjang by literatures. J. Korean Soc. Food Sci. Nutr. 2002, 31, 343–350. [Google Scholar]
- Sharma, G. Pros and cons of different sampling techniques. Int. J. Appl. Res. 2017, 3, 749–752. [Google Scholar]
- Korean Food Code. Standards and Specifications for Food Product; Ministry of Food and Drug Safety: Seoul, Korea, 2018. [Google Scholar]
- Andrade, G.; Pimpinato, R.F.; Francisco, J.G.; Monteiro, S.; Calori-Domingues, M.A.; Tornisielo, V.L. Evaluation of mycotoxins and their estimated daily intake in popcorn and cornflakes using LC-MS techniques. LWT Food Sci. Technol. 2018, 95, 240–246. [Google Scholar] [CrossRef]
- Ellison, S.; Williams, A. Eurachem/CITAC Guide. Quantifying Uncertainty in Analytical Measurement, 3rd ed.; EURACHEM/CITAC Guide CG 4: London, UK, 2012; Available online: https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf (accessed on 10 October 2019).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Molar Mass (g/mol) | Precursor Ion | Molecular Ion | Product Ion | Retention Time (min) | |
|---|---|---|---|---|---|---|
| Quantifier | Qualifier | |||||
| AFB1 | 312.277 | 313.2 | [M + H]+ | 285.0 | 284.0 | 10.8 |
| AFB2 | 314.293 | 315.2 | [M + H]+ | 287.0 | 297.0 | 10.4 |
| AFG1 | 328.276 | 329.2 | [M + H]+ | 311.0 | 301.0 | 10.2 |
| AFG2 | 330.292 | 331.2 | [M + H]+ | 313.0 | 303.0 | 9.8 |
| OTA | 403.815 | 404.8 | [M + H]+ | 358.0 | 387.0 | 13.3 |
| ZEN | 318.369 | 319.3 | [M + H]+ | 301.0 | 283.0 | 13.2 |
| ZAN | 320.385 | 321.0 | [M + H]+ | 303.1 | 277.1 | 13.1 |
| α-ZEL | 320.385 | 321.0 | [M + H]+ | 285.1 | 259.1 | 12.9 |
| α-ZAL | 322.401 | 305.0 | [M + H − H2O]+ | 287.1 | 277.1 | 12.8 |
| β-ZEL | 320.385 | 303.0 | [M + H − H2O]+ | 285.1 | 267.1 | 12.4 |
| β-ZAL | 322.401 | 305.0 | [M + H − H2O]+ | 287.1 | 277.0 | 12.1 |
| T-2 | 466.527 | 489.5 | [M + Na]+ | 387.09 | 327.09 | 12.7 |
| HT-2 | 424.49 | 447.4 | [M + Na]+ | 345.0 | 285.0 | 12.2 |
| NIV | 312.318 | 357.2 | [M + HCOO-]- | 281.0 | 311.0 | 3.4 |
| DON | 296.319 | 341.3 | [M + HCOO-]- | 265.0 | 295.0 | 6.1 |
| 3ADON | 338.356 | 339.3 | [M + H]+ | 231.0 | 279.0 | 9.4 |
| D3G | 458.46 | 503.4 | [M + HCOO-]- | 457.1 | 427.1 | 6.6 |
| FB1 | 721.838 | 722.8 | [M + H]+ | 704.2 | 705.2 | 11.8 |
| FB2 | 705.839 | 706.8 | [M + H]+ | 688.3 | 530.3 | 12.8 |
