Contribution and Interaction of Shiga Toxin Genes to Escherichia coli O157:H7 Virulence

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Risk of HUS
2.2. Risk of HUS in Children
2.3. Risk of Renal Replacement Therapy
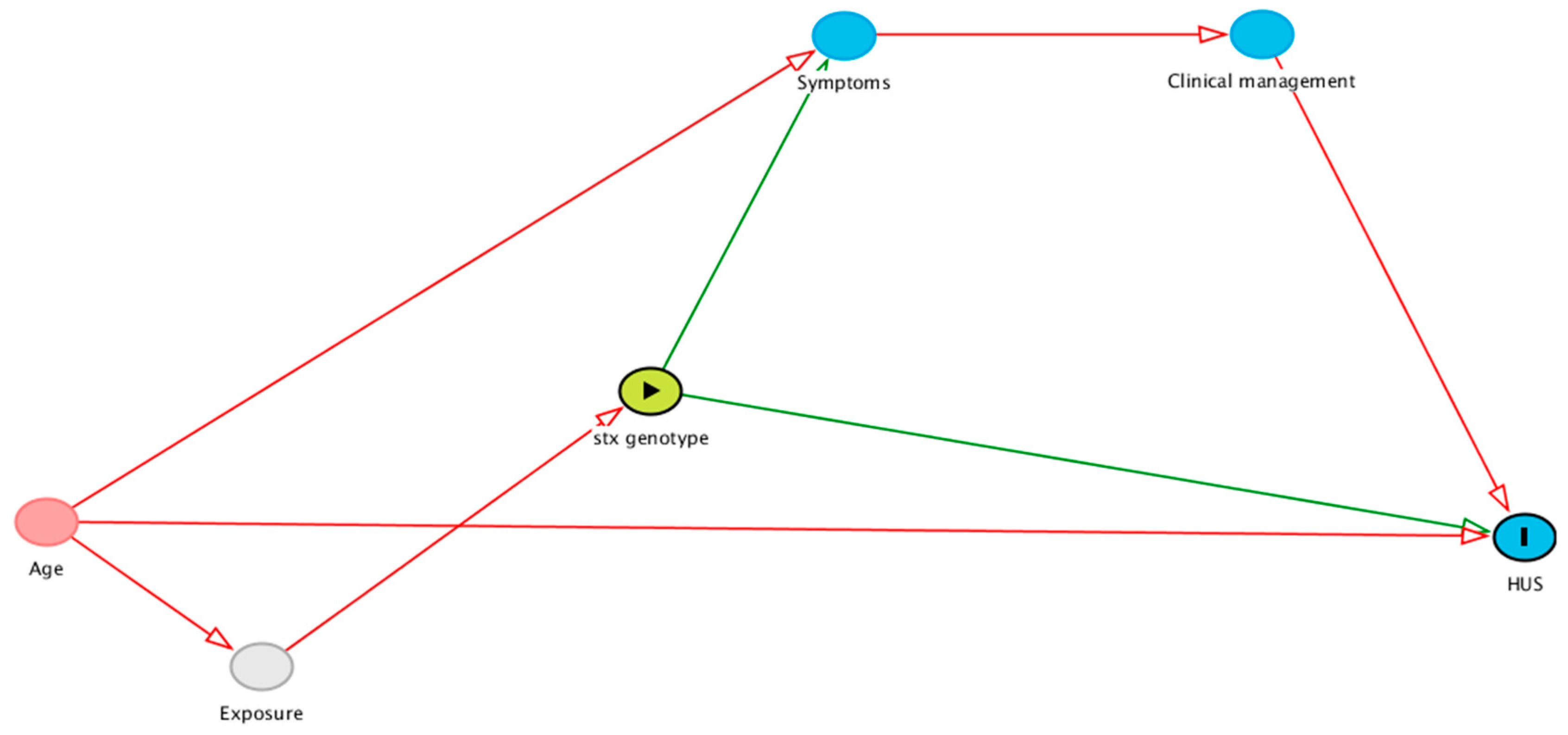
2.4. stx Allelic Interaction
2.5. Sensitivity Analysis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Study Population
5.2. Genotyping
5.3. Statistical Analysis
Sensitivity Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banatvala, N.; Griffin, P.M.; Greene, K.D.; Barrett, T.J.; Bibb, W.F.; Green, J.H.; Wells, J.G. The United States National Prospective Hemolytic Uremic Syndrome Study: Microbiologic, serologic, clinical, and epidemiologic findings. J. Infect. Dis. 2001, 183, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000–2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Mody, R.K.; Gu, W.; Griffin, P.M.; Jones, T.F.; Rounds, J.; Shiferaw, B.; Tobin-D’Angelo, M.; Smith, G.; Spina, N.; Hurd, S.; et al. Postdiarrheal hemolytic uremic syndrome in United States children: Clinical spectrum and predictors of in-hospital death. J. Pediatr. 2015, 166, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- McKee, R.S.; Schnadower, D.; Tarr, P.I.; Xie, J.; Finkelstein, Y.; Desai, N.; Lane, R.D.; Bergmann, K.R.; Kaplan, R.L.; Hariharan, S.; et al. Predicting Hemolytic Uremic Syndrome and Renal Failure in Shiga Toxin-Producing Escherichia coli Infected Children. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Persson, S.; Olsen, K.E.; Ethelberg, S.; Scheutz, F. Subtyping method for Escherichia coli shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef]
- Eklund, M.; Leino, K.; Siitonen, A. Clinical Escherichia coli strains carrying stx genes: Stx variants and stx-positive virulence profiles. J. Clin Microbiol. 2002, 40, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef]
- Luna-Gierke, R.E.; Griffin, P.M.; Gould, L.H.; Herman, K.; Bopp, C.A.; Strockbine, N.; Mody, R.K. Outbreaks of non-O157 Shiga toxin-producing Escherichia coli infection: USA. Epidemiol. Infect. 2014, 142, 2270–2280. [Google Scholar] [CrossRef]
