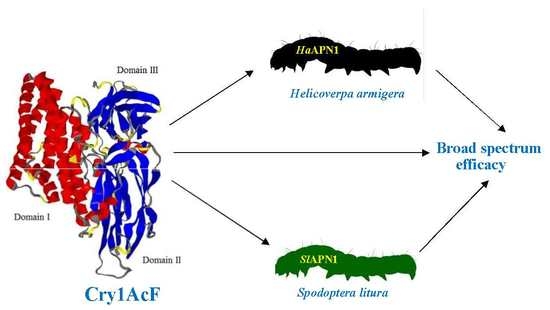
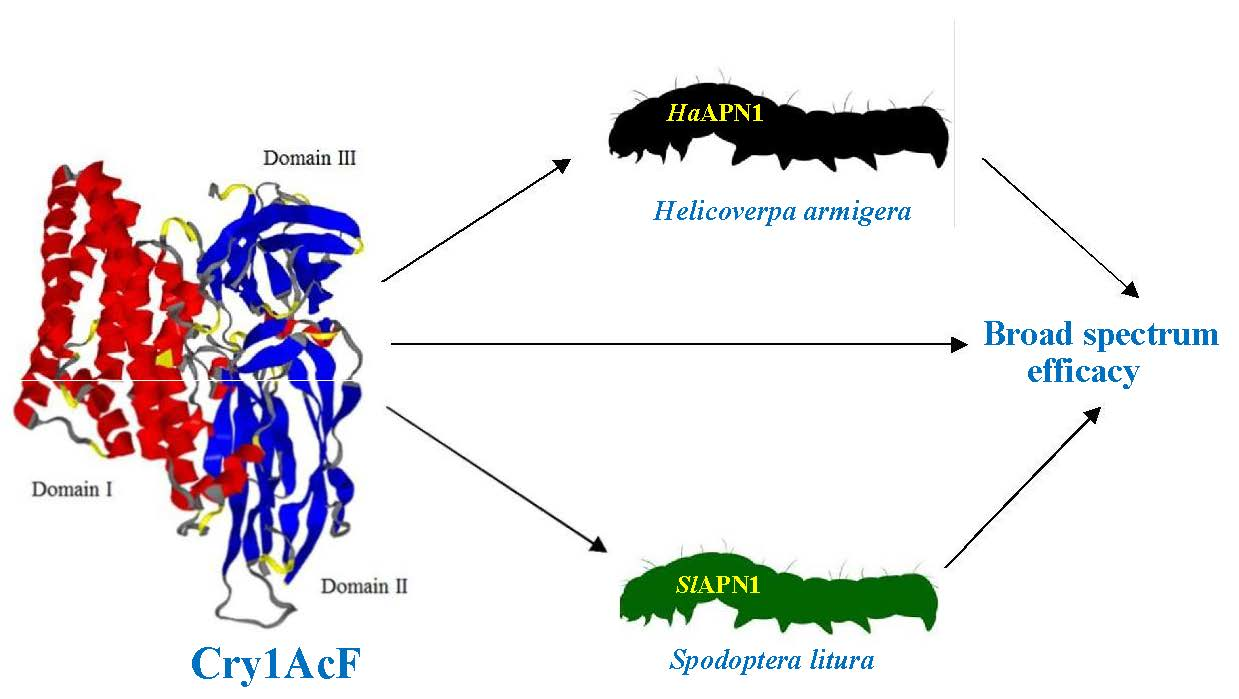
Molecular Interaction-Based Exploration of the Broad Spectrum Efficacy of a Bacillus thuringiensis Insecticidal Chimeric Protein, Cry1AcF

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
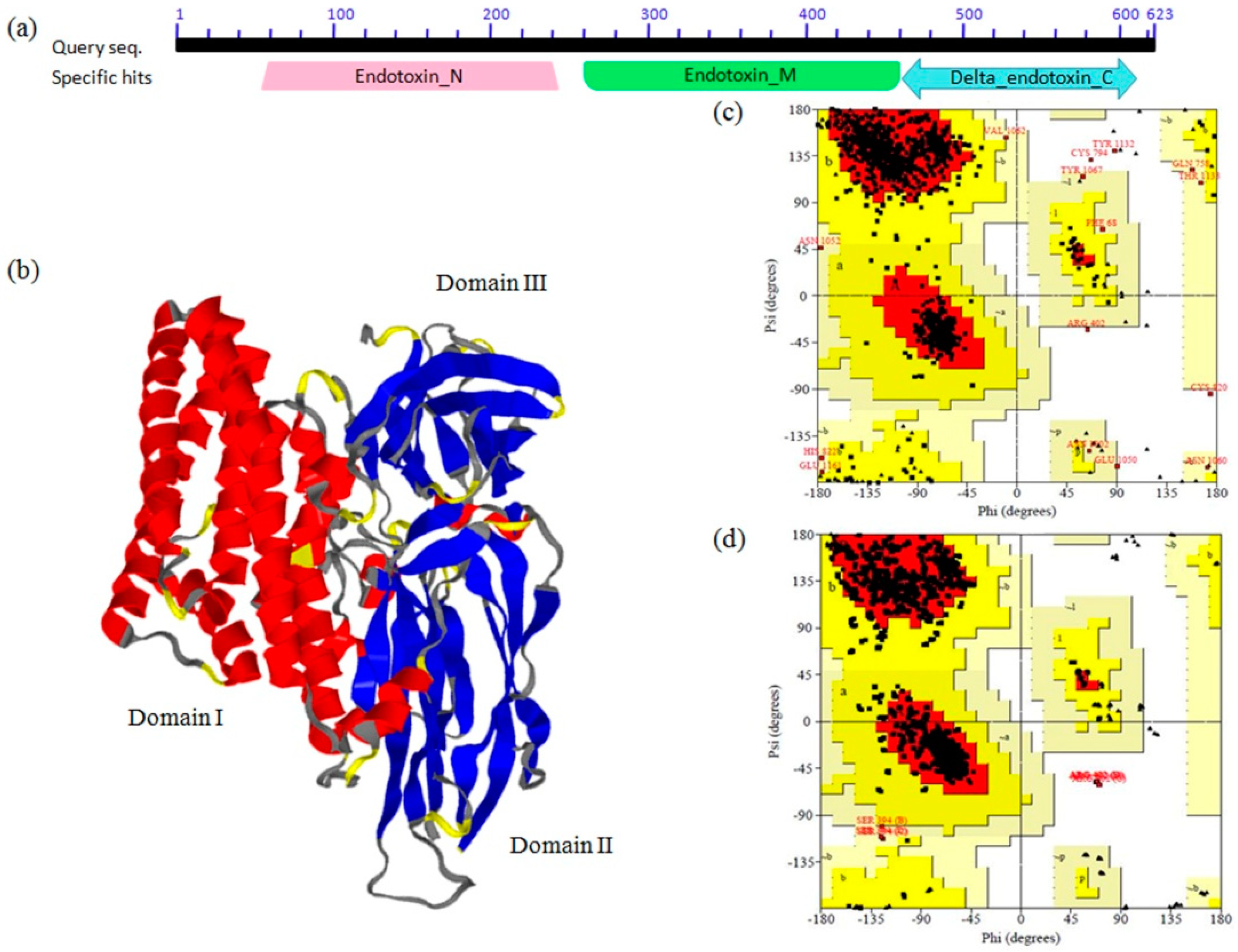
2.1. Molecular Modeling and Validation