| FB3 | 705.839 | 706.8 | [M + H]+ | 688.3 | 530.3 | 12.4 |
| Mycotoxin | Calibration Curve | R2 | LOD (μg/kg) | LOQ (μg/kg) | SSE (%) | ||
|---|---|---|---|---|---|---|---|
| Calibration Range (μg/kg) | Slope | Intercept | |||||
| AFB1 | 0.5−20 | 10,644.3 | −142.4 | 0.9999 | 0.08 | 0.26 | 89.7 |
| AFB2 | 0.5−20 | 8025.6 | −834.4 | 0.9998 | 0.14 | 0.43 | 94.6 |
| AFG1 | 0.5−20 | 8873.9 | 394.8 | 0.9999 | 0.10 | 0.30 | 100.8 |
| AFG2 | 0.5−20 | 6878.5 | −722.1 | 0.9999 | 0.15 | 0.45 | 98.7 |
| OTA | 0.5−20 | 20,887.2 | 8984.6 | 0.9998 | 0.06 | 0.17 | 87.0 |
| ZEN | 25−1000 | 4638.2 | −5420.4 | 0.9999 | 4.68 | 14.18 | 96.7 |
| ZAN | 25−1000 | 11,972.7 | −8964.7 | 1.0000 | 1.66 | 5.03 | 95.5 |
| α-ZEL | 25−1000 | 700.0 | −716.6 | 0.9999 | 1.47 | 4.44 | 94.2 |
| α-ZAL | 25−1000 | 2098.0 | 2773.7 | 0.9998 | 2.29 | 6.94 | 95.9 |
| β-ZEL | 25−1000 | 3649.0 | −12,582.2 | 1.0000 | 1.72 | 5.23 | 99.5 |
| β-ZAL | 25−1000 | 2042.0 | 8338.2 | 0.9997 | 2.13 | 6.45 | 101.9 |
| T-2 | 25−1000 | 23,047.8 | 411,783.3 | 0.9995 | 2.55 | 7.73 | 103.8 |
| HT-2 | 25−1000 | 18,293.4 | 257,935.3 | 0.9994 | 1.86 | 5.65 | 93.1 |
| NIV | 25−1000 | 1423.3 | −6891.8 | 0.9999 | 3.21 | 9.74 | 98.2 |
| DON | 25−1000 | 941.3 | −3171.0 | 1.0000 | 4.37 | 13.24 | 98.0 |
| 3ADON | 25−1000 | 821.9 | −5844.9 | 0.9999 | 0.9 | 2.73 | 100.4 |
| D3G | 25−1000 | 6246.2 | −12,849.3 | 0.9999 | 2.72 | 8.24 | 100.3 |
| FB1 | 25−1000 | 22,786.0 | −127,037.2 | 0.9998 | 2.57 | 7.78 | 98.1 |
| FB2 | 25−1000 | 24,992.8 | −211,390.0 | 1.0000 | 2.16 | 6.56 | 87.1 |
| FB3 | 25−1000 | 19,609.8 | −21,763.0 | 1.0000 | 2.22 | 6.72 | 86.5 |
| Mycotoxin | %Recovery (%RSD) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Day (n = 9) | Inter-Day (n = 9) | |||||||||||||||
| 2 × LOQ * | 5 × LOQ | 10 × LOQ | 20 × LOQ | 2 × LOQ | 5 × LOQ | 10 × LOQ | 20 × LOQ | |||||||||
| AFB1 | 98.4 | (5.9) | 97.8 | (2.7) | 97.5 | (4.3) | 101.5 | (3.1) | 96.5 | (9.1) | 93.2 | (4.8) | 94.7 | (4.9) | 97.8 | (5.6) |
| AFB2 | 100.5 | (4.7) | 97.0 | (3.9) | 99.1 | (2.2) | 101.1 | (2.3) | 100.6 | (3.0) | 95.8 | (2.8) | 97.6 | (3.7) | 99.1 | (3.8) |
| AFG1 | 100.0 | (4.0) | 95.6 | (3.9) | 95.4 | (2.8) | 95.7 | (2.8) | 94.7 | (5.3) | 94.7 | (4.3) | 93.0 | (5.1) | 93.0 | (5.2) |
