A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners



Abstract
:1. Introduction
2. Results and Discussion
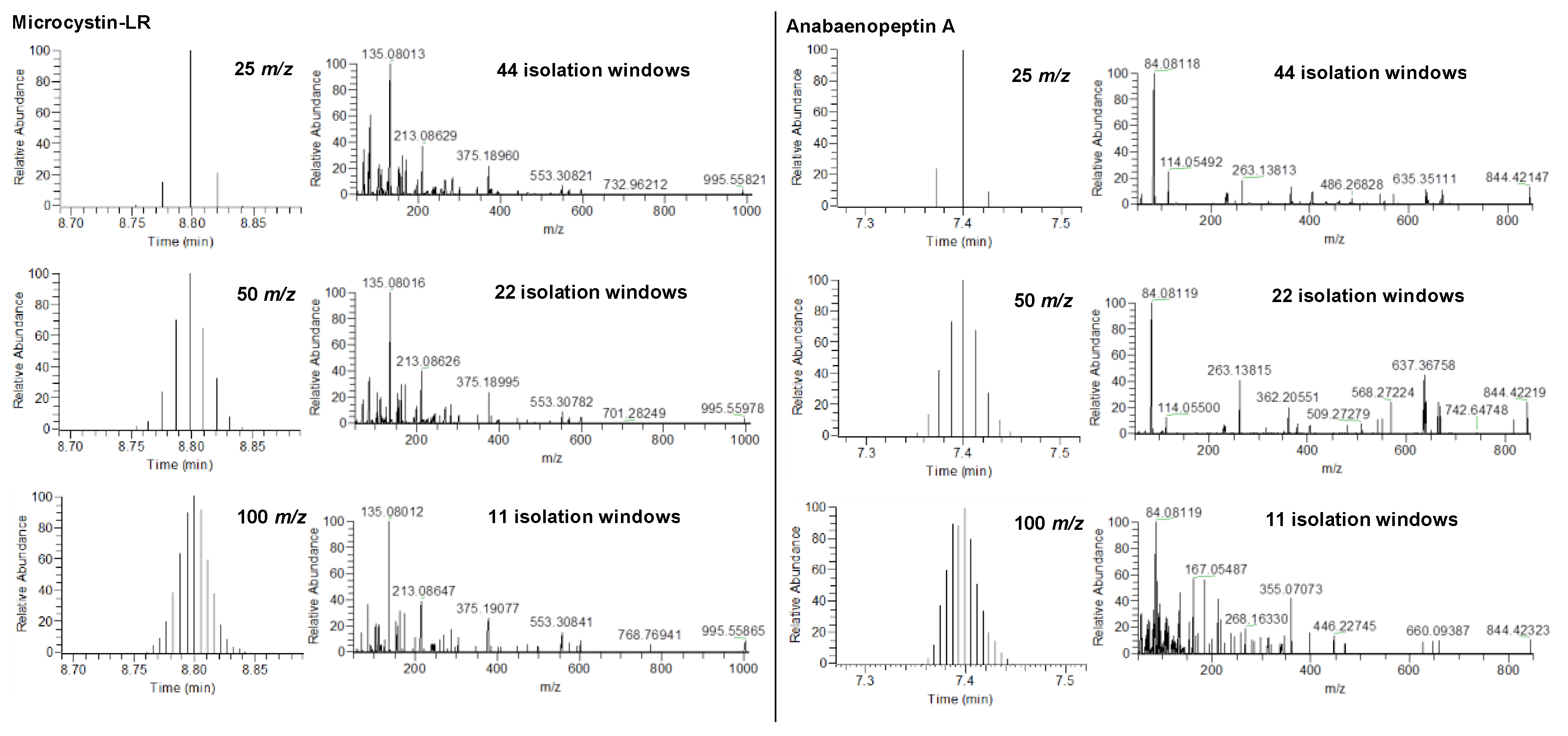
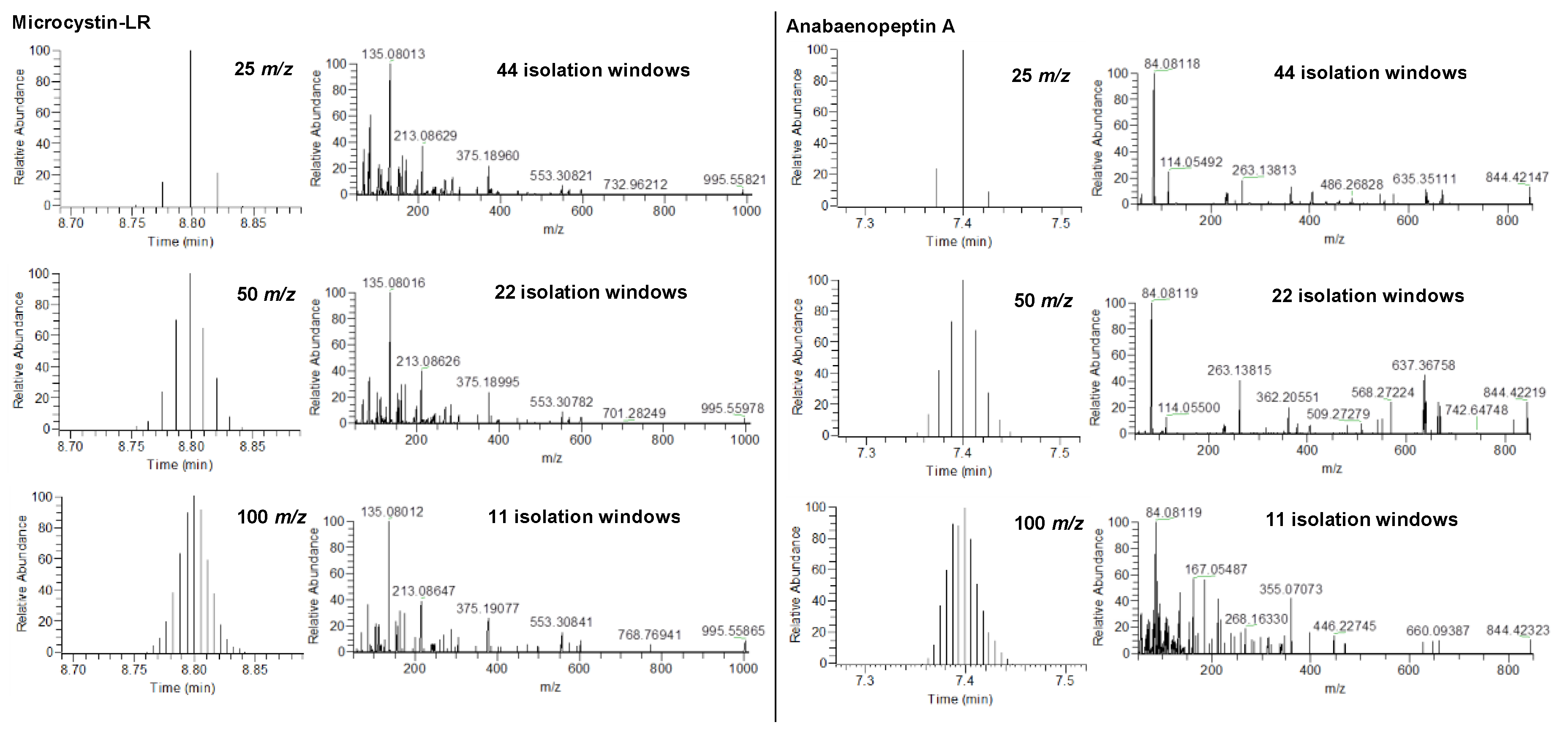
2.1. HRMS Parameters for Suspect Screening via DIA
2.2. Building in-House Databases
2.3. First Features Selection with Compound Discoverer
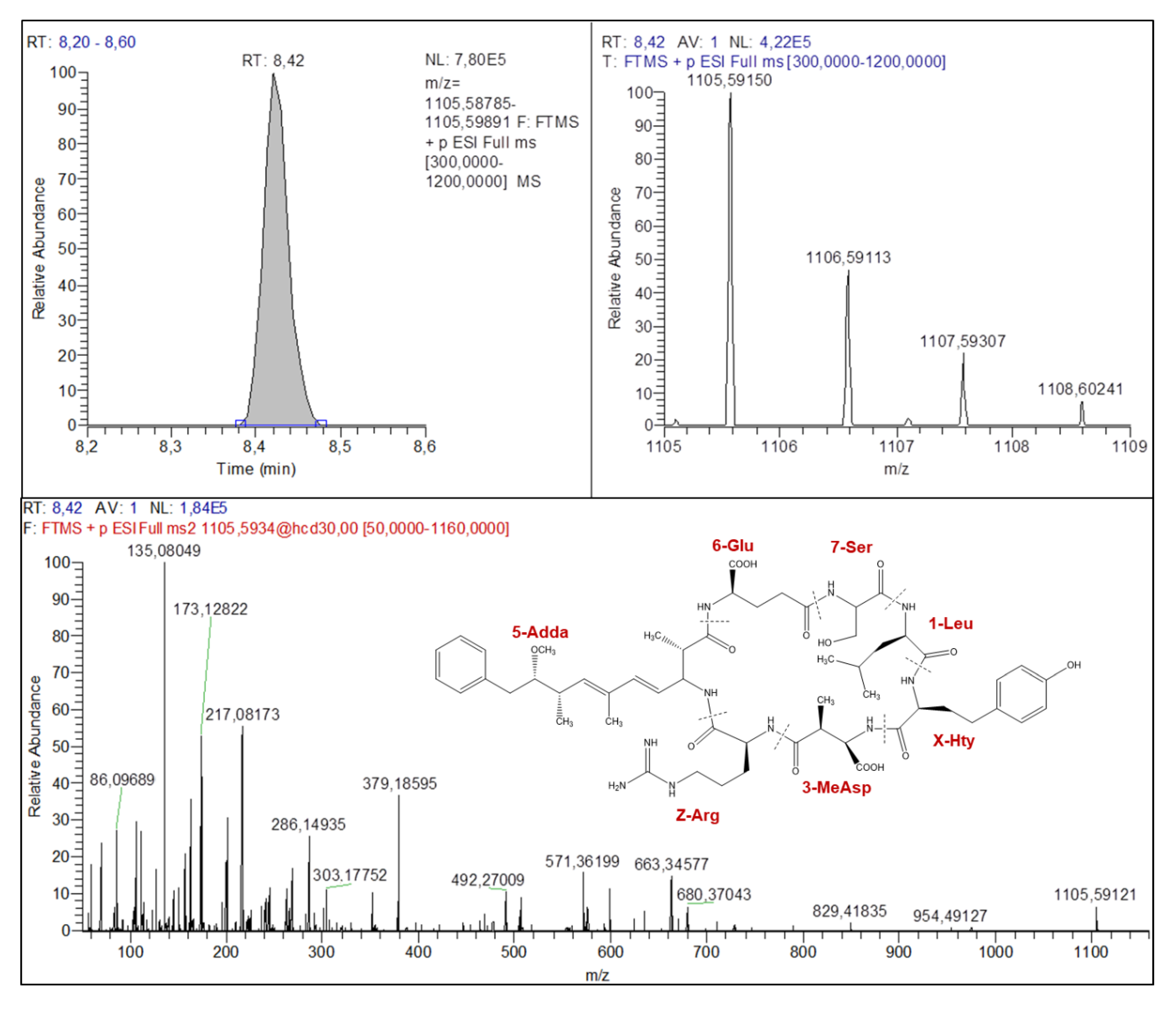
