Development of Sensitive and Reliable UPLC-MS/MS Methods for Food Analysis of Emerging Mycotoxins in China Total Diet Study

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Tandem Mass Parameters
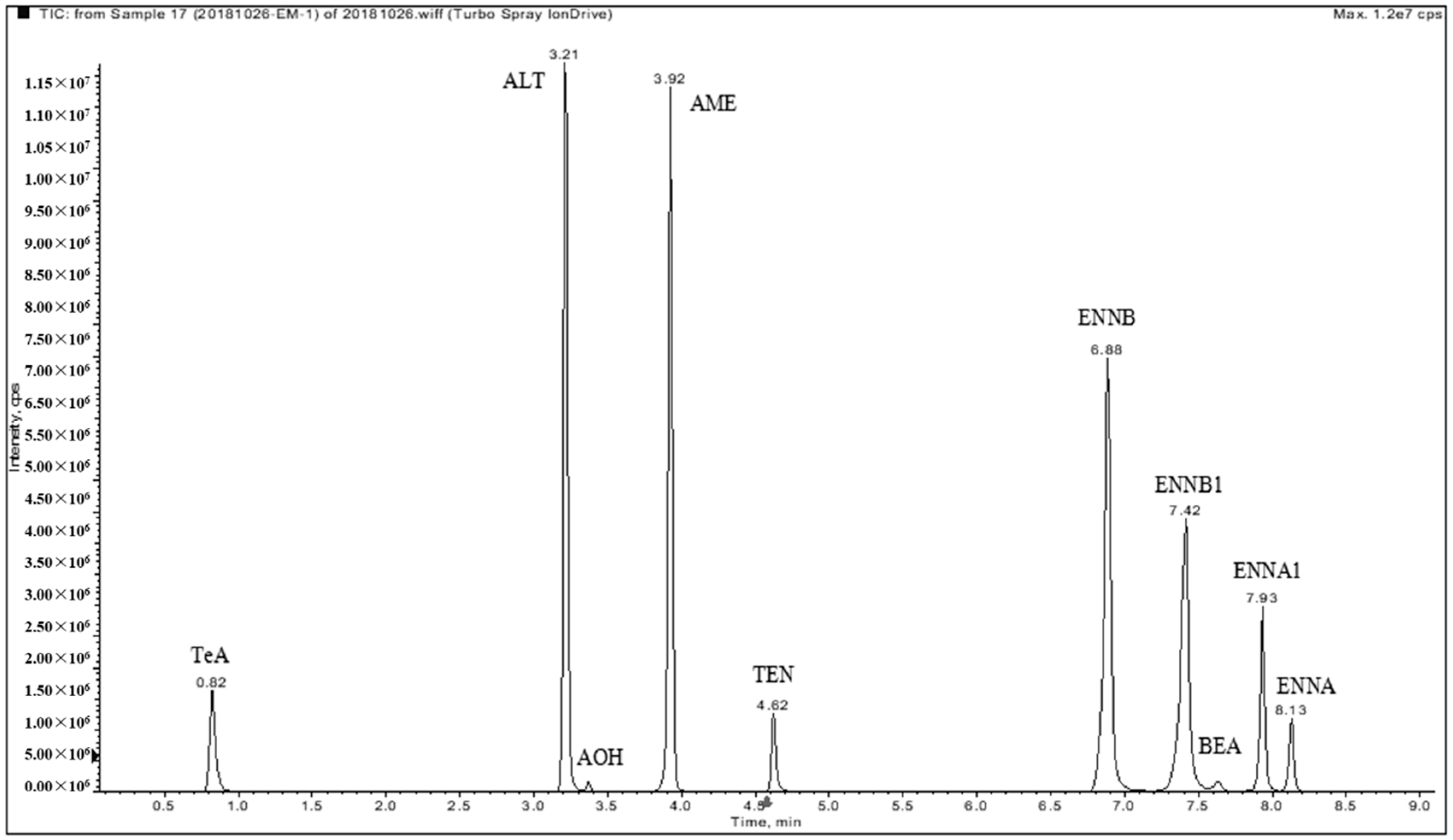
2.2. Chromatographic Separation
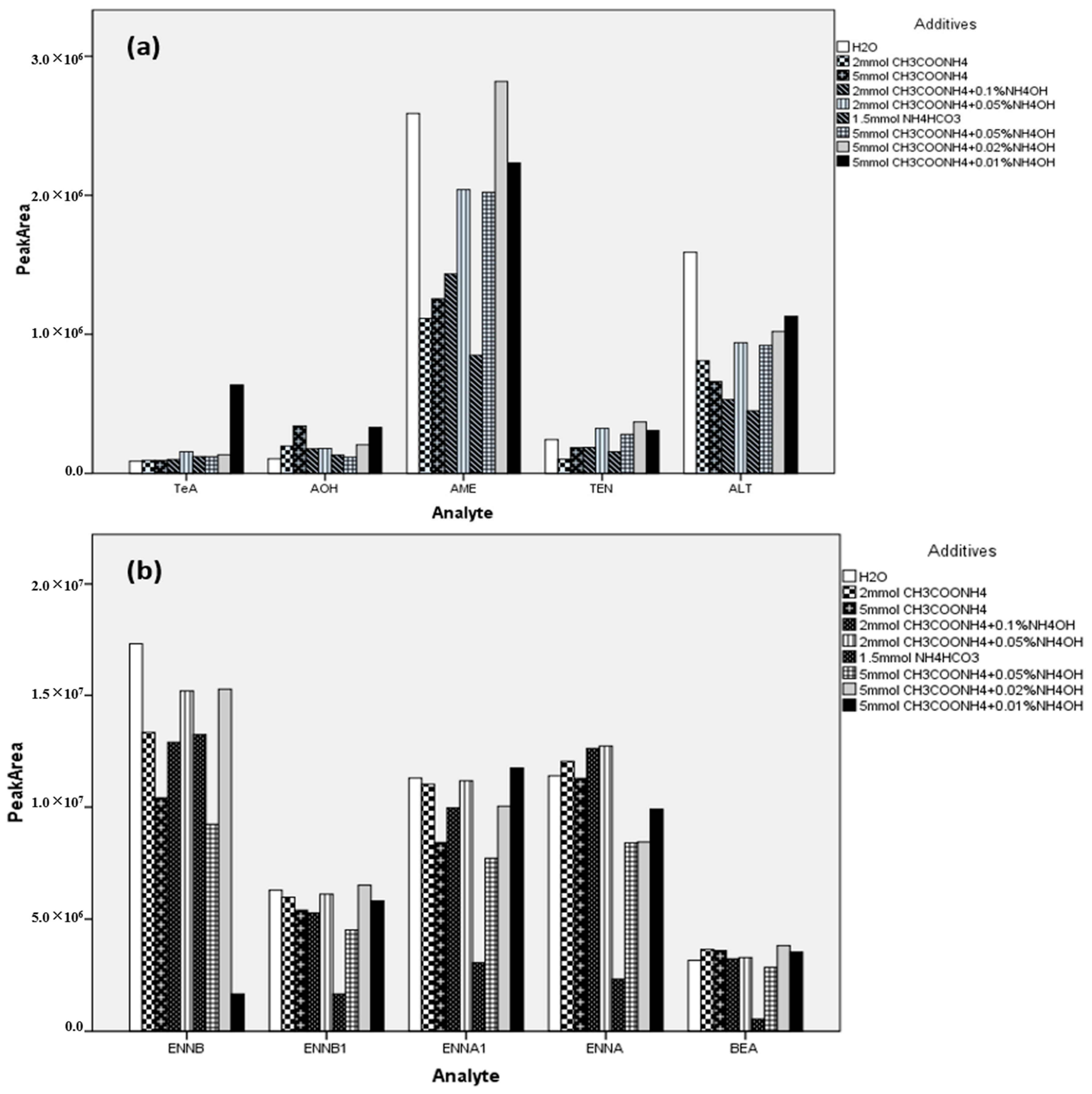
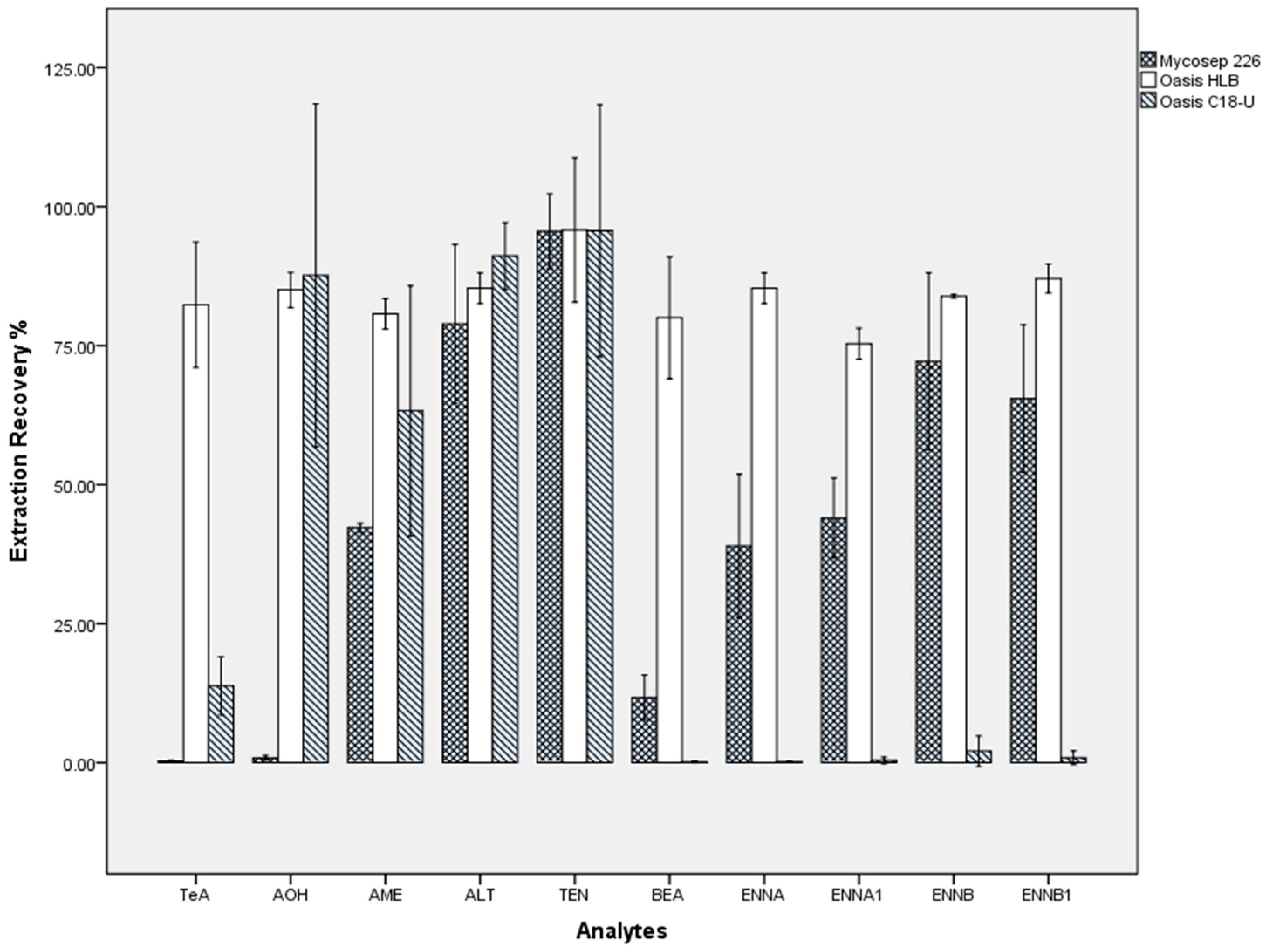
2.3. Sample Preparation
2.4. Method Validation
2.5. Application of the Method in the 6th China TDS
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Food Consumption and Sampling
4.3. Preparation of Standard Serials and Quality Control Samples
4.4. Sample Preparation
4.5. LC-MS/MS Analysis
4.5.1. Chromatographic Condition
4.5.2. Mass Spectrometry Condition
4.6. Method Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chełkowski, J.; Visconti, A. Alternaria: Biology, Plant Diseases and Metabolites; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Scott, P.M.; Kanhere, S.R. Chromatographic method for Alternaria toxins in apple juice. Methods Mol. Biol. 2001, 157, 225–234. [Google Scholar] [PubMed]
- Carson, M.L. Epidemiology and yield losses associated with Alternaria blight of sunflower. Phytopathology 1985, 75, 1151–1156. [Google Scholar] [CrossRef]
- De Berardis, S.; De Paola, E.L.; Montevecchi, G.; Garbini, D.; Masino, F.; Antonelli, A.; Melucci, D. Determination of four Alternaria alternata mycotoxins by QuEChERs approach coupled with liquid chromatography-tandem mass spectrometry in tomato-based and fruit-based products. Food Res. Int. 2018, 106, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Rang, J.; Crous, P.W.; Mchau, G.R.A.; Serdani, M.; Song, S. Phylogenetic analysis of Alternaria spp. associated with apple core rot and citrus black rot in South Africa. Mycol. Res. 2002, 106, 1151–1162. [Google Scholar] [CrossRef]
- Kosiak, B.; Torp, M.; Skjerve, E.; Andersen, B. Alternaria and Fusarium in Norwegian grains of reduced quality-a matched pair sample study. Int. J. Food Microbiol. 2004, 93, 51–62. [Google Scholar] [CrossRef]
- Asam, S.; Konitzer, K.; Schieberle, P.; Rychlik, M. Stable isotope dilution assays of alternariol and alternariol monomethyl ether in beverages. J. Agric. Food Chem. 2009, 57, 5152–5160. [Google Scholar] [CrossRef]
- Solfrizzo, M.; De Girolamo, A.; Vitti, C.; Visconti, A. Liquid chromatographic determination of Alternaria toxins in carrots. J. AOAC Int. 2004, 87, 101–106. [Google Scholar] [PubMed]
- Siegel, D.; Feist, M.; Proske, M.T.; Koch, M.; Nehls, I. Quantification of the Alternaria mycotoxin tenuazonic acid in beer. Food Chem. 2010, 120, 902–906. [Google Scholar] [CrossRef]