| AFG2 | 96.6 | (2.2) | 94.0 | (2.0) | 95.1 | (2.2) | 96.3 | (4.4) | 95.7 | (6.4) | 92.3 | (5.5) | 93.4 | (5.3) | 89.6 | (9.2) |
| OTA | 80.9 | (7.2) | 76.1 | (5.3) | 83.0 | (6.5) | 79.9 | (6.1) | 86.0 | (5.0) | 71.7 | (2.7) | 76.9 | (6.4) | 74.6 | (3.3) |
| ZEN | 89.4 | (3.6) | 85.7 | (5.0) | 83.1 | (7.3) | 84.1 | (5.1) | 90.8 | (5.4) | 83.3 | (5.1) | 83.8 | (8.7) | 85.2 | (6.5) |
| ZAN | 99.3 | (2.2) | 85.0 | (3.1) | 79.3 | (6.8) | 77.6 | (4.8) | 100.2 | (2.2) | 85.1 | (3.7) | 80.3 | (6.8) | 80.0 | (4.8) |
| α-ZEL | 104.5 | (3.3) | 93.4 | (5.0) | 88.6 | (6.2) | 85.1 | (4.6) | 103.6 | (4.0) | 92.0 | (4.5) | 87.9 | (9.1) | 86.7 | (5.8) |
| α-ZAL | 89.4 | (8.2) | 85.8 | (11.3) | 88.0 | (6.1) | 90.8 | (4.2) | 88.0 | (7.5) | 85.7 | (8.1) | 87.0 | (8.7) | 90.8 | (6.3) |
| β-ZEL | 105.6 | (3.2) | 89.4 | (3.9) | 83.5 | (7.4) | 82.0 | (4.0) | 109.6 | (3.1) | 91.0 | (3.3) | 85.0 | (7.3) | 84.1 | (3.6) |
| β-ZAL | 92.7 | (5.0) | 90.1 | (4.8) | 87.9 | (6.6) | 90.2 | (4.5) | 93.6 | (5.3) | 90.5 | (5.8) | 89.7 | (6.5) | 91.6 | (4.4) |
| T-2 | 75.7 | (16.2) | 92.0 | (10.4) | 96.0 | (12.6) | 94.7 | (12.6) | 75.0 | (4.5) | 91.9 | (6.0) | 96.4 | (8.2) | 97.6 | (6.6) |
| HT-2 | 79.2 | (5.3) | 74.3 | (13.7) | 76.6 | (12.5) | 83.0 | (11.5) | 86.8 | (5.8) | 72.6 | (13.2) | 78.9 | (12.7) | 82.1 | (15.2) |
| NIV | 95.2 | (15.2) | 89.5 | (7.4) | 89.5 | (12.6) | 84.3 | (8.9) | 112.3 | (5.9) | 97.1 | (7.6) | 86.7 | (13.4) | 85.5 | (8.1) |
| DON | 100.3 | (6.6) | 94.9 | (6.3) | 95.5 | (8.2) | 89.7 | (6.6) | 109.0 | (6.4) | 100.1 | (6.5) | 90.6 | (9.7) | 89.6 | (5.1) |
| 3ADON | 103.2 | (4.0) | 91.5 | (6.3) | 90.5 | (7.4) | 87.0 | (10.0) | 107.2 | (1.8) | 95.1 | (4.9) | 96.1 | (3.8) | 93.6 | (5.1) |
| D3G | 95.2 | (11.7) | 93.2 | (5.2) | 93.2 | (13.0) | 85.8 | (8.8) | 106.4 | (6.4) | 95.8 | (5.1) | 88.1 | (13.0) | 85.3 | (8.0) |
| FB1 | 94.3 | (11.0) | 99.5 | (8.0) | 95.1 | (5.1) | 83.8 | (6.7) | 93.2 | (8.2) | 103.5 | (7.4) | 92.3 | (13.2) | 90.3 | (7.9) |
| FB2 | 100.3 | (4.7) | 87.6 | (11.5) | 89.1 | (6.9) | 81.1 | (9.6) | 103.5 | (13.5) | 88.9 | (11.8) | 86.5 | (12.8) | 84.8 | (10.0) |
| FB3 | 96.6 | (4.1) | 87.0 | (11.2) | 91.2 | (6.8) | 82.2 | (5.8) | 97.6 | (11.7) | 88.6 | (9.4) | 88.8 | (12.9) | 84.2 | (10.3) |