2.4. Confirmation of Suspects Using MS/MS Spectra
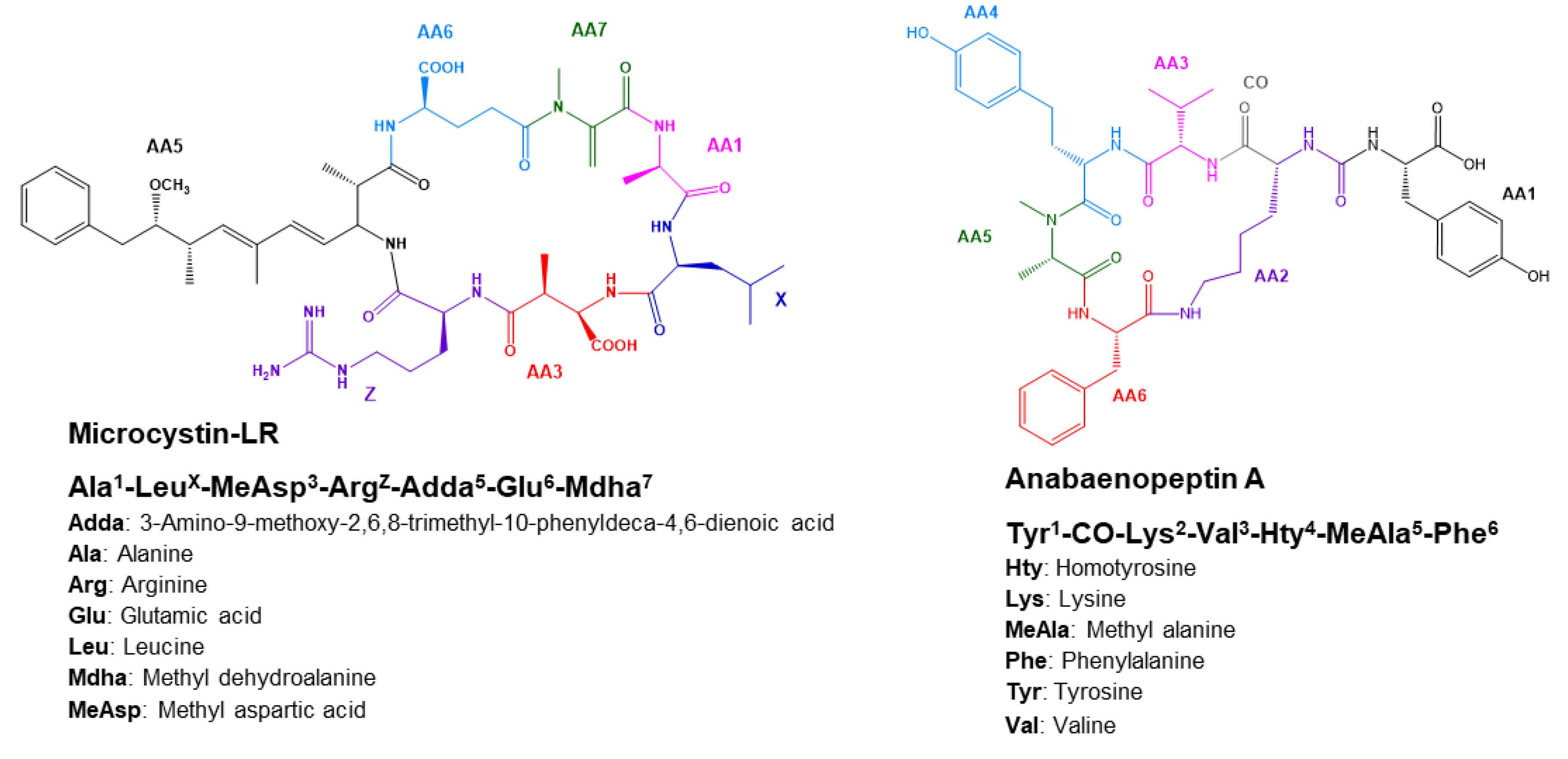
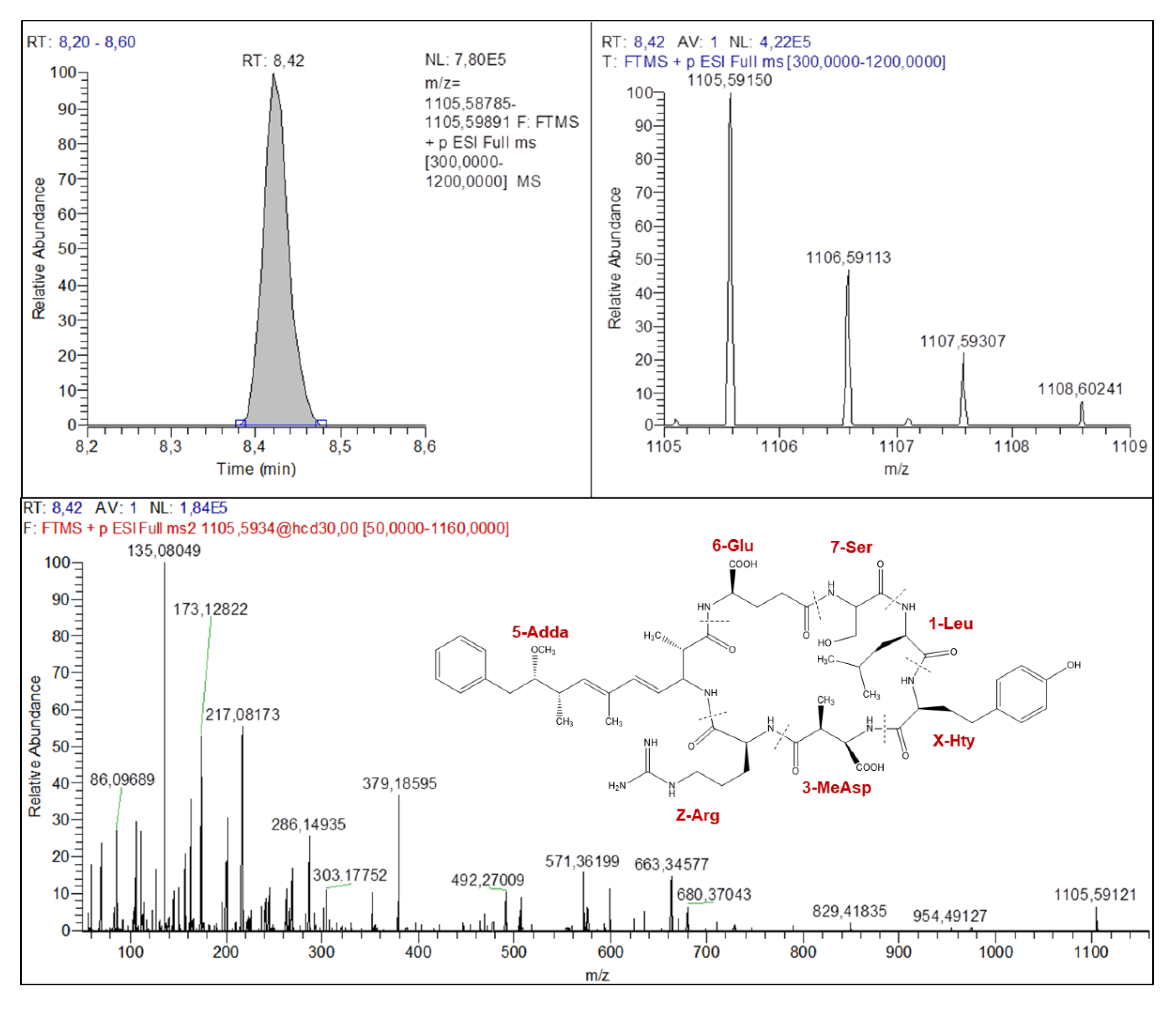
2.4.1. Microcystins Structures Elucidation
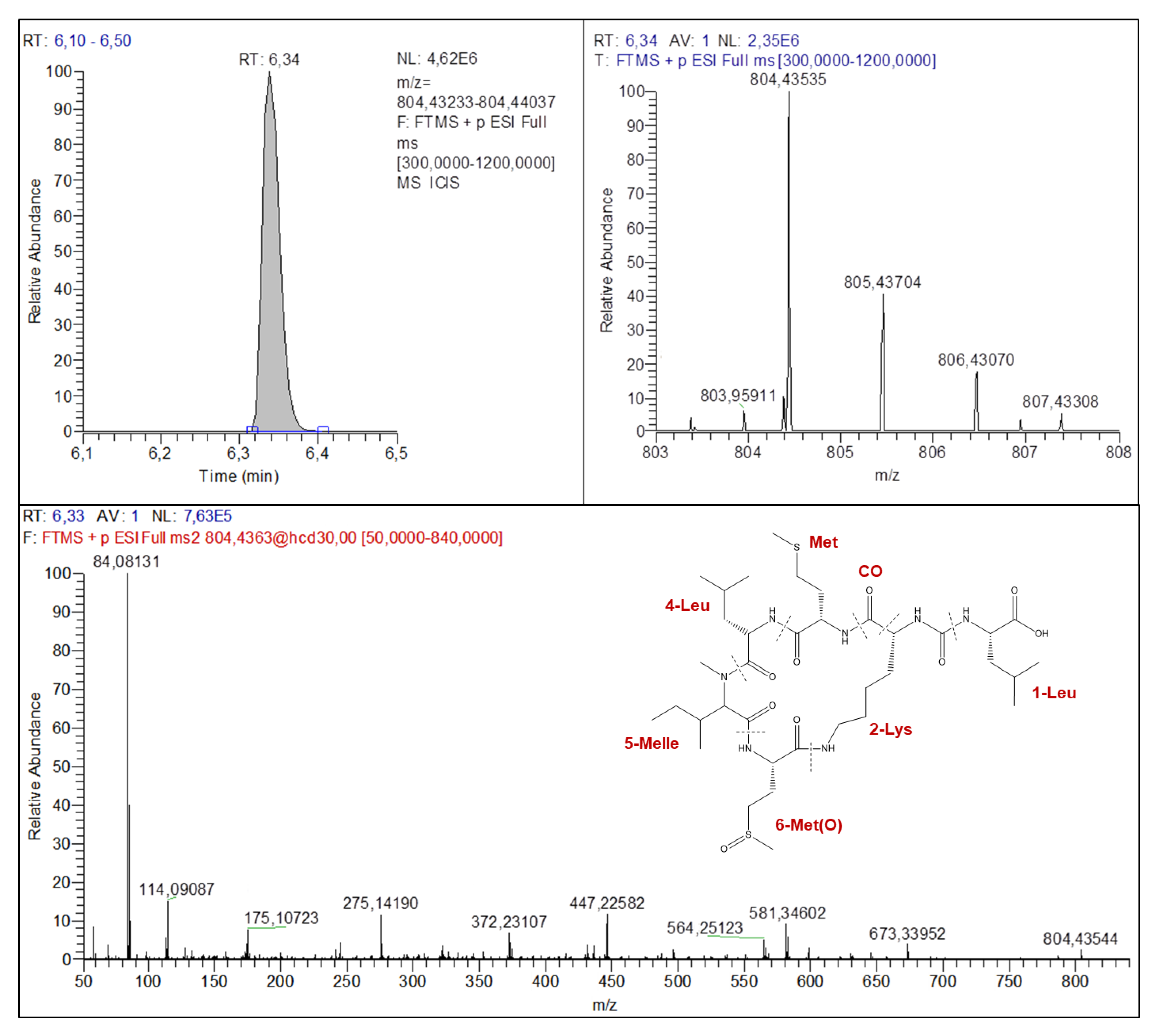
2.4.2. Anabaenopeptins Structures Elucidation
2.5. Quantification and Semi-Quantification
3. Conclusions
4. Materials and Methods
4.1. Chemicals, Reagents and Stock Solutions
4.2. Sample Collection, Preparation and Quantification
4.3. Instrumental Conditions
4.3.1. On-Line Solid Phase Extraction and Chromatographic Conditions
4.3.2. HRMS Conditions
4.4. Suspect Screening Using DIA Methodology
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartram, J.; Chorus, I. Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring and Management; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Carmichael, W.W.; Azevedo, S.; An, J.S.; Molica, R.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- USEPA. Guidelines and Recommandations. Available online: https://www.epa.gov/nutrient-policy-data/guidelines-and-recommendations#what2 (accessed on 2 February 2019).