- Ostry, V. Alternaria Mycotoxins: An overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008, 1, 175–188. [Google Scholar] [CrossRef]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging Mycotoxins: Beyond Traditionally Determined Food Contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.W.; Posner, H.; Blois, M.; Harvan, D.; Spalding, J.W. Toxicity of metabolites produced by the Alternaria. Environ. Health Perspect. 1973, 4, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Eschbach, S.; Metzler, M. Alternaria toxins: DNA strand-breaking activity in mammalian cells in vitro. Mycotoxin Res. 2007, 3, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Brugger, E.M.; Wagner, J.; Schumacher, D.M.; Koch, K.; Podlech, J.; Metzler, M.; Lehmann, L. Mutagenicity of the mycotoxin alternariol in cultured mammalian cells. Toxicol. Lett. 2006, 3, 221–230. [Google Scholar] [CrossRef]
- Liu, G.T.; Qian, Y.Z.; Zhang, P.; Dong, W.H.; Qi, Y.M.; Guo, H.T. Etiological role of Alternaria alternata in human esophageal cancer. Chin. Med. J. 1992, 105, 394–400. [Google Scholar]
- EFSA. Scientific opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 2407. [Google Scholar] [CrossRef]
- EFSA. Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, 4654. [Google Scholar] [CrossRef]
- Gaumann, E.; Roth, S.M.; Ettlinger, L.; Plattner, P.A.; Nager, U. Enniatin, ein neues gegen Mykobakterien wirksames Antibiotikum. Experimentia 1947, 3, 202–203. [Google Scholar] [CrossRef]
- Bernardini, M.; Carillia, A.; Pacioni, G.; Santurbano, B. Isolation of beauvericin from Paecilomyces fumoso-roseus. Phytochemistry 1975, 14, 1865. [Google Scholar] [CrossRef]
- Deol, B.S.; Ridley, D.D.; Singh, P. Isolation of cyclodepsipeptides from plant pathogenic fungi. Aust. J. Chem. 1978, 31, 1397–1399. [Google Scholar] [CrossRef]
- Nilanonta, C.; Isaka, M.; Chanphen, R.; Thong, O.N.; Tanticharoen, M.; Thebtaranonth, Y. Unusual enniatins produced by the insect pathogenic fungus Verticillium hemipterigenum: Isolation and studies on precursor-directed biosynthesis. Tetrahedron 2003, 59, 1015–1020. [Google Scholar] [CrossRef]
- Santini, A.; Meca, G.; Uhlig, S.; Ritieni, A. Fusaproliferin, beauvericin and enniatins: Occurrence in food-a review. World Mycotoxin J. 2012, 5, 71–81. [Google Scholar] [CrossRef]
- Zinedine, A.; Meca, G.; Manes, J.; Font, G. Further data on the occurrence of Fusarium emerging mycotoxins enniatins (A, A1, B, B1), fusaproliferin and beauvericin in raw cereals commercialized in Morocco. Food Control 2011, 22, 1–5. [Google Scholar] [CrossRef]
