Marine Toxins and Nociception: Potential Therapeutic Use in the Treatment of Visceral Pain Associated with Gastrointestinal Disorders

 , , , and
, , , and
Abstract
:1. Introduction
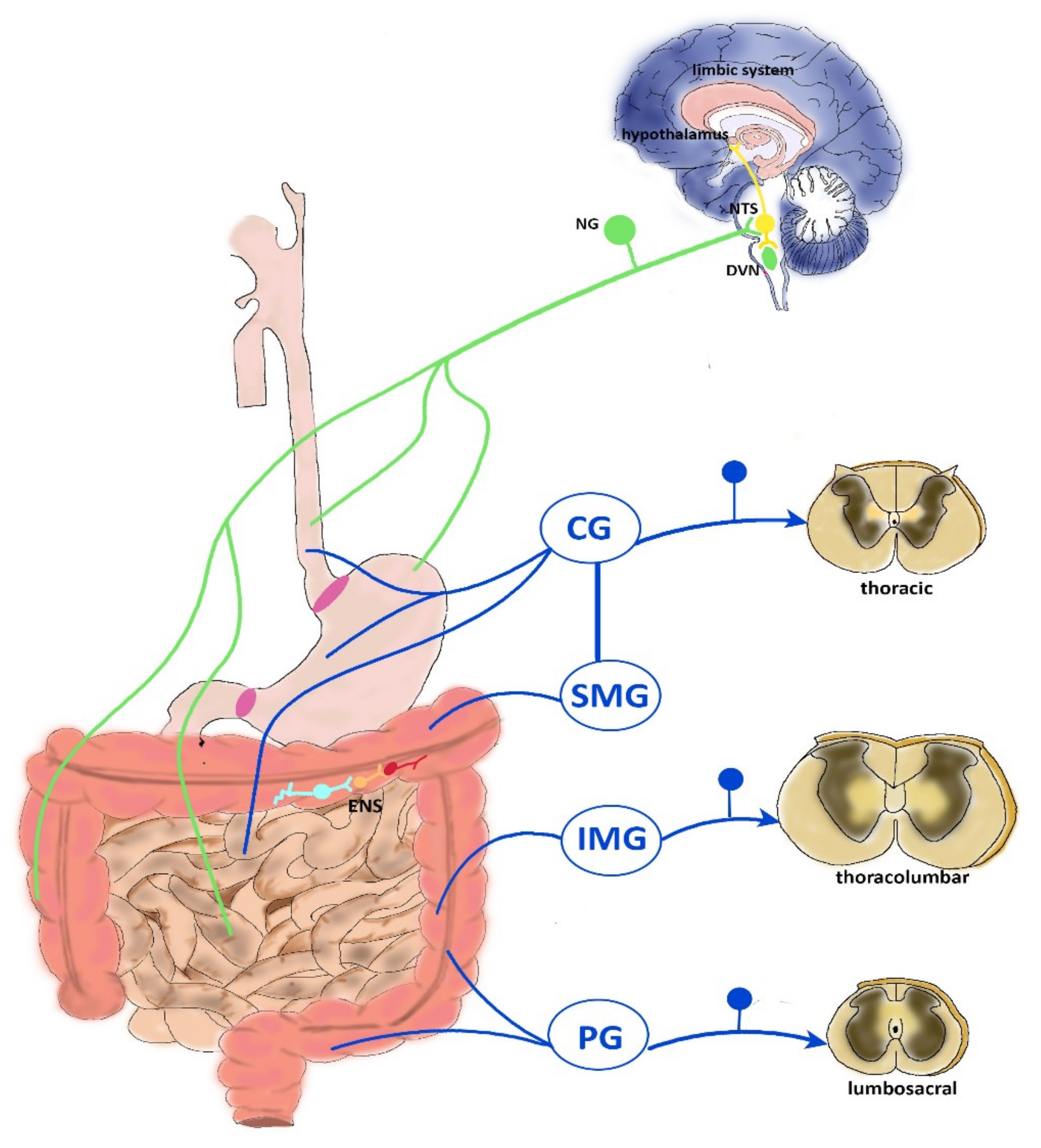
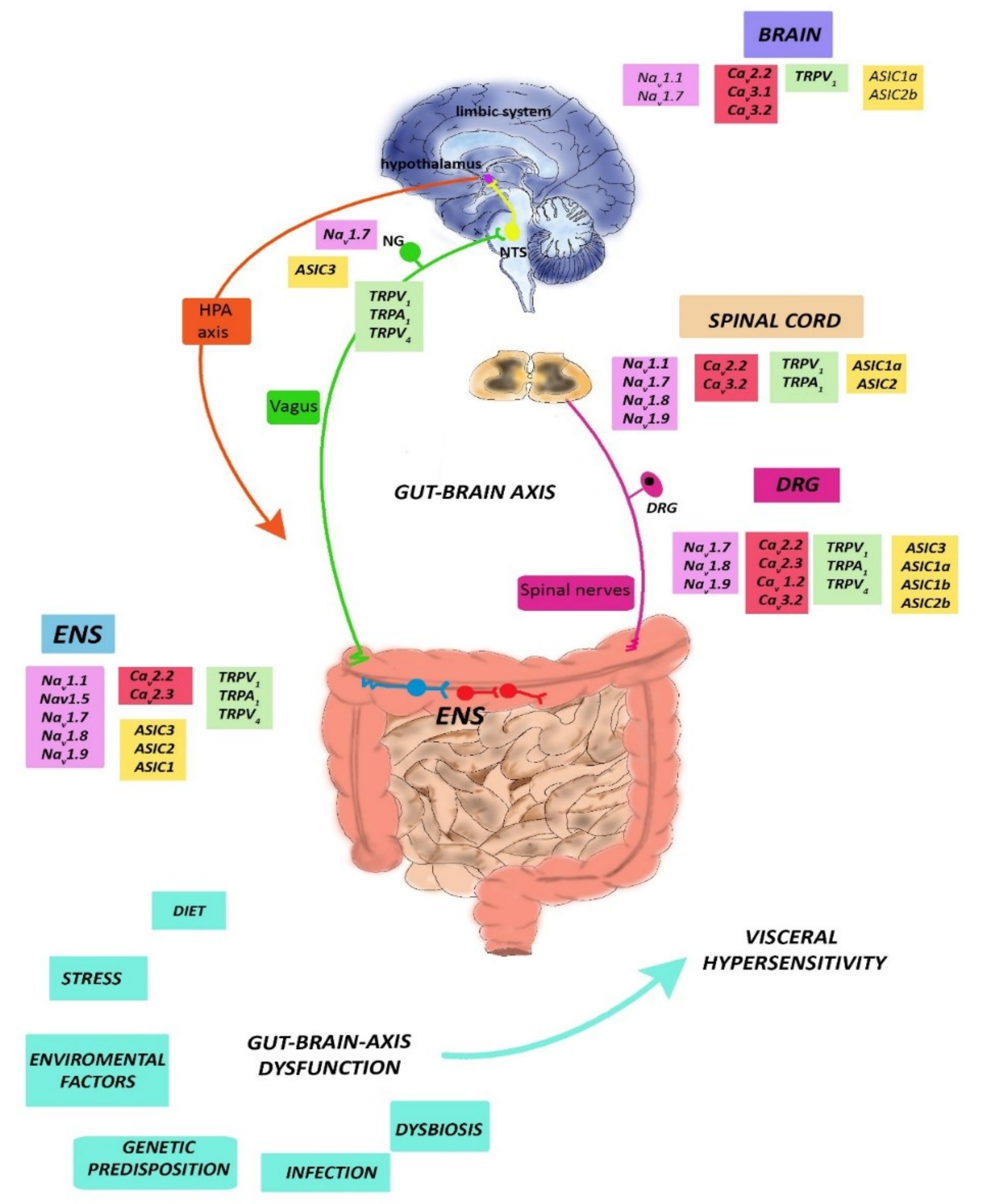
2. Neurophysiology of Visceral Pain
2.1. CNS Modulation of Visceral Pain
2.2. Vagal Innervation
2.3. Thoracolumbar Innervation
2.4. Pelvic Innervation
2.5. ENS Reflexes
3. Chronic Visceral Pain: The Paradigms of IBS and IBD
4. Chronic Abdominal Visceral Pain: Focus on Marine Toxins as Possible Therapeutic Tools
4.1. Toxins Active at VGSCs
4.1.1. VGSCs
4.1.2. Tetrodotoxin and Saxitoxin
4.2. Toxins Active at VGCC
4.2.1. VGCCs
4.2.2. ω-Conotoxin
4.2.3. α-Conotoxin
4.3. Toxins Active at TRPs
4.4. Toxins Acting at ASICs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kannampalli, P.; Sengupta, J.N. Role of principal ionotropic and metabotropic receptors in visceral pain. J. Neurogastroenterol. Motil. 2015, 21, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Enck, P.; Aziz, Q.; Barbara, G.; Farmer, A.D.; Fukudo, S.; Mayer, E.A.; Niesler, B.; Quigley, E.M.M.; Rajilić-Stojanović, M.; Schemann, M.; et al. Irritable bowel syndrome. Nat. Rev. Dis. Primers 2016, 2, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielińska, A.; Sałaga, M.; Włodarczyk, M.; Fichna, J. Focus on current and future management possibilities in inflammatory bowel disease-related chronic pain. Int. J. Colorectal Dis. 2019, 34, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, S.; Dickenson, A.H. Visceral pain: The ins and outs, the ups and downs. Curr. Opin. Support. Palliat. Care 2012, 6, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Moloney, R.D.; O’Mahony, S.M.; Dinan, T.G.; Cryan, J.F. Stress-induced visceral pain: Toward animal models of irritable-bowel syndrome and associated comorbidities. Front. Psychiatry 2015, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic Signaling Along The Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2019, 20, 1482. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Maatuf, Y.; Geron, M.; Priel, A. The Role of Toxins in the Pursuit for Novel Analgesics. Toxins 2019, 11, 131. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.B.; Rash, L.D.; Mobli, M.; King, G.F. Production of Recombinant Disulfide-Rich Venom Peptides for Structural and Functional Analysis via Expression in the Periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef]