- Orth, D.; Grif, K.; Khan, A.B.; Naim, A.; Dierich, M.P.; Wurzner, R. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 2007, 59, 235–242. [Google Scholar] [CrossRef]
- Ostroff, S.M.; Tarr, P.I.; Neill, M.A.; Lewis, J.H.; Hargrett-Bean, N.; Kobayashi, J.M. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 1989, 160, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Cornick, N.A.; Jelacic, S.; Ciol, M.A.; Tarr, P.I. Escherichia coli O157:H7 infections: Discordance between filterable fecal shiga toxin and disease outcome. J. Infect. Dis. 2002, 186, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Donohue-Rolfe, A.; Kondova, I.; Oswald, S.; Hutto, D.; Tzipori, S. Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J. Infect. Dis. 2000, 181, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Petro, C.D.; Trojnar, E.; Sinclair, J.; Liu, Z.M.; Smith, M.; O’Brien, A.D.; Melton-Celsa, A. Shiga Toxin Type 1a (Stx1a) Reduces the Toxicity of the More Potent Stx2a In Vivo and In Vitro. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [PubMed]
- Jelacic, S.; Wobbe, C.L.; Boster, D.R.; Ciol, M.A.; Watkins, S.L.; Tarr, P.I.; Stapleton, A.E. ABO and P1 blood group antigen expression and stx genotype and outcome of childhood Escherichia coli O157:H7 infections. J. Infect. Dis. 2002, 185, 214–219. [Google Scholar] [CrossRef]
- Gouveia, S.; Proctor, M.E.; Lee, M.S.; Luchansky, J.B.; Kaspar, C.W. Genomic comparisons and Shiga toxin production among Escherichia coli O157:H7 isolates from a day care center outbreak and sporadic cases in southeastern Wisconsin. J. Clin. Microbiol. 1998, 36, 727–733. [Google Scholar]
- Iyoda, S.; Manning, S.D.; Seto, K.; Kimata, K.; Isobe, J.; Etoh, Y.; Ichihara, S.; Migita, Y.; Ogata, K.; Honda, M.; et al. Phylogenetic Clades 6 and 8 of Enterohemorrhagic Escherichia coli O157:H7 With Particular stx Subtypes are More Frequently Found in Isolates From Hemolytic Uremic Syndrome Patients Than From Asymptomatic Carriers. Open Forum Infect. Dis. 2014, 1, ofu061. [Google Scholar] [CrossRef]