2.2. Docking and Interaction Analysis
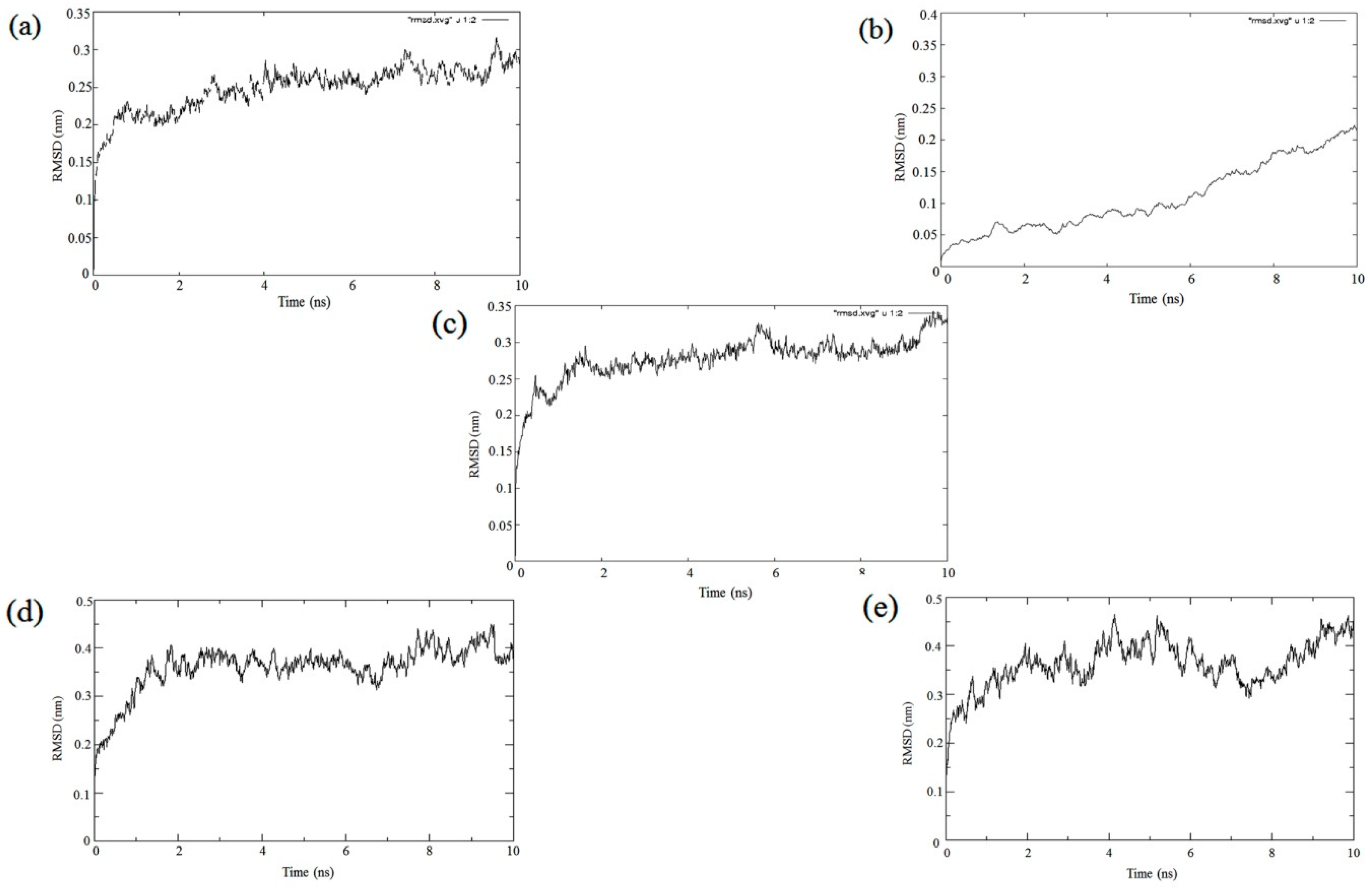
2.3. Molecular Dynamic Simulations
3. Materials and Methods
3.1. Source of Sequence and Primary Analysis
3.2. Molecular Modeling and Validation
3.3. Docking and Interaction Analysis
3.4. Molecular Dynamics (MD) Simulations of Cry1AcF and APN1 Receptors
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Service for the Acquisition of Agri-Biotech Applications (ISAAA). Executive Summary: Global Status of Commercialized Biotech/GM Crops: 2016; ISAAA: Ithaca, NY, USA, 2016; Available online: https://www.isaaa.org/resources/publications/briefs/52/executivesummary/pdf/B52-ExecSum-English.pdf (accessed on 15 October 2018).