- Undheim, E.A.B.; Jenner, R.A.; King, G.F. Centipede venoms as a source of drug leads. Expert Opin. Drug Discov. 2016, 11, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Bohlen, C.J.; Julius, D. Receptor-targeting mechanisms of pain-causing toxins: How ow? Toxicon 2012, 60, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The Enteric Nervous System and Gastrointestinal Innervation: Integrated Local and Central Control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [PubMed]
- Blackshaw, L.A.; Brookes, S.J.H.; Grundy, D.; Schemann, M. Sensory transmission in the gastrointestinal tract. Neurogastroenterol. Motil. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y. Differential Ascending Projections From the Male Rat Caudal Nucleus of the Tractus Solitarius: An Interface between Local Microcircuits and Global Macrocircuits. Front. Neuroanat. 2018, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.F.; Roizenblatt, S.; Tufik, S. Afferent pain pathways: A neuroanatomical review. Brain Res. 2004, 1000, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.P.; Dilley, J.B.; Drossman, D.; Crowell, M.D. Brain-gut connections in functional GI disorders: Anatomic and physiologic relationships. Neurogastroenterol. Motil. 2006, 18, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, W.; De Man, J.G.; Pelckmans, P.A.; De Winter, B.Y. Neuroanatomy of lower gastrointestinal pain disorders. World J. Gastroenterol. 2014, 20, 1005–1020. [Google Scholar] [CrossRef]
- Grundy, D. Neuroanatomy of visceral nociception: Vagal and splanchnic afferent. Gut 2002, 51 (Suppl. 1), i2–i5. [Google Scholar] [CrossRef]
- Page, A.J.; Martin, C.M.; Blackshaw, L.A. Vagal mechanoreceptors and chemoreceptors in mouse stomach and esophagus. J. Neurophysiol. 2002, 87, 2095–2103. [Google Scholar] [CrossRef]
- Holzer, P. Afferent signalling of gastric acid challenge. J. Physiol. Pharmacol. 2003, 54 (Suppl. 4), 43–53. [Google Scholar]