- Kouri, K.; Lemmens, M.; Lemmens, G.R. Beauvericin-induced channels in ventricular myocytes and liposomes. Biochim. Biophys. Acta 2003, 1609, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Kamyar, M.; Rawnduzi, P.; Studenik, C.R. Investigation of the electrophysiological properties of enniatins. Arch. Biochem. Biophys. 2004, 429, 215–223. [Google Scholar] [CrossRef]
- Ojcius, D.M.; Zychlinsky, A.; Zheng, L.M.; Young, J.D. Ionophore-induced apoptosis: Role of DNA fragmentation and calcium fluxes. Exp. Cell Res. 1991, 197, 43–49. [Google Scholar] [CrossRef]
- Fornelli, F.; Minervini, F.; Logrieco, A. Cytotoxicity of fungal metabolites to lepidopteran (Spodoptera frugiperda) cell line (SF-9). J. Invertebr. Pathol. 2004, 85, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Logrieco, A.; Moretti, A.; Fornelli, F.; Fogliano, V.; Ritieni, A.; Caiaffa, M.F. Fusaproliferin production by Fusarium subglutinans and its toxicity to Artemia salina SF-9 insect cells and IARC/LCL171 human B lymphocytes. Appl. Environ. Microbiol. 1996, 62, 3378–3384. [Google Scholar]
- Josephs, R.D.; Krska, R.; Schuhmacher, R.; Grasserbauer, M.A. Rapid method for the determination of the Fusarium mycotoxin beauvericin in maize. Fresenius J. Anal. Chem. 1999, 363, 130–131. [Google Scholar] [CrossRef]
- Logrieco, A.; Rizzo, A.; Ferracane, R.; Ritieni, A. Occurrence of beauvericin and enniatins in wheat affected by Fusarium avenaceum head blight. Appl. Environ. Microbiol. 2002, 68, 82–85. [Google Scholar] [CrossRef]
- Krska, R.; Schuhmache, R.; Grasserbauer, M. Determination of the Fusarium mycotoxin beauvericin at micrograms/kg levels in corn by high-performance liquid chromatography with diode-array detection. J. Chromatogr. A 1996, 746, 233–238. [Google Scholar] [CrossRef]
- Hasan, H.A. Alternaria mycotoxins in black rot lesion of tomato fruit: Conditions and regulation of their production. Mycopathologia 1995, 130, 171–177. [Google Scholar] [CrossRef]
- Pavón, M.Á.; Luna, A.; de la Cruz, S.; González, I.; Martín, R.; García, T. PCR-based assay for the detection of Alternaria species and correlation with HPLC determination of altenuene, alternariol and alternariol monomethyl ether production in tomato products. Food Control 2012, 25, 45–52. [Google Scholar] [CrossRef]