- Schnepf, E.; Crickmore, N.V.; Van Rie, J.; Lereclus, D.; Baum, J.; Feitelson, J.; Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar] [PubMed]
- Van Rie, J. Bacillus thuringiensis and its use in transgenic insect control technologies. Int. J. Med. Microbiol. 2000, 290, 463–469. [Google Scholar] [CrossRef]
- Kumar, P.A.; Bambawale, O.M. Insecticidal Proteins of Bacillus thuringiensis and their Application in Agriculture. In Advances in Microbial Toxin Research and Its Biotechnological Exploitation; Upadhyay, R.K., Ed.; Springer: Boston, MA, USA, 2002; pp. 259–280. [Google Scholar]
- Pardo-Lopez, L.; Soberon, M.; Bravo, A. Bacillus thuringiensis insecticidal three-domain Cry toxins: Mode of action, insect resistance and consequences for crop protection. FEMS Microbiol. Rev. 2012, 37, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Tabashnik, B.E.; Brévault, T.; Carrière, Y. Insect resistance to Bt crops: Lessons from the first billion acres. Nature biotech. 2013, 31, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; Crickmore, N. Structural classification of insecticidal proteins–towards an in silico characterization of novel toxins. J. Invert. Pathol. 2017, 142, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Devine, G.J.; Furlong, M.J. Insecticide use: Contexts and ecological consequences. Agric. Human Values 2007, 24, 281–306. [Google Scholar] [CrossRef]
- Tajne, S.; Boddupally, D.; Sadumpati, V.; Vudem, D.R.; Khareedu, V.R. Synthetic fusion-protein containing domains of Bt Cry1Ac and Allium sativumlectin (ASAL) conferred enhanced insecticidal activity against major lepidopteran pests. J. Biotech. 2014, 171, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.H.; Baisakh, N.; Oliva, N.; Datta, K.; Frutos, R.; Datta, S.K. Translational fusion hybrid Bt genes confer resistance against Yellow Stem Borer in transgenic elite Vietnamese rice (Oryza sativa L.) cultivars. Crop Sci. 2006, 46, 781–789. [Google Scholar] [CrossRef]
- Honée, G.; Vriezen, W.; Visser, B. A translation fusion product of two different insecticidal crystal protein genes of Bacillus thuringiensis exhibits an enlarged insecticidal spectrum. Appl. Environ. Microbiol. 1990, 56, 823–825. [Google Scholar] [PubMed]
- Harper, B.K.; Mabon, S.A.; Leffel, S.M.; Halfhill, M.D.; Richards, H.A.; Moyer, K.A.; Stewart, C.N. Green fluorescent protein as a marker for expression of a second gene in transgenic plants. Nature Biotech. 1999, 17, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Zghal, R.Z.; Elleuch, J.; Ali, M.B.; Darriet, F.; Rebaï, A.; Chandre, F.; Tounsi, S. Towards novel Cry toxins with enhanced toxicity/broader: A new chimeric Cry4Ba/Cry1Ac toxin. Appl. Microbiol. Biotech. 2017, 101, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.K.; Mandaokar, A.D.; Ananda Kumar, P.; Sharma, R.P. Synergistic effect of Cry1Ac and Cry1F delta-endotoxons of Bacillus thuringiensis on cotton bollworm, Helicoverpa armigera. Curr. Sci. 1998, 75, 663–664. [Google Scholar]
- Kumar, A.M.; Sreevathsa, R.; Reddy, K.N.; Ganesh, P.T.; Udayakumar, M. Amenability of castor to an Agrobacterium-mediated in planta transformation strategy using a cry1AcF gene for insect tolerance. J. Crop. Sci. Biotech. 2011, 14, 125–132. [Google Scholar] [CrossRef]
- Gowri Neelima, M.; Ramu, S.V.; Sreevathsa, R.; Rani, A.; Kumar, A.R.V.; Gayatri, M.C. In planta transformation strategy to generate transgenic plants in chickpea: Proof of concept with a cry gene. J. Plant. Biol. 2008, 35, 201–206. [Google Scholar]
- Keshavareddy, G.; Rohini, S.; Ramu, S.V.; Sundaresha, S.; Kumar, A.R.V.; Kumar, P.A.; Udayakumar, M. Transgenics in groundnut (Arachis hypogaea L.) expressing cry1AcF gene for resistance to Spodoptera litura (F.). Physiol. Mol. Biol. Plants. 2013, 19, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Keshamma, E.; Sreevathsa, R.; Kumar, A.M.; Reddy, K.N.; Manjulatha, M.; Shanmugam, N.B.; Udayakumar, M. Agrobacterium-mediated in planta transformation of field bean (Lablab purpureus L.) and recovery of stable transgenic plants expressing the cry1AcF gene. Plant Mol. Biol. Report. 2012, 30, 67–78. [Google Scholar] [CrossRef]