| Mycotoxin | Spiking Level (μg/kg) | Expanded Uncertainty | Uncertainty/Result (%) | Mycotoxin | Spiking Level (μg/kg) | Expanded Uncertainty | Uncertainty/Result (%) |
|---|---|---|---|---|---|---|---|
| AFB1 | 1 | 0.23 | 22.7 | β-ZAL | 15 | 1.18 | 15.8 |
| 2 | 0.31 | 15.4 | 37 | 3.28 | 17.7 | ||
| 3 | 0.73 | 14.6 | 74 | 7.53 | 20.3 | ||
| 6 | 1.26 | 12.6 | 148 | 12.93 | 17.5 | ||
| AFB2 | 1 | 0.20 | 19.8 | T-2 | 15 | 2.38 | 31.7 |
| 2 | 0.31 | 15.3 | 37 | 2.40 | 13.0 | ||
| 3 | 0.69 | 13.8 | 74 | 2.92 | 7.9 | ||
| 6 | 1.32 | 13.2 | 148 | 6.02 | 8.1 | ||
| AFG1 | 1 | 0.20 | 20.3 | HT-2 | 15 | 2.86 | 19.1 |
| 2 | 0.31 | 15.5 | 37 | 7.23 | 39.1 | ||
| 3 | 0.72 | 14.4 | 74 | 9.85 | 26.6 | ||
| 6 | 1.39 | 13.9 | 148 | 18.92 | 25.6 | ||
| AFG2 | 1 | 0.20 | 19.6 | NIV | 15 | 1.10 | 14.7 |
| 2 | 0.32 | 15.8 | 37 | 1.76 | 9.5 | ||
| 3 | 0.72 | 14.3 | 74 | 4.33 | 11.7 | ||
| 6 | 1.35 | 13.5 | 148 | 11.86 | 16.0 | ||
| OTA | 1 | 0.41 | 41.3 | DON | 15 | 0.69 | 9.2 |
| 2 | 0.83 | 41.6 | 37 | 1.89 | 10.2 | ||
| 3 | 1.42 | 28.4 | 74 | 3.88 | 10.5 | ||
| 6 | 3.11 | 31.1 | 148 | 10.49 | 14.2 | ||
| ZEN | 15 | 2.02 | 13.4 | 3ADON | 15 | 0.85 | 11.4 |
| 37 | 7.21 | 19.5 | 37 | 2.18 | 11.8 | ||
| 74 | 16.66 | 22.5 | 74 | 4.30 | 11.6 | ||
| 148 | 29.22 | 19.7 | 148 | 10.16 | 13.7 | ||
| ZAN | 15 | 1.95 | 13.0 | D3G | 15 | 0.78 | 10.3 |
| 37 | 8.56 | 23.1 | 37 | 1.50 | 8.1 | ||
| 74 | 22.02 | 29.8 | 74 | 3.09 | 8.3 | ||
| 148 | 47.40 | 32.0 | 148 | 11.45 | 15.5 | ||
| α-ZEL | 15 | 2.01 | 13.4 | FB1 | 15 | 1.00 | 6.7 |
| 37 | 5.84 | 15.8 | 37 | 1.22 | 6.6 | ||
| 74 | 14.41 | 19.5 | 74 | 2.40 | 6.5 | ||
| 148 | 33.21 | 22.4 | 148 | 10.74 | 14.5 | ||
| α-ZAL | 15 | 2.66 | 17.8 | FB2 | 15 | 0.50 | 6.7 |
| 37 | 7.80 | 21.1 | 37 | 2.86 | 15.4 | ||
| 74 | 14.86 | 20.1 | 74 | 4.80 | 13.0 | ||
| 148 | 25.70 | 17.4 | 148 | 17.58 | 23.8 | ||
| β-ZEL | 15 | 2.25 | 15.0 | FB3 | 15 | 0.53 | 7.0 |
| 37 | 6.57 | 17.7 | 37 | 2.77 | 15.0 | ||
| 74 | 17.36 | 23.5 | 74 | 3.28 | 8.9 | ||
| 148 | 38.32 | 25.9 | 148 | 16.22 | 21.9 |
| Mycotoxin | Commercial doenjang (μg/kg), n = 30 | Homemade doenjang (μg/kg), n = 30 | ||||||
|---|---|---|---|---|---|---|---|---|
| Incidence a | Mean (Positive Mean) | Median | Range | Incidence a | Mean (Positive Mean) | Median | Range | |