- Brierley, S.M.; Jones, R.C.W.; Gebhart, G.F.; Blackshaw, L.A. Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology 2004, 127, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Green, T.; Dockray, G.J. Characterization of the peptidergic afferent innervation of the stomach in the rat, mouse and guinea-pig. Neuroscience 1988, 25, 181–193. [Google Scholar] [PubMed]
- Tan, L.L.; Bornstein, J.C.; Anderson, C.R. Distinct chemical classes of medium-sized transient receptor potential channel vanilloid 1-immunoreactive dorsal root ganglion neurons innervate the adult mouse jejunum and colon. Neuroscience 2008, 156, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.A.; Traub, R.J.; Davis, B.M. Differences in spinal distribution and neurochemical phenotype of colonic afferents in mouse and rat. J. Comp. Neurol. 2006, 494, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Lynn, P.; Zagorodnyuk, V.; Hennig, G.; Costa, M.; Brookes, S. Mechanical activation of rectal intraganglionic laminar endings in the guinea pig distal gut. J. Physiol. 2005, 564, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyloh, M.; Nicholas, S.; Zagorodnyuk, V.P.; Brookes, S.J.; Spencer, N.J. Identification of the visceral pain pathway activated by noxious colorectal distension in mice. Front. Neurosci. 2011, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Macefield, V.G.; Schemann, M.; Gautron, L.; Brierley, S.M.; Hibberd, T.J.; Spencer, N.J. Spinal Afferent Innervation of the Colon and Rectum. Front. Cell. Neurosci. 2018, 12, 467. [Google Scholar]
- Giaroni, C.; De Ponti, F.; Cosentino, M.; Lecchini, S.; Frigo, G. Plasticity in the enteric nervous system. Gastroenterology 1999, 117, 1438–1458. [Google Scholar] [CrossRef]
- Furness, J.B.; Kunze, W.A.; Bertrand, P.P.; Clerc, N.; Bornstein, J.C. Intrinsic primary afferent neurons of the intestine. Prog. Neurobiol. 1998, 54, 1–18. [Google Scholar] [CrossRef]