- Kawano, K.; Okada, M.; Haga, T.; Maeda, K.; Goto, Y. Relationship between pathogenicity for humans and stx genotype in Shiga toxin-producing Escherichia coli serotype O157. Eur. J. Clin. Microbiol. Infect. Dis. 2008, 27, 227–232. [Google Scholar] [CrossRef]
- Tostes, R.; Goji, N.; Amoako, K.; Chui, L.; Kastelic, J.; DeVinney, R.; Stanford, K.; Reuter, T. Subtyping Escherichia coli Virulence Genes Isolated from Feces of Beef Cattle and Clinical Cases in Alberta. Foodborne Pathog. Dis. 2017, 14, 35–42. [Google Scholar] [CrossRef]
- Ashton, P.M.; Perry, N.; Ellis, R.; Petrovska, L.; Wain, J.; Grant, K.A.; Jenkins, C.; Dallman, T.J. Insight into Shiga toxin genes encoded by Escherichia coli O157 from whole genome sequencing. PeerJ 2015, 3, e739. [Google Scholar] [CrossRef]
- Leotta, G.A.; Miliwebsky, E.S.; Chinen, I.; Espinosa, E.M.; Azzopardi, K.; Tennant, S.M.; Robins-Browne, R.M.; Rivas, M. Characterisation of Shiga toxin-producing Escherichia coli O157 strains isolated from humans in Argentina, Australia and New Zealand. BMC Microbiol. 2008, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Tarr, G.A.; Shringi, S.; Phipps, A.I.; Besser, T.E.; Mayer, J.; Oltean, H.N.; Wakefield, J.; Tarr, P.I.; Rabinowitz, P. Geogenomic Segregation and Temporal Trends of Human Pathogenic Escherichia coli O157:H7, Washington, USA, 2005–2014(1). Emerg. Infect. Dis. 2018, 24, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.M.; Melton-Celsa, A.R.; O’Brien, A.D. Shiga Toxin (Stx) Type 1a Reduces the Oral Toxicity of Stx Type 2a. J. Infect. Dis. 2016, 213, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarr, G.A.; Shringi, S.; Oltean, H.N.; Mayer, J.; Rabinowitz, P.; Wakefield, J.; Tarr, P.I.; Besser, T.E.; Phipps, A.I. Importance of case age in the purported association between phylogenetics and hemolytic uremic syndrome in Escherichia coli O157:H7 infections. Epidemiol. Infect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.; Tarr, P.I. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: Integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 2003, 185, 3596–3605. [Google Scholar] [CrossRef] [PubMed]