- World Health Organization. Guidelines for Drinking-Water Quality; World Health Organization: Geneva, Switzerland, 2017; p. 541. [Google Scholar]
- Janssen, E.M.-L. Cyanobacterial peptides beyond microcystins—A review on co-occurrence, toxicity, and challenges for risk assessment. Water Res. 2019, 151, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Spoof, L.; Błaszczyk, A.; Meriluoto, J.; Cegłowska, M.; Mazur-Marzec, H. Structures and activity of new anabaenopeptins produced by Baltic Sea cyanobacteria. Mar. Drugs 2016, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Sedmak, B.; Carmeli, S.; Eleršek, T. “Non-toxic” cyclic peptides induce lysis of cyanobacteria—An effective cell population density control mechanism in cyanobacterial blooms. Microb. Ecol. 2008, 56, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Welker, M.; Von Döhren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed]
- Meriluoto, J.; Spoof, L.; Codd, G.A. Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Ortiz, X.; Korenkova, E.; Jobst, K.J.; MacPherson, K.A.; Reiner, E.J. A high throughput targeted and non-targeted method for the analysis of microcystins and anatoxin—A using on-line solid phase extraction coupled to liquid chromatography–quadrupole time-of-flight high resolution mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 4959–4969. [Google Scholar] [CrossRef]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Crimmins, B.S.; Holsen, T.M. Non-targeted Screening in Environmental Monitoring Programs. In Advancements of Mass Spectrometry in Biomedical Research; Springer: Basel, Switzerland, 2019; pp. 731–741. [Google Scholar]
- Teta, R.; Della Sala, G.; Glukhov, E.; Gerwick, L.; Gerwick, W.H.; Mangoni, A.; Costantino, V. Combined LC–MS/MS and molecular networking approach reveals new cyanotoxins from the 2014 cyanobacterial bloom in green lake, seattle. Environ. Sci. Technol. 2015, 49, 14301–14310. [Google Scholar] [CrossRef]
- Bogialli, S.; Bortolini, C.; Di Gangi, I.M.; Di Gregorio, F.N.; Lucentini, L.; Favaro, G.; Pastore, P. Liquid chromatography-high resolution mass spectrometric methods for the surveillance monitoring of cyanotoxins in freshwaters. Talanta 2017, 170, 322–330. [Google Scholar] [CrossRef]
- Yilmaz, M.; Foss, A.J.; Miles, C.O.; Özen, M.; Demir, N.; Balcı, M.; Beach, D.G. Comprehensive multi-technique approach reveals the high diversity of microcystins in field collections and an associated isolate of Microcystis aeruginosa from a Turkish lake. Toxicon 2019, 167, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Singer, H.P.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ogawa, K.; Matsuura, K.; Murata, H.; Suzuki, M.; Watanabe, M.F.; Itezono, Y.; Nakayama, N. Structural determination of geometrical isomers of microcystins LR and RR from cyanobacteria by two-dimensional NMR spectroscopic techniques. Chem. Res. Toxicol. 1990, 3, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Shin, H.J.; Matsuda, H.; Ishida, K.; Yamaguchi, K. A cyclic peptide, anabaenopeptin B, from the cyanobacterium Oscillatoria agardhii. Phytochemistry 1997, 44, 449–452. [Google Scholar] [CrossRef]
- Grach-Pogrebinsky, O.; Sedmak, B.; Carmeli, S. Seco [d-Asp3] microcystin-RR and [d-Asp3, d-Glu (OMe)6] microcystin-RR, two new microcystins from a toxic water bloom of the cyanobacterium planktothrix rubescens. J. Nat. Prod. 2004, 67, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.D.; Strangman, W.K.; Barbera, A.E.; Mallin, M.A.; McIver, M.R.; Wright, J.L. Microcystins and two new micropeptin cyanopeptides produced by unprecedented Microcystis aeruginosa blooms in North Carolina’s Cape Fear River. Harmful Algae 2014, 31, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Welker, M.; Fastner, J.; Erhard, M.; von Döhren, H. Applications of MALDI-TOF MS analysis in cyanotoxin research. Environ. Toxicol. Int. J. 2002, 17, 367–374. [Google Scholar] [CrossRef]
- Czarnecki, O.; Henning, M.; Lippert, I.; Welker, M. Identification of peptide metabolites of Microcystis (Cyanobacteria) that inhibit trypsin—Like activity in planktonic herbivorous Daphnia (Cladocera). Environ. Microbiol. 2006, 8, 77–87. [Google Scholar] [CrossRef]
- Chorus, I. Cyanotoxins: Occurrence, Causes, Consequences; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Haruštiaková, D.; Welker, M. Chemotype diversity in P lanktothrix rubescens (cyanobacteria) populations is correlated to lake depth. Environ. Microbiol. Rep. 2017, 9, 158–168. [Google Scholar] [CrossRef]
- Bouhaddada, R.; Nélieu, S.; Nasri, H.; Delarue, G.; Bouaïcha, N. High diversity of microcystins in a Microcystis bloom from an Algerian lake. Environ. Pollut. 2016, 216, 836–844. [Google Scholar] [CrossRef]
- Di Gregorio, F.N.; Bogialli, S.; Ferretti, E.; Lucentini, L. First evidence of MC-HtyR associated to a Plankthothrix rubescens blooming in an Italian lake based on a LC-MS method for routinely analysis of twelve microcystins in freshwaters. Microchem. J. 2017, 130, 329–335. [Google Scholar] [CrossRef]