- Sørensen, J.L.; Nielsen, K.F.; Rasmussen, P.H. Development of a LC-MS/MS Method for the Analysis of Enniatins and Beauvericin in Whole Fresh and Ensiled Maize. J. Agric. Food Chem. 2008, 56, 10439–10443. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M.; Rokka, M.; Eila, J. Determination of Fusarium mycotoxins beauvericin and enniatins (A, A1, B, B1) in eggs of laying hens using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Food Chem. 2009, 115, 1120–1127. [Google Scholar] [CrossRef]
- Prelle, A.; Spadaro, D.; Garibaldi, A.; Gullino, M.L. A new method for detection of five alternaria toxins in food matrices based on LC-APCI-MS. Food Chem. 2013, 140, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Suzuki, Y.; Sugitakonishi, Y. Occurrence of beauvericin and enniatins in wheat flour and corn grits on the Japanese market and their co-contamination with type B trichothecene mycotoxins. Food Addit. Contam. A 2016, 33, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Asam, S.; Konitzer, K.; Rychlik, M. Precise determination of the Alternaria mycotoxins alternariol and alternariol monomethyl ether in cereal, fruit and vegetable products using stable isotope dilution assays. Mycotoxin Res. 2011, 27, 23–28. [Google Scholar] [CrossRef]
- Noser, J.; Schneider, P.; Rother, M.; Schmutz, H. Determination of six Alternaria toxins with UPLC-MS/MS and their occurrence in tomatoes and tomato products from the Swiss market. Mycotoxin Res. 2011, 27, 265–271. [Google Scholar] [CrossRef]
- Walravens, J.; Mikula, H.; Rychlik, M.; Asam, S.; Devos, T.; Njumbe, E.E. Validated UPLC-MS/MS methods to quantitate free and conjugated Alternaria toxins in commercially available tomato products and fruit and vegetable juices in Belgium. J. Agric. Food Chem. 2016, 64, 5101–5109. [Google Scholar] [CrossRef]
- Walravens, J.; Mikula, H.; Rychlik, M.; Asam, S.; Ediage, E.N.; Di Mavungu, J.D. Development and validation of an ultrahigh-performance liquid chromatography tandem mass spectrometric method for the simultaneous determination of free and conjugated Alternaria toxins in cereal-based foodstuffs. J. Chromatogr. A 2014, 1372, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Puntscher, H.; Kütt, M.L.; Skrinjar, P.; Mikula, H.; Podlech, J.; Fröhlich, J.; Marko, D.; Warth, B. Tracking emerging mycotoxins in food: Development of an LC-MS/MS method for free and modified alternaria toxins. Anal. Bioanal. Chem. 2018, 410, 4481–4494. [Google Scholar] [CrossRef] [PubMed]
- Sophie, F.; Siska, C.; Mathias, D.; Gunther, A. Emerging fusarium and alternaria mycotoxins: Occurrence, toxicity and toxicokinetics. Toxins 2017, 9, 228. [Google Scholar]
- WHO. Total Diet Studies: A Recipe for Safer Food. 2005. Available online: http://www.who.int/foodsafety/chem/TDS_recipe_2005_en.pdf (accessed on 17 March 2019).