- Ceccotti, C.; Giaroni, C.; Bistoletti, M.; Viola, M.; Crema, F.; Terova, G. Neurochemical characterization of myenteric neurons in the juvenile gilthead sea bream (Sparus aurata) intestine. PLoS ONE 2018, 13, e0201760. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Giaroni, C. Purinergic signalling and development of the autonomic nervous system. Auton. Neurosci. Basic Clin. 2015, 191, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Giaroni, C.; Marchet, S.; Carpanese, E.; Prandoni, V.; Oldrini, R.; Bartolini, B.; Moro, E.; Vigetti, D.; Crema, F.; Lecchini, S.; et al. Role of neuronal and inducible nitric oxide synthases in the guinea pig ileum myenteric plexus during in vitro ischemia and reperfusion. Neurogastroenterol. Motil. 2013, 25, e114–e126. [Google Scholar] [CrossRef] [PubMed]
- Filpa, V.; Carpanese, E.; Marchet, S.; Pirrone, C.; Conti, A.; Rainero, A.; Moro, E.; Chiaravalli, A.M.; Zucchi, I.; Moriondo, A.; et al. Nitric oxide regulates homeoprotein OTX1 and OTX2 expression in the rat myenteric plexus after intestinal ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G374–G389. [Google Scholar] [CrossRef] [PubMed]
- Bistoletti, M.; Caputi, V.; Baranzini, N.; Marchesi, N.; Filpa, V.; Marsilio, I.; Cerantola, S.; Terova, G.; Baj, A.; Grimaldi, A.; et al. Antibiotic treatment-induced dysbiosis differently affects BDNF and TrkB expression in the brain and in the gut of juvenile mice. PLoS ONE 2019, 14, e0212856. [Google Scholar] [CrossRef] [PubMed]
- Bin, A.; Caputi, V.; Bistoletti, M.; Montopoli, M.; Colucci, R.; Antonioli, L.; De Martin, S.; Castagliuolo, I.; Orso, G.; Giaroni, C.; et al. The ecto-enzymes CD73 and adenosine deaminase modulate 5′-AMP-derived adenosine in myofibroblasts of the rat small intestine. Purinergic Signal. 2018, 14, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Caputi, V.; Marsilio, I.; Filpa, V.; Cerantola, S.; Orso, G.; Bistoletti, M.; Paccagnella, N.; De Martin, S.; Montopoli, M.; Dall’Acqua, S.; et al. Antibiotic-induced dysbiosis of the microbiota impairs gut neuromuscular function in juvenile mice. Br. J. Pharmacol. 2017, 174, 3623–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbransen, B.D.; Sharkey, K.A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef]
- Buéno, L.; Fioramonti, J.; Garcia-Villar, R. Pathobiology of visceral pain: Molecular mechanisms and therapeutic implications. III. Visceral afferent pathways: A source of new therapeutic targets for abdominal pain. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G670–G676. [Google Scholar] [CrossRef]