- Leopold, S.R.; Shaikh, N.; Tarr, P.I. Further evidence of constrained radiation in the evolution of pathogenic Escherichia coli O157:H7. Infect. Genet. Evol. 2010, 10, 1282–1285. [Google Scholar] [CrossRef]
- Trine, M.L.; Jørgensen, H.J.; O’Sullivan, K.; Bohlin, J.; Ligård, G.; Granum, P.E.; Lindbäck, T. The highly virulent 2006 Norwegian EHEC O103:H25 outbreak strain is related to the 2011 German O104:H4 outbreak strain. PLoS ONE 2012, 7, e31413. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Prager, R.; Köck, R.; Mellmann, A.; Zhang, W.; Tschäpe, H.; Tarr, P.I.; Karch, H. Shiga toxin gene loss and transfer in vitro and in vivo during enterohemorrhagic Escherichia coli O26 infection in humans. Appl. Environ. Microbiol. 2007, 73, 3144–3150. [Google Scholar] [CrossRef]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.J.; O’Brien, A.D. Comparisons of native Shiga toxins (Stxs) type 1 and 2 with chimeric toxins indicate that the source of the binding subunit dictates degree of toxicity. PLoS ONE 2014, 9, e93463. [Google Scholar] [CrossRef]
- Meites, S.; Buffone, G.J. Pediatric Clinical Chemistry: Reference (Normal) Values; AACC Press: Washington, DC, USA, 1989. [Google Scholar]
- Jung, W.K.; Bono, J.L.; Clawson, M.L.; Leopold, S.R.; Shringi, S.; Besser, T.E. Lineage and genogroup-defining single nucleotide polymorphisms of Escherichia coli O157:H7. Appl. Environ. Microbiol. 2013, 79, 7036–7041. [Google Scholar] [CrossRef]
- Shringi, S.; Schmidt, C.; Katherine, K.; Brayton, K.A.; Hancock, D.D.; Besser, T.E. Carriage of stx2a differentiates clinical and bovine-biased strains of Escherichia coli O157. PLoS ONE 2012, 7, e51572. [Google Scholar] [CrossRef] [PubMed]
- Chui, L.; Couturier, M.R.; Chiu, T.; Wang, G.; Olson, A.B.; McDonald, R.R.; Antonishyn, N.A.; Horsman, G.; Gilmour, M.W. Comparison of Shiga toxin-producing Escherichia coli detection methods using clinical stool samples. J. Mol. Diagn. 2010, 12, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Zhi, S.; Szelewicki, J.; Ziebell, K.; Parsons, B.; Chui, L. General detection of Shiga toxin 2 and subtyping of Shiga toxin 1 and 2 in Escherichia coli using qPCR. J. Microbiol. Methods 2019, 159, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| stx1a2a (n = 439) | stx2a-only (n = 225) | stx2a2c (n = 190) | Other (n = 82) | Overall (n = 936) | |