- Ramu, S.V.; Rohini, S.; Keshavareddy, G.; Gowri, N.M.; Shanmugam, N.B.; Kumar, A.R.V.; Udayakumar, M. Expression of a synthetic cry1AcF gene in transgenic Pigeon pea confers resistance to Helicoverpa armigera. J. Appl. Entomol. 2012, 136, 675–687. [Google Scholar] [CrossRef]
- Dehury, B.; Sahu, M.; Sahu, J.; Sarma, K.; Sen, P.; Modi, M.K.; Choudhury, M.D. Structural analysis and molecular dynamics simulations of novel δ-endotoxin Cry1Id from Bacillus thuringiensis to pave the way for development of novel fusion proteins against insect pests of crops. J. Mol. Model. 2013, 19, 5301–5316. [Google Scholar] [CrossRef] [PubMed]
- Plácido, A.; Coelho, A.; Abreu, N.L.; Gomes, V.A.; Fátima, B.M.; Ramos, J.J.; Marani, M.M. Cry1A(b)16 toxin from Bacillus thuringiensis: Theoretical refinement of three-dimensional structure and prediction of peptides as molecular markers for detection of genetically modified organisms. Proteins 2017, 85, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, S.; Gómez, I.; Sánchez, J.; García-Gómez, B.I.; Soberón, M.; Bravo, A. An intramolecular salt bridge in Bacillus thuringiensis Cry4Ba toxin is involved in the stability of Helix α-3, which is needed for oligomerization and insecticidal activity. Appl. Environ. Microbiol. 2017, 83, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Javed, M.R.; Rao, A.; Khan, M.A.; Ahad, A.; Shahid, A.A.; Husnain, T. In-silico determination of insecticidal potential of Vip3Aa-Cry1Ac fusion protein against Lepidopteran targets using molecular docking. Front. Plant Sci. 2015, 6, 1081. [Google Scholar] [CrossRef] [PubMed]
- Pigott, C.R.; Ellar, D.J. Role of receptors in Bacillus thuringiensis crystal toxin activity. Microbiol. Mol. Biol. Rev. 2007, 71, 255–281. [Google Scholar] [CrossRef] [PubMed]
- Knight, P.J.; Crickmore, N.; Ellar, D.J. The receptor for Bacillus thuringiensis CrylA(c) delta-endotoxin in the brush border membrane of the lepidopteran Manduca sexta is aminopeptidase N. Mol. Microbiol. 1994, 11, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Elleuch, J.; Zghal, R.Z.; Jemaà, M.; Azzouz, H.; Tounsi, S.; Jaoua, S. New Bacillus thuringiensis toxin combinations for biological control of lepidopteran larvae. Int. J. Biol. Macromol. 2014, 65, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Dammak, M.; Jaoua, S.; Tounsi, S. Construction of a Bacillus thuringiensis genetically-engineered strain harbouring the secreted Cry1Ia delta-endotoxin in its crystal. J. Biotechnol. Lett. 2011, 33, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Pazos, S.A.; Salamanca, J.A. Minireview and hypothesis: Homology modeling of Spodoptera litura (Lepidoptera: Noctuidae) Aminopeptidase N receptor. Rev. Acad. Colomb. Cien. 2008, 32, 139–144. [Google Scholar]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- O’donoghue, S.I.; Goodsell, D.S.; Frangakis, A.S.; Jossinet, F.; Laskowski, R.A.; Nilges, M.; Saibil, H.R.; Schafferhans, A.; Wade, R.C.; Westhof, E.; et al. Visualization of macromolecular structures. Nat. Methods 2010, 7 (Suppl. 3), S42–S55. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Rajamohan, F.; Jenkins, J.L.; Curtiss, A.S.; Dean, D.H. Role of two arginine residues in domain II, loop 2 of Cry1Ab and Cry1Ac Bacillus thuringiensis delta-endotoxin in toxicity and binding to Manduca sexta and Lymantria dispar aminopeptidase N. Mol. Microbiol. 2000, 38, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Jenkins, J.L.; You, T.H.; Curtiss, A.; Son, J.J.; Adang, M.J.; Dean, D.H. Mutations at the arginine residues in alpha8 loop of Bacillus thuringiensis delta-endotoxin Cry1Ac affect toxicity and binding to Manduca sexta and Lymantria dispar aminopeptidase N. FEBS Lett. 2001, 497, 108–112. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.L.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schaffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCBS protein data bank (PDB). Available online: http://www.rcsb.org/ (accessed on 1 September 2018).
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of non-bonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, S. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PATCHDOCK Molecular docking algorithm based on shape complementarity principles. Available online: https://bioinfo3d.cs.tau.ac.il/PatchDock/ (accessed on 14 October 2018).