- Ballot, A.; Sandvik, M.; Rundberget, T.; Botha, C.J.; Miles, C.O. Diversity of cyanobacteria and cyanotoxins in Hartbeespoort Dam, South Africa. Mar. Freshw. Res. 2014, 65, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.; Andreote, A.; Fiore, M.; Dörr, F.; Pinto, E. Structural characterization of new peptide variants produced by cyanobacteria from the Brazilian Atlantic coastal Forest using liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry. Mar. Drugs 2015, 13, 3892–3919. [Google Scholar] [CrossRef] [PubMed]
- Diehnelt, C.W.; Dugan, N.R.; Peterman, S.M.; Budde, W.L. Identification of microcystin toxins from a strain of Microcystis aeruginosa by liquid chromatography introduction into a hybrid linear ion trap-fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 2006, 78, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Benke, P.; Kumar, M.V.; Pan, D.; Swarup, S. A mass spectrometry-based unique fragment approach for the identification of microcystins. Analyst 2015, 140, 1198–1206. [Google Scholar] [CrossRef]
- Qi, Y.; Rosso, L.; Sedan, D.; Giannuzzi, L.; Andrinolo, D.; Volmer, D.A. Seven new microcystin variants discovered from a native Microcystis aeruginosa strain–unambiguous assignment of product ions by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 220–224. [Google Scholar] [CrossRef]
- Moschet, C.; Lew, B.M.; Hasenbein, S.; Anumol, T.; Young, T.M. LC-and GC-QTOF-MS as complementary tools for a comprehensive micropollutant analysis in aquatic systems. Environ. Sci. Technol. 2017, 51, 1553–1561. [Google Scholar] [CrossRef]
- Roy-Lachapelle, A.; Duy, S.V.; Munoz, G.; Dinh, Q.T.; Bahl, E.; Simon, D.F.; Sauvé, S. Analysis of multiclass cyanotoxins (microcystins, anabaenopeptins, cylindrospermopsin and anatoxins) in lake waters using on-line SPE liquid chromatography high-resolution Orbitrap mass spectrometry. Anal. Methods 2019, 11, 3126–3133. [Google Scholar] [CrossRef]
- Letzel, T.; Lucke, T.; Schulz, W.; Sengl, M.; Letzel, M. OMI (Organic Molecule Identification) in water using LC-MS (/MS): Steps from “unknown” to “identified”: A contribution to the discussion In a class of its own. Lab More Int. 2014, 4, 24–28. [Google Scholar]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V. Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10, 744. [Google Scholar] [CrossRef]
- Doerr, A. DIA mass spectrometry. Nat. Methods 2014, 12, 35. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Chen, X.; Zhong, L.; Yin, Y. Development of data-independent acquisition workflows for metabolomic analysis on a quadrupole-orbitrap platform. Talanta 2017, 164, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Ang, J.E.; Revell, V.; Mann, A.; Mäntele, S.; Otway, D.T.; Johnston, J.D.; Thumser, A.E.; Skene, D.J.; Raynaud, F. Identification of human plasma metabolites exhibiting time-of-day variation using an untargeted liquid chromatography–mass spectrometry metabolomic approach. Chronobiol. Int. 2012, 29, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Nikolskiy, I.; Mahieu, N.G.; Chen, Y.J.; Tautenhahn, R.; Patti, G.J. An untargeted metabolomic workflow to improve structural characterization of metabolites. Anal. Chem. 2013, 85, 7713–7719. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted metabolomics strategies—Challenges and emerging directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.; Caixach, J. An integrated strategy for rapid and accurate determination of free and cell-bound microcystins and related peptides in natural blooms by liquid chromatography–electrospray-high resolution mass spectrometry and matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry using both positive and negative ionization modes. J. Chromatogr. A 2015, 1407, 76–89. [Google Scholar] [PubMed]
- Tsuji, K.; Naito, S.; Kondo, F.; Ishikawa, N.; Watanabe, M.F.; Suzuki, M.; Harada, K.I. Stability of microcystins from cyanobacteria: Effect of light on decomposition and isomerization. Environ. Sci. Technol. 1994, 28, 173–177. [Google Scholar] [CrossRef]
- Sano, T.; Kaya, K. Oscillamide Y, a chymotrypsin inhibitor from toxic Oscillatoria agardhii. Tetrahedron Lett. 1995, 36, 5933–5936. [Google Scholar] [CrossRef]
- Gkelis, S.; Lanaras, T.; Sivonen, K. Cyanobacterial toxic and bioactive peptides in freshwater bodies of Greece: Concentrations, occurrence patterns, and implications for human health. Mar. Drugs 2015, 13, 6319–6335. [Google Scholar] [CrossRef]
- Beversdorf, L.; Weirich, C.; Bartlett, S.; Miller, T. Variable cyanobacterial toxin and metabolite profiles across six eutrophic lakes of differing physiochemical characteristics. Toxins 2017, 9, 62. [Google Scholar] [CrossRef]
- Kokociński, M.; Dziga, D.; Spoof, L.; Stefaniak, K.; Jurczak, T.; Mankiewicz-Boczek, J.; Meriluoto, J. First report of the cyanobacterial toxin cylindrospermopsin in the shallow, eutrophic lakes of western Poland. Chemosphere 2009, 74, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Pick, F.R. Blooming algae: A Canadian perspective on the rise of toxic cyanobacteria. Can. J. Fish. Aquat. Sci. 2016, 73, 1149–1158. [Google Scholar] [CrossRef]