|---|---|---|---|---|---|
| Age (years) | |||||
| Median (IQR) | 18.0 (5.00, 42.5) | 11.0 (4.00, 30.0) | 12.0 (4.00, 33.0) | 23.0 (8.00, 46.8) | 16.0 (5.00, 39.3) |
| Comorbidity | |||||
| Present | 50 (11.4%) | 26 (11.6%) | 18 (9.5%) | 7 (8.5%) | 101 (10.8%) |
| Absent | 350 (79.7%) | 184 (81.8%) | 158 (83.2%) | 69 (84.1%) | 761 (81.3%) |
| Missing | 39 (8.9%) | 15 (6.7%) | 14 (7.4%) | 6 (7.3%) | 74 (7.9%) |
| Outbreak-related | |||||
| Yes | 42 (9.6%) | 32 (14.2%) | 14 (7.4%) | 1 (1.2%) | 89 (9.5%) |
| No | 397 (90.4%) | 193 (85.8%) | 176 (92.6%) | 81 (98.8%) | 847 (90.5%) |
| Diarrhea | |||||
| Present | 433 (98.6%) | 221 (98.2%) | 187 (98.4%) | 81 (98.8%) | 922 (98.5%) |
| Absent | 2 (0.5%) | 3 (1.3%) | 1 (0.5%) | 0 (0%) | 6 (0.6%) |
| Missing | 4 (0.9%) | 1 (0.4%) | 2 (1.1%) | 1 (1.2%) | 8 (0.9%) |
| Blood in stool | |||||
| Present | 398 (90.7%) | 189 (84.0%) | 161 (84.7%) | 61 (74.4%) | 809 (86.4%) |
| Absent | 31 (7.1%) | 32 (14.2%) | 22 (11.6%) | 20 (24.4%) | 105 (11.2%) |
| Missing | 10 (2.3%) | 4 (1.8%) | 7 (3.7%) | 1 (1.2%) | 22 (2.4%) |
| Vomiting | |||||
| Present | 204 (46.5%) | 121 (53.8%) | 102 (53.7%) | 28 (34.1%) | 455 (48.6%) |
| Absent | 223 (50.8%) | 103 (45.8%) | 82 (43.2%) | 52 (63.4%) | 460 (49.1%) |
| Missing | 12 (2.7%) | 1 (0.4%) | 6 (3.2%) | 2 (2.4%) | 21 (2.2%) |
| Abdominal pain | |||||
| Present | 408 (92.9%) | 204 (90.7%) | 175 (92.1%) | 71 (86.6%) | 858 (91.7%) |
| Absent | 16 (3.6%) | 13 (5.8%) | 8 (4.2%) | 8 (9.8%) | 45 (4.8%) |
| Missing | 15 (3.4%) | 8 (3.6%) | 7 (3.7%) | 3 (3.7%) | 33 (3.5%) |
| Fever | |||||
| Present | 167 (38.0%) | 78 (34.7%) | 70 (36.8%) | 20 (24.4%) | 335 (35.8%) |
| Absent | 245 (55.8%) | 132 (58.7%) | 107 (56.3%) | 53 (64.6%) | 537 (57.4%) |
| Missing | 27 (6.2%) | 15 (6.7%) | 13 (6.8%) | 9 (11.0%) | 64 (6.8%) |
| Hospitalized | |||||