- Sirot, V.; Fremy, J.M.; Leblanc, J.C. Dietary exposure to mycotoxins and health risk assessment in the second French total diet study. Food Chem. Toxicol. 2013, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eduardo, B.; María, I.; Tania, P.; Cristina, R.; Félix, H. Development of sensitive and rapid analytical methodology for food analysis of 18 mycotoxins included in a total diet study. Anal. Chim. Acta 2013, 783C, 39–48. [Google Scholar]
- Raad, F.; Nasreddine, L.; Hilan, C.; Bartosik, M.; Parent-Massin, D. Dietary exposure to aflatoxins, ochratoxin a and deoxynivalenol from a total diet study in an adult urban lebanese population. Food Chem. Toxicol. 2014, 73, 35–43. [Google Scholar] [CrossRef]
- Sprong, R.C.; De, W.L.; Te, B.J.D.; Alewijn, M.; Lopez, P.; Mengelers, M.J.B. A mycotoxin-dedicated total diet study in the netherlands in 2013: Part iii—Exposure and risk assessment. World Mycotoxin J. 2016, 9, 109–128. [Google Scholar] [CrossRef]
- Wu, Y.N.; Li, X.W. The Fourth China Total Diet Study; Chemical Industry Press: Beijing, China, 2015. [Google Scholar]
- Wu, Y.N.; Zhao, Y.F.; Li, J.G. The Fifth China Total Diet Study; Science Press: Beijing, China, 2018. [Google Scholar]
- Jesus, B.; Juan-Carlos, M.; Samira, E.A.; Jordi, M.; Andellah, Z. Simultaneous determination of Fusarium mycotoxins in wheat grain from Morocco by liquid chromatography coupled to triple quadrupole mass spectrometry. Food Control 2014, 46, 1–5. [Google Scholar]
- Sebastià, N.; Meca, G.; Soriano, J.M.; Mañes, J. Presence of Fusarium emerging mycotoxins in tiger-nuts commercialized in Spain. Food Control 2012, 25, 631–635. [Google Scholar] [CrossRef]
- Josefa, T.; Guillermina, F.; Jorge, M.; Emilia, F. Nuts and dried fruits: Natural occurrence of emerging Fusarium mycotoxins. Food Control 2013, 33, 215–220. [Google Scholar]
- Katharina, H.; Marina, G.; Jan, S.; Michael, R. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in beer. Food Chem. 2017, 218, 447–454. [Google Scholar]
- Antonia, G.F.; Roberto, R.; María, L.G.; José, L.M.V. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar]
- Souheib, O.; Guiseppe, M.; Ahmed, M.; Abdelwahed, G.; Jordi, M. Determination of Fusarium mycotoxins enniatins, beauvericin and fusaproliferin in cereals and derived products from Tunisia. Food Control 2011, 22, 1373–1377. [Google Scholar]
- Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF (accessed on 17 March 2019).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 12 January 2016).