| AFB1 * | 5/30 | 0.07 (0.40) | 0.33 | 0.11−0.96 | 11/30 | 0.54 (1.48) | 0.65 | 0.19−4.45 |
| AFB2 | 3/30 | 0.08 (0.77) | 0.77 | 0.70−0.84 | 5/30 | 0.14 (0.82) | 0.87 | 0.62−0.98 |
| AFG1 * | 0/30 | - b | - | - | 5/30 | 0.10 (0.60) | 0.57 | 0.49−0.77 |
| AFG2 | 0/30 | - | - | - | 2/30 | 0.03 (0.47) | 0.47 | 0.21−0.73 |
| OTA | 5/30 | 0.80 (4.80) | 0.19 | 0.16−23.27 | 18/30 | 3.05 (5.08) | 3.78 | 0.20−19.68 |
| ZEN ** | 4/30 | 2.79 (20.90) | 17.47 | 5.16−43.48 | 17/30 | 17.82 (31.45) | 19.40 | 5.86−85.50 |
| ZAN * | 0/30 | - | - | - | 5/30 | 0.94 (5.67) | 6.18 | 3.28−7.11 |
| α-ZEL | 0/30 | - | - | - | 1/30 | 0.28 (8.29) | 8.29 | 8.29 |
| α-ZAL | 2/30 | 0.34 (5.17) | 5.17 | 5.13−5.21 | 6/30 | 1.04 (5.21) | 5.01 | 4.05−7.18 |
| β-ZEL * | 0/30 | - | - | - | 5/30 | 1.56 (9.39) | 9.91 | 7.38−11.56 |
| β-ZAL | 5/30 | 0.93 (5.56) | 5.64 | 4.67−6.19 | 3/30 | 1.66 (16.56) | 15.59 | 8.40−25.70 |
| T-2 | 0/30 | - | - | - | 0/30 | - | - | - |
| HT-2 | 1/30 | 0.88 (26.52) | 26.52 | 26.52 | 1/30 | 0.06 (1.69) | 1.69 | 1.69 |
| NIV | 1/30 | 0.26 (7.67) | 7.67 | 7.67 | 6/30 | 0.89 (4.43) | 4.31 | 3.27−6.03 |
| DON | 4/30 | 1.72 (12.88) | 14.25 | 4.78−18.25 | 1/30 | 0.51 (15.35) | 15.35 | 15.35 |
| 3ADON | 0/30 | - | - | - | 0/30 | - | - | - |
| D3G | 0/30 | - | - | - | 1/30 | 0.09 (2.72) | 2.72 | 2.72 |
| FB1 ** | 8/30 | 1.50 (5.61) | 6.64 | 2.21−8.13 | 16/30 | 5.04 (9.44) | 9.39 | 5.52−17.26 |
| FB2 * | 8/30 | 1.57 (5.90) | 6.20 | 1.85−9.11 | 16/30 | 7.00 (13.33) | 6.03 | 3.86−56.19 |
| FB3 | 9/30 | 1.68 (5.60) | 6.10 | 2.48−9.22 | 12/30 | 2.00 (5.00) | 4.96 | 2.37−9.36 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, S.Y.; Ryu, S.Y.; Tian, F.; Lee, S.Y.; Park, S.B.; Chun, H.S. Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup. Toxins 2019, 11, 594. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100594
Woo SY, Ryu SY, Tian F, Lee SY, Park SB, Chun HS. Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup. Toxins. 2019; 11(10):594. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100594
Chicago/Turabian StyleWoo, So Young, So Young Ryu, Fei Tian, Sang Yoo Lee, Su Been Park, and Hyang Sook Chun. 2019. "Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup" Toxins 11, no. 10: 594. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100594