| Yes | 167 (38.0%) | 93 (41.3%) | 85 (44.7%) | 20 (24.4%) | 365 (39.0%) |
| No | 263 (59.9%) | 130 (57.8%) | 105 (55.3%) | 60 (73.2%) | 558 (59.6%) |
| Missing | 9 (2.1%) | 2 (0.9%) | 0 (0%) | 2 (2.4%) | 13 (1.4%) |
| HUS | |||||
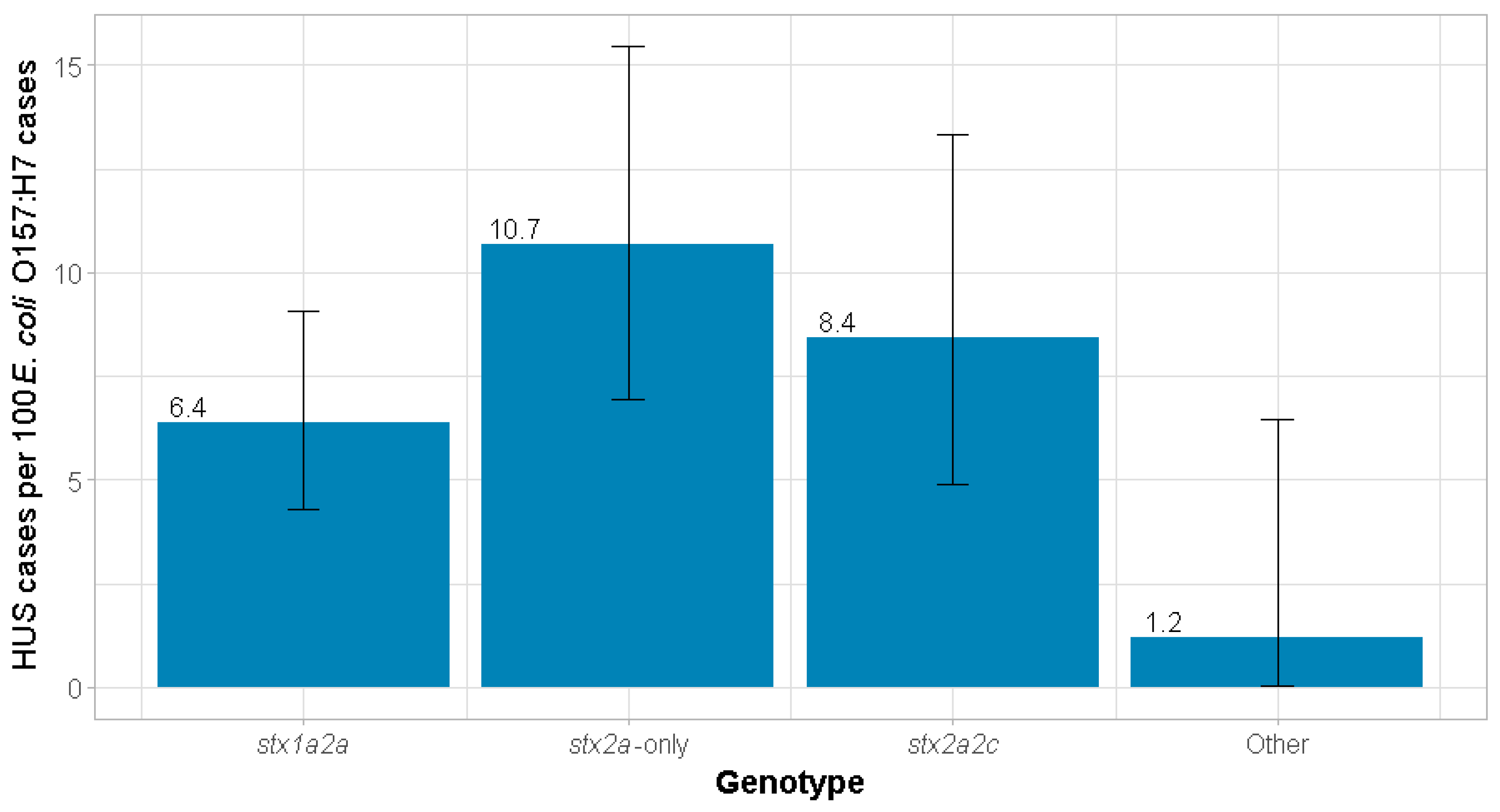
| Yes | 28 (6.4%) | 24 (10.7%) | 16 (8.4%) | 1 (1.2%) | 69 (7.4%) |
| No | 411 (93.6%) | 201 (89.3%) | 174 (91.6%) | 81 (98.8%) | 867 (92.6%) |
| RRT | |||||
| Yes | 11 (2.5%) | 14 (6.2%) | 10 (5.3%) | 1 (1.2%) | 36 (3.8%) |
| No | 428 (97.5%) | 211 (93.8%) | 180 (94.7%) | 81 (98.8%) | 900 (96.2%) |
| stx2a vs. | RD (95% CI) | RR (95% CI) | ||||
|---|---|---|---|---|---|---|
| Crude | Age-Adjusted | Fully Adjusted | Crude | Age-Adjusted | Fully Adjusted | |
| stx1a2a | 0.043 (−0.003, 0.089) | 0.036 (−0.010, 0.082) | 0.044 (−0.003, 0.091) | 1.67 (0.99, 2.82) | 1.52 (0.9, 2.54) | 1.58 (0.98, 2.56) |
| stx2a2c | 0.022 (−0.034, 0.079) | 0.021 (−0.035, 0.077) | 0.03 (−0.026, 0.087) | 1.27 (0.69, 2.31) | 1.24 (0.68, 2.25) | 1.43 (0.83, 2.45) |
| Other | 0.095 (0.048, 0.141) | 0.082 (0.037, 0.127) | 0.058 (0.013, 0.103) | 8.96 (1.23, 65.2) | 7.28 (0.98, 54.14) | 4.65 (0.7, 31.08) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarr, G.A.M.; Stokowski, T.; Shringi, S.; Tarr, P.I.; Freedman, S.B.; Oltean, H.N.; Rabinowitz, P.M.; Chui, L. Contribution and Interaction of Shiga Toxin Genes to Escherichia coli O157:H7 Virulence. Toxins 2019, 11, 607. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100607
Tarr GAM, Stokowski T, Shringi S, Tarr PI, Freedman SB, Oltean HN, Rabinowitz PM, Chui L. Contribution and Interaction of Shiga Toxin Genes to Escherichia coli O157:H7 Virulence. Toxins. 2019; 11(10):607. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100607
Chicago/Turabian StyleTarr, Gillian A.M., Taryn Stokowski, Smriti Shringi, Phillip I. Tarr, Stephen B. Freedman, Hanna N. Oltean, Peter M. Rabinowitz, and Linda Chui. 2019. "Contribution and Interaction of Shiga Toxin Genes to Escherichia coli O157:H7 Virulence" Toxins 11, no. 10: 607. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100607