- Kataoka, H.; Itano, M.; Ishizaki, A. Determination of patulin in fruit juice and dried fruit samples by in-tube solid-phase microextraction coupled with liquid chromatography-mass spectrometry. J. Chromatogr. A 2009, 1216, 3746–3750. [Google Scholar] [CrossRef] [PubMed]
- Fente, C.A.; Jaimez, J.; Vazquez, B.I. Determination of alternariol in tomato paste using solid phase extraction and high-performance liquid chromatography with fluorescence detection. Analyst 1998, 123, 2277–2280. [Google Scholar] [CrossRef]
- Rodrigo, E.M.; Gezimar, D.; Edson, R. Analysis of Alternariol and Alternariol Monomethyl Ether on Flavedo and Albedo Tissues of Tangerines (Citrus reticulata) with Symptoms of Altemaria Brown Spot. J. Agric. Food Chem. 2007, 55, 4980–4986. [Google Scholar]
- Herebian, D.; Zühlke, S.; Lamshöft, M.; Spiteller, M. Multi-Mycotoxin Analysis in Complex Biological Matrices Using LC-ESI/MS: Experimental Study Using Triple Stage Quadrupole and LTQ-Orbitrap. J. Sep. Sci. 2009, 32, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Nazari, F.; Sulyok, M.; Kobarfard, F.; Yazdanpanah, H.; Krska, R. Evaluation of Emerging Fusarium mycotoxins beauvericin, Enniatin, Fusaproliferin and Moniliformin in Domestic Rice in Iran. Iran. J. Pharm. Res. 2015, 14, 505–512. [Google Scholar]
- Michael, S.; Rudolf, K.; Rainer, S. Application of an LC-MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010, 119, 408–416. [Google Scholar]
- Hamler, K.; Rychlik, M. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in cereals. Anal. Bioanal. Chem. 2016, 218, 447–454. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- European Commission. Method Validation and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed. Document No. SANTE/11813/2017. 2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 1 January 2018).




{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor | Quantification Ion | DP/CE 1 | Confirmation Ion | DP/CE 1 | Ion Ratio |
|---|---|---|---|---|---|---|
| AOH | 258.8 | 185.1 | 150/43 | 213.0 | 150/37 | 0.88 |
| AME | 270.9 | 256.0 | −110/−29 | 228.0 | −110/−39 | 0.30 |
| TeA | 196.2 | 139.0 | −50/−28 | 112.2 | −50/−34 | 0.71 |
| TEN | 415.3 | 312.2 | 120/29 | 301.9 | 120/19 | 0.23 |
| ALT | 292.9 | 275.1 | 30/13 | 257.0 | 30/25 | 0.70 |
| BEA | 784.5 | 244.2 | 220/38 | 262.3 | 220/34 | 0.69 |
| ENNA1 | 668.2 | 210.0 | 200/32 | 228.2 | 200/33 | 0.45 |
| ENNA | 682.3 | 210.0 | 220/34 | 228.2 | 220/37 | 0.41 |
| ENNB1 | 654.4 | 196.0 | 180/33 | 214.1 | 180/35 | 0.76 |
| ENNB | 640.3 | 196.4 | 180/34 | 214.2 | 180/33 | 0.62 |
| 13C-TeA | 198.2 | 141.0 | −50/−28 | 114.0 | −50/−36 | 0.50 |
| d2-TEN | 440.2 | 404.4 | 140/35 | 412.4 | 140/37 | 0.31 |
| 13C-AFB2 | 332.0 | 303.2 | 100/38 | 273.1 | 100/45 | 0.71 |
| Analyte | RE 1 (%) | Matrix Effect (%) | RA 2 (%) | LOQ (μg kg−1) | LOD (μg kg−1) |
|---|---|---|---|---|---|
| AME | 62.4~78.2 | 71.4~134 | 55.8~100.8 | 0.01~0.08 | 0.003~0.03 |
| TeA | 75.6~100.8 | 65.6~122 | 61.6~98.6 | 0.1~0.9 | 0.04~0.3 |
| TEN | 78.2~99.5 | 99.7~162.7 | 94.2~140.9 | 0.05~0.2 | 0.02~0.05 |
| ALT | 73.2~120.7 | 98.5~190.7 | 77.3~158 | 0.2~0.9 | 0.04~0.3 |
| AOH | 53.7~107.5 | 73.9~143.5 | 51.5~111.3 | 0.4~0.9 | 0.1~0.3 |
| BEA | 51.3~113.3 | 74.5~174.8 | 53.3~152.3 | 0.01~0.08 | 0.002~0.02 |
| ENNA1 | 55.5~110.7 | 71.4~188.2 | 63.8~146.6 | 0.007~0.06 | 0.002~0.1 |
| ENNA | 54.4~117.0 | 72.3~155.66 | 48.4~182.1 | 0.007~0.06 | 0.002~0.02 |
| ENNB1 | 56~89.0 | 114.4~196.7 | 76.5~139 | 0.007~0.06 | 0.002~0.02 |
| ENNB | 54~110.7 | 115.7~170.0 | 74.4~135.1 | 0.002~0.04 | 0.0004~0.01 |