- USEPA. EPA Drinking Water Health Advisories for Cyanotoxins. Available online: https://www.epa.gov/cyanohabs/epa-drinking-water-health-advisories-cyanotoxins (accessed on 26 September 2019).
- Biosynthesis. Amino Acid Masses Tables. Available online: https://www.biosyn.com/tew/amino-acid-masses-tables.aspx (accessed on 16 September 2019).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent and Fragment Ions | Known MC (Certified Standard) | Known MC (No Certified Standard) | Unknown MC | |||
|---|---|---|---|---|---|---|
| MC-LR | [GluOMe6] MC-LR | [M(O)1] MC-LR | [M(O)1, GluOMe6] MC-LR | [Asp3]MC-RHar | [Leu1, Ser7] MC-HtyR | |
| M+H+ | 995.55927 | 1009.57104 | 1071.55340 | 1085.56928 | 1038.57291 | 1105.59150 |
| Isotope #1 | 996.55629 | 1010.56808 | 1072.55682 | 1086.5709 | 1039.57413 | 1106.59113 |
| Isotope #2 | 997.56067 | 1011.57490 | 1073.55194 | 1087.57513 | 1107.59307 | |
| Isotope #3 | 998.56488 | 1074.56341 | 1088.56762 | 1108.60241 | ||
| M+2H2+ | 536.27992 | 519.78065 | ||||
| Isotope #1 | 536.77931 | 520.29199 | ||||
| Isotope #2 | 537.28075 | 520.79363 | ||||
| Isotope #3 | 521.29156 | |||||
| M+H+-H2O | 977.56032 | 991.56022 | 1053.54316 | 1067.55874 | ||
| M+H+-CO | 967.54996 | 981.57442 | 1077.59665 | |||
| M+H+-CH2NHC(NH)NH2) (Arg) | 999.49677 | |||||
| M+H+-AA6 | 866.51198 | 976.54852 | ||||
| M+H+-134 (Adda) | 861.47956 | 875.49571 | 937.48531 | 951.49270 | 904.49915 | 971.51668 |
| M+H+-134 (Adda)-NH3 | 844.44971 | 858.46904 | 920.45409 | 934.46807 | 887.47064 | 954.49127 |
| Z+Adda+AA6+AA3+AA1-CO+H+ | 847.43655 | 861.45323 | ||||
| Z+Adda+AA6+AA3+H+ | 728.39793 | 742.41144 | 728.39593 | 742.41195 | 728.39601 | 728.39614 |
| AA3+Z+Adda+AA6-H2O+H+ | 710.38705 | 724.40291 | 710.38457 | 724.40284 | 710.38447 | |
| Z+Adda+AA6+CO+H+ | 625.33379 | 639.34874 | 625.33299 | 639.34839 | 625.33274 | |
| AA3+Z+Adda+H+ | 599.35556 | 599.35522 | 599.35471 | 599.35514 | 599.35420 | 599.36213 |
| Z+Adda+AA6+H+ | 599.35556 | 613.36953 | 599.35471 | 613.36946 | 613.37004 | 599.36213 |
| AA3+Z+Adda-CO+H+ | 571.35843 | 571.35829 | 571.35844 | 571.35823 | 571.35963 | 571.36199 |
| Z+Adda+AA6-CO+H+ | 571.35843 | 585.37421 | 571.35844 | 585.37418 | 585.37388 | 571.36199 |
| [AA7+AA1+X+AA3+Z+NH2+2H]+ | 570.33513 | 570.33402 | 646.33282 | 646.33296 | 613.35189 | 680.37043 |
| AA7+AA1+X+AA3+Z+H+ | 553.31097 | 553.30853 | 629.30526 | 629.30531 | 596.32510 | 663.34577 |
| AA7+AA1+X+AA3+Z-H2O+H+ | 535.29685 | 535.29715 | 611.29594 | 611.29603 | 578.31403 | |
| AA7+AA1+X+AA3+Z-CO+H+ | 525.31401 | 525.31395 | 601.31109 | 601.31177 | 568.33044 | 635.35001 |
| [AA1+X+AA3+Z+NH2+2H]+ | 487.29752 | 563.29411 | 416.26012 | 593.34189 | ||
| AA1+X+AA3+Z+H+ | 470.26987 | 470.26974 | 546.26878 | 546.27001 | 513.28930 | 576.31199 |
| AA1+X+AA3+Z-NH3+H+ | 453.23973 | 453.23985 | 529.24225 | 529.24229 | 496.26170 | 559.28760 |
| AA1+X+AA3+Z-H2O+H+ | 452.25983 | 452.25967 | 528.25943 | 528.25977 | 495.26546 | |
| AA1+X+AA3+Z-CO-NH3+H+ | 468.26542 | |||||
| Z+Adda-134+AA6-NH3+H+ | 448.25002 | 462.27024 | 448.25379 | 462.27020 | 462.27011 | |
| Adda-134+AA6+AA7+AA1-NH3+H+ | 446.22694 | 460.24297 | 522.22418 | 536.24198 | 492.27009 | |
| [X+AA3+Z+NH2+2H]+ | 416.26101 | 416.26113 | ||||
| X+AA3+Z+H+ | 399.23512 | 399.23409 | 399.23411 | 399.23417 | 442.25185 | 463.22949 |
| AA7+AA1+X+AA3+H+ | 397.20653 | 397.20649 | 426.20885 | 507.24592 | ||
| AA6+AA7+AA1+X+H+ | 397.20653 | 411.22257 | 473.20550 | 487.22124 | 440.22463 | 507.24592 |
| X+AA3+Z-NH3+H+ | 382.20854 | 382.20868 | 382.20836 | 382.20855 | 425.22561 | 446.20174 |
| Adda-134+AA6+AA7-NH3+H+ | 375.19117 | 389.20689 | 375.19028 | 389.20690 | 375.19269 | 379.18595 |
| Adda-134+AA6+AA7-NH3-CO+H+ | 347.19498 | 361.21108 | 361.21113 | 347.19155 | 351.19024 | |
| Adda-134+AA6-NH3+H+ | 292.15384 | 306.16894 | 292.15371 | 306.16887 | 292.15414 | |
| [AA3+Z+NH2+2H]+ | 303.17697 | 303.17668 | 303.17683 | 303.17739 | 303.17752 | |
| AA7+AA1+X-NH3+H+ | 294.15521 | |||||
| X+AA3+H+ | 272.13442 | 307.12806 | ||||
| AA3+Z-NH2+H+ | 286.14888 | 286.14981 | 286.14997 | 286.14989 | 286.14832 | 286.14935 |
| AA7+AA1+X+H+ | 268.16531 | 268.16581 | 344.16287 | 344.16366 | 311.18244 | 378.20111 |
| AA1+Z+H+ | 242.16093 | |||||
| AA6+AA7+CO+ | 253.08124 | 253.08129 | 239.06653 | 243.06043 | ||
| AA6+AA7+H+ | 213.08659 | 227.10269 | 213.08735 | 227.10254 | 213.08693 | 217.08173 |
| AA6+AA7-CO+H+ | 185.09586 | 189.08683 | ||||
| [Z+NH2+2H]+ | 174.13423 | 174.13431 | 174.13459 | |||
| Adda-134-NH3+H+ | 163.11149 | 163.11151 | 163.11156 | 163.11148 | 163.11138 | 163.11150 |
| AA7+AA1+H+ | 155.08136 | 155.08127 | 231.07989 | 231.07983 | 155.08138 | 201.12293 |
| AA7+AA1-CO+H+ | 127.08639 | 127.08636 | 203.08461 | 203.08484 | 127.08664 | 173.12822 |
| Adda frag (Ph-CH2-CH(O+Me) | 135.08040 | 135.08041 | 135.08073 | 135.08055 | 135.08049 | 135.08049 |
| X Immonium ion | 86.09695 | 86.09680 | 86.09682 | 129.11388 | 129.11358 | |
| Ser Immonium ion | 60.04481 | |||||
| Leu Immonium ion | 86.09677 | 86.09689 | ||||
| Parent and Fragment Ions | Known AP (Certified Standard) | Known AP (No Certified Standard) | Unknown AP | ||||