- Lorenzen, S.; Zhang, Y. Monte Carlo refinement of rigid-body protein docking structures with backbone displacement and side-chain optimization. Prot. Sci. 2007, 16, 2716–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.P.S.; Philippe, H.H.; Ilario, G.T.; Alan, E.M.; Salomon, R.B.; Jens, F.; Andrew, E.T.; Thomas, H.; Peter, K.; Wilfred, F.G. The GROMOS biomolecular simulation program package. J. Phys. Chem. 1999, 103, 3596–3607. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ramachandran Plot Analysis Parameters | Template 4ARY_A1 | Cry1AcF | ||
|---|---|---|---|---|
| Number of Residues | Percentage (%) | Number of Residues | Percentage (%) | |
| Template 4ARY 1A | ||||
| Residues in most favored regions [A,B,L] | 1843 | 91.3% | 938 | 89.8% |
| Residues in additional allowed regions [a,b,l,p] | 168 | 8.3% | 91 | 8.7% |
| Residues in generously allowed regions [~a,~b,~l,~p] | 3 | 0.1% | 10 | 1% |
| Residues in disallowed regions | 4 | 0.2% | 5 | 0.5% |
| Total | 100% | 100% | ||
| Number of non-glycine and non-proline residues | 2018 | 1044 | ||
| Number of end-residues (excl. Gly and Pro) | 5 | 2 | ||
| Number of glycine residues (shown as triangles) | 172 | 83 | ||
| Number of proline residues | 120 | 53 | ||
| Total number of residues | 2315 | 1182 | ||
| Homology Modeled Protein | Verify-3D (IDScore > 0.2) | ERRAT Score | ProSA (Z Score) |
|---|---|---|---|
| Cry1AcF | 92.56% | 77.193 | −9.69 |
| Helicoverpa armigera (HaAPN1) | 79.86% | 65.8 | −10.21 |
| Spodoptera litura (SlAPN1) | 83.40% | 67 | −10.89 |
| Donor | Chain | Amino Acid Number | Molecules Involved in H Bonding | Acceptor | Chain | Amino Acid Number | Molecules Involved in H Bonding | Distance |
|---|---|---|---|---|---|---|---|---|
| SER | A * | 443 | OG | ASN | B | 494 | OD1 | 3.19 |
| SER | A | 443 | OG | ALA | B | 492 | O | 1.86 |
| ASN | B # | 494 | ND2 | SER | A | 443 | O | 2.80 |
| LYS | B | 463 | NZ | ASN | A | 442 | OD1 | 2.60 |
| ASN | B | 494 | N | ASN | A | 442 | O | 2.89 |
| ASN | B | 494 | ND2 | ASN | A | 376 | O | 3.22 |
| GLN | A | 347 | N | GLU | B | 399 | OE1 | 2.05 |
| HSD | B | 454 | NDI | ALA | A | 344 | O | 3.29 |
| ASN | B | 459 | ND2 | THR | A | 340 | OG1 | 3.16 |
| THR | A | 340 | OG1 | ASN | B | 459 | OD1 | 2.47 |
| HSD | B | 454 | NE2 | TYR | A | 314 | OH | 2.96 |
| Donor | Chain | Amino Acid Number | Molecules Involved in H Bonding | Acceptor | Chain | Amino Acid Number | Molecules Involved in H Bonding | Distance |
|---|---|---|---|---|---|---|---|---|
| SER | B # | 220 | OG | TYR | A | 306 | OH | 2.98 |
| SER | B | 220 | OG | GLN | A | 320 | OE1 | 2.54 |
| TYR | B | 213 | OH | GLN | A | 347 | O | 2.99 |
| ARG | A * | 349 | NH1 | THR | B | 208 | O | 2.99 |
| ARG | A | 349 | NH2 | SER | B | 248 | O | 2.21 |
| ARG | B | 269 | NH1 | VAL | A | 351 | O | 3.05 |
| ARG | B | 269 | NE | PRO | A | 397 | O | 2.92 |
| ARG | B | 269 | NH2 | PR0 | A | 397 | O | 2.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathinam, M.; Kesiraju, K.; Singh, S.; Thimmegowda, V.; Rai, V.; Pattanayak, D.; Sreevathsa, R. Molecular Interaction-Based Exploration of the Broad Spectrum Efficacy of a Bacillus thuringiensis Insecticidal Chimeric Protein, Cry1AcF. Toxins 2019, 11, 143. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030143
Rathinam M, Kesiraju K, Singh S, Thimmegowda V, Rai V, Pattanayak D, Sreevathsa R. Molecular Interaction-Based Exploration of the Broad Spectrum Efficacy of a Bacillus thuringiensis Insecticidal Chimeric Protein, Cry1AcF. Toxins. 2019; 11(3):143. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030143
Chicago/Turabian StyleRathinam, Maniraj, Karthik Kesiraju, Shweta Singh, Vinutha Thimmegowda, Vandna Rai, Debasis Pattanayak, and Rohini Sreevathsa. 2019. "Molecular Interaction-Based Exploration of the Broad Spectrum Efficacy of a Bacillus thuringiensis Insecticidal Chimeric Protein, Cry1AcF" Toxins 11, no. 3: 143. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030143