| Analyte | Matrices | Sample Preparation | Analysis Method | LOQ | References |
|---|---|---|---|---|---|
| ATs | Fruit and fruit juices | SPE | UPLC-MS/MS | 0.6~3 μg L−1 | [59] |
| ATs | Juices, beers, and tomato sauces | SPE | LC-APCI-MS | 0.16~12.31 μg kg−1 | [36] |
| ATs | Tomato paste | SPE | HPLC-PDA | 1.93 μg kg−1 | [60] |
| AOH, AME | Tangerines | SPE | HPLC-MS/MS | 0.13 μg kg−1 | [61] |
| ALT, AOH, AME, ENNB, BEA | Maize and Wheat | Extraction solvent (ACN/water/acetic acid, 79:20:1 v/v/v) | HPLC-MS/MS | 0.5~1 μg kg−1 | [62] |
| ENNs, BEA | Cereals (wheat, barley, maize, and sorghum) | Extracted by methanol | LC-DAD | 400~600 μg kg−1 | [60] |
| ENNs | wheat flour and corn grits | SPE | LC-MS/MS | 2~3 μg kg−1 | [37] |
| ENNs | Rice | Extraction solvent (ACN/water/glacial acetic acid, 79:20:1 v/v/v) | HPLC-MS/MS | 0.06 μg kg−1 | [63] |
| ENNs | Bread, fruits, vegetables, cheeses, nuts, and jam | Extraction solvent (ACN/water/acetic acid, 79:20:1 v/v/v) | HPLC-MS/MS | 0.3~0.9 μg kg−1 | [64] |
| BEA and ENNs | Egg | QuEChERS | UPLC-MS/MS | 2~10 μg kg−1 | [55] |
| BEA and ENNs | Cereals (wheat, Barley, maize, malt and oat) | Bond Elute cartridge | LC-MS/MS | 0.9~4.2 μg kg−1 | [65] |
| Analyte | Spiked Level (ng/mL) | Measured Value (ng/mL) | RM 1 (%) | RSD (%) | |
|---|---|---|---|---|---|
| Intra-Day | Inter-Day | ||||
| AME | 2 | 1.68 (1.26~2.63) | 84.04 (62.8~131.1) | 1.2~11.8 | 3.6~12.2 |
| 20 | 16.08 (14.36~21.86) | 80.38 (71.8~109.3) | 1.2~5.2 | 3.6~8.7 | |
| 200 | 158.93 (131.00~188) | 79.46 (65.5~94) | 2.1~7.6 | 2.8~10.3 | |
| TeA | 2 | 2.07 (1.62~2.58) | 103.28 (81~129.1) | 1.2~6.9 | 3.3~9.3 |
| 20 | 18.63 (17.07~20.25) | 93.18 (85.3~101.3) | 1.4~8.2 | 3.2~10.9 | |
| 200 | 179.08 (171.4~194) | 89.53 (85.7~97) | 0.9~7.8 | 3.2~10.5 | |
| TEN | 2 | 2.27 (1.87~2.52) | 113.58 (93.5~125.9) | 2.2~9.5 | 4.4~11.8 |
| 20 | 21.60 (16.35~26.34) | 107.11 (81.8~131.7) | 1.2~9.2 | 3.2~10.8 | |
| 200 | 203.21 (167.3~232.9) | 99.38 (75.8~116.4) | 1.6~6.3 | 2.3~9.8 | |
| ALT | 2 | 2.28 (1.71~2.69) | 113.92 (85.5~134.6) | 1.6~8.1 | 3.6~10.5 |
| 20 | 21.96 (15.98~25.42) | 109.79 (80~127.1) | 1.2~8.4 | 6.3~11.3 | |
| 200 | 205.13 (151.5~257.2) | 102.58 (75.8~128.6) | 1.6~7.8 | 4.3~10.2 | |
| AOH | 2 | 2.06 (1.41~2.88) | 102.98 (70.7~143.8) | 2.1~8.2 | 4.8~11.2 |
| 20 | 17.19 (13.59~24.16) | 85.98 (67.9~120.8) | 2.4~11.1 | 5.1~12.3 | |
| 200 | 176.87 (136.2~257.6) | 88.43 (68.1~128.8) | 1.5~9.8 | 3.2~10.2 | |
| BEA | 0.2 | 0.22 (0.13~0.3) | 110.79 (63.1~149.1) | 1.5~9.6 | 4.8~11.2 |
| 2 | 1.90 (1.31~2.84) | 94.89 (65.3~141.9) | 2.0~7.7 | 4.1~9.6 | |
| 20 | 16.77 (12.31~24.75) | 83.88 (61.5~123.8) | 2.1~9.1 | 5.3~10.9 | |
| ENNA1 | 0.2 | 0.23 (0.13~0.31) | 114.33 (64.5~154.3) | 1.2~8.1 | 2.4~10.2 |
| 2 | 1.78 (1.21~2.57) | 88.83 (60.6~128.5) | 1.3~5.6 | 2.5~8.8 | |
| 20 | 18.12 (12.22~26.81) | 90.62 (61.1~134.1) | 2.3~9.2 | 4.4~11.5 | |
| ENNA | 0.2 | 0.22 (0.12~0.33) | 107.68 (60.3~164.4) | 2.6~6.5 | 4.8~10.1 |
| 2 | 1.92 (1.23~2.7) | 98.38 (61.6~145.3) | 1.3~4.8 | 2.3~8.3 | |
| 20 | 18.75 (13.96~27.64) | 93.73 (69.8~138.1) | 1.2~10.1 | 2.1~12.1 | |
| ENNB1 | 0.2 | 0.21 (0.15~0.31) | 104.08 (72.7~154.9) | 1.1~5.9 | 3.8~10.2 |
| 2 | 1.76 (1.32~3.14) | 88.23 (66.2~156.8) | 1.7~9.1 | 3.6~10.2 | |
| 20 | 17.64 (12.94~26.66) | 88.23 (64.7~133.3) | 1.4~8.4 | 4.3~11.3 | |
| ENNB | 0.2 | 0.20 (0.14~0.31) | 101.13 (71.9~153.4) | 2.4~5.6 | 4.2~10.8 |
| 2 | 1.81 (1.32~2.53) | 90.26 (65.9~126.5) | 1.3~10.3 | 4.3~11.2 | |
| 20 | 19.31 (12.16~35.19) | 96.54 (60.8~175.9) | 1.9~9.1 | 2.3~11.5 | |