|---|---|---|---|---|---|---|---|
| AP-A | AP-C | AP-F | Ferintoic acid A | Oscillamide Y | AP731 | AP803 | |
| M+H+ | 844.42399 | 809.45396 | 851.47649 | 867.43760 | 858.43789 | 732.39224 | 804.43535 |
| Isotope #1 | 845.42487 | 810.45755 | 852.47948 | 868.44117 | 859.44270 | 733.39499 | 805.43704 |
| Isotope #2 | 846.42939 | 811.46099 | 853.48112 | 869.44461 | 860.44575 | 734.39684 | 806.43070 |
| Isotope #3 | 837.43126 | 861.44882 | 735.39821 | 807.43308 | |||
| M+H+-NH3 | 792.42755 | ||||||
| M+H+-H2O | 826.41253 | 791.44322 | 833.46495 | 849.42755 | 840.42805 | 714.38127 | 786.42485 |
| M+H+-H2O-CO | 821.43177 | 758.42793 | |||||
| M+H+-AA6residue | 603.34925 | 657.39861 | |||||
| M+H+-AA1 | 681.36103 | 695.37553 | |||||
| M+H+-AA1-H2O | 663.34863 | 663.34859 | 677.36401 | 663.34841 | 677.36411 | 567.31335 | 673.33952 |
| M+H+-CO-AA1-H2O | 635.35366 | 635.35363 | 635.35349 | 649.37013 | 539.31810 | 645.34572 | |
| M+H+-AA4-AA5 | 528.28961 | 547.32259 | 589.34485 | 605.30624 | 596.30578 | 548.26999 | 564.25123 |
| M+H+-AA3-AA4 | 591.29012 | ||||||
| M+H+-AA3-AA4-H2O | 550.26467 | 515.29601 | 573.28032 | 550.26427 | 502.22842 | 542.29930 | |
| M+H+-AA3-AA4-CO | 540.28143 | 563.29597 | 540.28113 | 532.31511 | |||
| M+H+-AA1-CO-AA6residue-H2O | 479.29701 | 581.34602 | |||||
| M+H+-AA1-CO-AA6resisue | 428.28596 | 516.32042 | |||||
| M+H+-AA1-AA4-AA6residue-H2O | 394.20841 | 496.25720 | |||||
| M+H+-AA1-AA3-AA4 | 405.21182 | 405.21189 | 405.21165 | 405.21192 | 405.21185 | 373.17124 | 447.22582 |
| Lys+AA3+AA5+AA6+H+ | 460.29013 | 474.30651 | 460.28997 | 474.30632 | 428.24910 | 534.27652 | |
| Lys+AA5+AA6+CO+H+ | 389.21756 | 389.21743 | 357.17623 | 431.23184 | |||
| AA6+Lys+CO+AA3+H+ | 403.23383 | 403.23379 | 385.20775 | 435.17195 | |||
| AA3+AA4+AA5+H+ | 362.20673 | 362.20676 | 376.22245 | 362.20651 | 376.22195 | 284.19651 | 372.23107 |
| AA5+AA6+H+ | 233.12808 | 233.12801 | 233.12811 | 233.12811 | 233.12809 | 201.08693 | 275.14190 |
| AA3+AA4+H+ | 277.15417 | 277.15409 | 277.15416 | 213.16084 | 245.13166 | ||
| AA4+AA5+H+ | 263.13861 | 263.13851 | 263.13865 | 263.13866 | 263.13866 | 185.12842 | 241.19079 |
| AA1+H+ | 175.11875 | ||||||
| AA1+CO+ | 201.09792 | ||||||
| [AA1+2H]+ | 130.11017 | ||||||
| Ph-CH2-OH | 107.04936 | 107.04913 | 107.04945 | 107.04961 | |||
| Lys Immonium ion | 84.08123 | 84.08122 | 84.08120 | 84.08119 | 84.08120 | 84.08134 | 84.08131 |
| AA1 Immonium Ion | 136.07545 | 129.11359 | 120.08070 | 86.09692 | |||
| Phe Immonium | 120.08100 | 120.08110 | 120.08110 | ||||
| Sample No. | CYN | [Asp3]MC-RR | MC-RR | MC-YR | MC-LR | [Asp3]MC-LR | MC-HiIR | MC-LA | MC-LY | AP-A | AP-B |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ND | ND | ND | ND | 90 ± 28 | ND | ND | 486 ± 105 | ND | ND | ND |
| 2 | ND | ND | ND | ND | ND | ND | ND | 364 ± 70 | ND | ND | 95 ± 17 |
| 3 | ND | ND | 491 ± 95 | 76 ± 6 | 1010 ± 21 | ND | ND | ND | ND | ND | ND |
| 4 | ND | ND | ND | ND | 106 ± 10 | ND | ND | 1165 ± 60 | ND | ND | ND |
| 5 | ND | ND | ND | ND | 47 ± 5* | ND | ND | ND | ND | 1290 ± 259 | 851 ± 116 |
| 6 | ND | ND | ND | ND | ND | ND | ND | ND | ND | 188 ± 66 | 348 ± 38 |
| 7 | ND | ND | ND | ND | 254 ± 29 | ND | ND | ND | 41 ± 13 * | ND | 124 ± 32 |
| 8 | 153 ± 66 | ND | ND | ND | 62 ± 5 | ND | ND | ND | ND | ND | ND |
| 9 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 10 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 11 | ND | 41,364 ± 3885 | 840 ± 87 | 259 ± 17 | 416 ± 39 | 1073 ± 116 | ND | ND | ND | 3178 ± 97 | 5836 ± 187 |
| 12 | ND | 123 ± 8 | 5691 ± 506 | 2692 ± 382 | 3263 ± 179 | ND | 321 ± 98 | ND | 39 ± 36 * | 137 ± 25 | 239 ± 46 |
| Sample No. | [GluOMe6] MC-LR | [M(O)1] MC-LR | [M(O)1, GluOMe6] MC-LR | [Asp3] MC-RHar | [Leu1, Ser7] MC-HtyR | AP-C | AP-F | Ferintoic acid A | Oscillamide Y | AP731 | AP803 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 596 ± 36 | 57 ± 11 | 197 ± 28 | ND | ND | ND | ND | ND | ND | ND | ND |
| 5 | ND | ND | ND | ND | ND | ND | 175 ± 34 | ND | 484 ± 55 | ND | 1035 ± 108 |
| 9 | ND | ND | ND | 201 ± 47 | ND | ND | ND | ND | ND | ND | ND |
| 11 | ND | ND | ND | ND | ND | 75 ± 9 | 221 ± 15 | ND | 88 ± 10 | 109 ± 7 | ND |
| 12 | ND | ND | ND | ND | 124 ± 23 | ND | ND | 60 ± 11 | ND | ND | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy-Lachapelle, A.; Solliec, M.; Sauvé, S.; Gagnon, C. A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners. Toxins 2019, 11, 619. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11110619
Roy-Lachapelle A, Solliec M, Sauvé S, Gagnon C. A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners. Toxins. 2019; 11(11):619. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11110619
Chicago/Turabian StyleRoy-Lachapelle, Audrey, Morgan Solliec, Sébastien Sauvé, and Christian Gagnon. 2019. "A Data-Independent Methodology for the Structural Characterization of Microcystins and Anabaenopeptins Leading to the Identification of Four New Congeners" Toxins 11, no. 11: 619. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11110619