| Food | AME | TeA | TEN | AOH | ALT | BEA | ENNA | ENNA1 | ENNB | ENNB1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cereals and cereal products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 4 | 5 | 4 | 0 | 0 | 4 | 3 | 3 | 5 | 4 | |
| Range (μg kg−1) | 0.14–10.92 | 2.60–15.11 | 0.22–2.28 | - | - | 0.55–1.95 | 0.12–0.83 | 0.10–0.22 | 0.37–1.05 | 0.10–0.35 | |
| Legume and related products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 1 | 3 | 3 | 0 | 0 | 4 | 2 | 2 | 4 | 2 | |
| Range (μg kg−1) | 1.77 | 0.33–2.79 | 0.10–0.28 | - | - | 0.47–5.46 | 1.03–1.26 | 0.26–0.29 | 0.16–0.37 | 0.36 | |
| Potatoes and potato products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 4 | 2 | 1 | 0 | 2 | 2 | 5 | 1 | 4 | 4 | |
| Range (μg kg−1) | 0.13–2.40 | 0.20–0.41 | 0.96 | - | 1.08–1.72 | 2.23–2.58 | 0.13–1.01 | 0.15 | 0.21–0.87 | 0.11–0.34 | |
| Meats and meat products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | |
| Range (μg kg−1) | - | - | - | - | - | - | - | - | 0.13–0.24 | - | |
| Eggs and egg products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 2 | 4 | 2 | 0 | 0 | 3 | 3 | 2 | 4 | 4 | |
| Range (μg kg−1) | 0.72–1.31 | 0.26–27.73 | 0.11–27.53 | - | - | 1.28–6.70 | 0.20–1.55 | 0.30–0.51 | 0.15–0.61 | 0.18–0.50 | |
| Aquatic foods and aquatic food products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 1 | 1 | 2 | 0 | 1 | 2 | 0 | 0 | 3 | 1 | |
| Range (μg kg−1) | 0.38 | 0.68 | 0.10–0.68 | - | 8.77 | 0.32–1.02 | - | - | 0.29–0.65 | 0.22 | |
| Milk and dairy products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 0 | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| Range (μg kg−1) | - | 1.93–3.00 | - | - | - | 0.16 | - | - | 0.26 | - | |
| Vegetables and vegetable products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 2 | 3 | 2 | 0 | 1 | 2 | 0 | 0 | 3 | 2 | |
| Range (μg kg−1) | 1.10–3.35 | 0.19–0.71 | 0.21–0.25 | - | 1.22 | 1.85–3.29 | - | - | 0.20–0.47 | 0.22–0.26 | |
| Fruits and fruit products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| Range (μg kg−1) | 0.2 | 6.62 | - | - | - | 0.21 | - | - | - | - | |
| Sugar and sugar products | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Range (μg kg−1) | 0.12 | - | - | - | - | - | - | - | - | - | |
| Beverages and water | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Range (μg kg−1) | 0.16 | - | - | - | - | - | - | - | - | - | |
| Alcohol beverages | Samples (n) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Positive (n) | 2 | 4 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| Range (μg kg−1) | 0.27–1.00 | 2.26–17.62 | 0.14–0.16 | - | - | 0.12 | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Qiu, N.; Zhou, S.; Lyu, B.; Zhang, S.; Li, J.; Zhao, Y.; Wu, Y. Development of Sensitive and Reliable UPLC-MS/MS Methods for Food Analysis of Emerging Mycotoxins in China Total Diet Study. Toxins 2019, 11, 166. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030166
Sun D, Qiu N, Zhou S, Lyu B, Zhang S, Li J, Zhao Y, Wu Y. Development of Sensitive and Reliable UPLC-MS/MS Methods for Food Analysis of Emerging Mycotoxins in China Total Diet Study. Toxins. 2019; 11(3):166. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030166
Chicago/Turabian StyleSun, Danlei, Nannan Qiu, Shuang Zhou, Bing Lyu, Shuo Zhang, Jingguang Li, Yunfeng Zhao, and Yongning Wu. 2019. "Development of Sensitive and Reliable UPLC-MS/MS Methods for Food Analysis of Emerging Mycotoxins in China Total Diet Study" Toxins 11, no. 3: 166. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030166