- van der Veek, P.P.J.; van Rood, Y.R.; Masclee, A.A.M. Symptom Severity but Not Psychopathology Predicts Visceral Hypersensitivity in Irritable Bowel Syndrome. Clin. Gastroenterol. Hepatol. 2008, 6, 321–328. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Bahramsoltani, R.; Abdollahi, M.; Rahimi, R. The role of visceral hypersensitivity in irritable bowel syndrome: Pharmacological targets and novel treatments. J. Neurogastroenterol. Motil. 2016, 22, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.J.; Simrén, M.; Ludidi, S.; Kruimel, J.W.; Conchillo, J.M.; Masclee, A.A. Revisiting concepts of visceral nociception in irritable bowel syndrome. Eur. J. Pain 2012, 16, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Wang, B.; Stanghellini, V.; de Giorgio, R.; Cremon, C.; Di Nardo, G.; Trevisani, M.; Campi, B.; Geppetti, P.; Tonini, M.; et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 2007, 132, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Li, Y.; Tang, D.; Huang, L.; Yuan, Y. Neuron-glial communication mediated by TNF-α and glial activation in dorsal root ganglia in visceral inflammatory hypersensitivity. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G788–G795. [Google Scholar] [CrossRef] [PubMed]
- Azpiroz, F.; Bouin, M.; Camilleri, M.; Mayer, E.A.; Poitras, P.; Serra, J.; Spiller, R.C. Mechanisms of hypersensitivity in IBS and functional disorders. Neurogastroenterol. Motil. 2007, 19, 62–88. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Rinaman, L.; Cryan, J.F. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol. Stress 2017, 7, 124–136. [Google Scholar] [Green Version]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef]
- De Winter, B.Y.; Deiteren, A.; De Man, J.G. Novel nervous system mechanisms in visceral pain. Neurogastroenterol. Motil. 2016, 28, 309–315. [Google Scholar] [CrossRef]
- Diaz-Horta, O.; Baj, A.; Maccari, G.; Salvatoni, A.; Toniolo, A. Enteroviruses and causality of type 1 diabetes: How close are we? Pediatric Diabetes 2012, 13, 92–99. [Google Scholar] [CrossRef]
- Morales-Soto, W.; Gulbransen, B.D. Enteric Glia: A New Player in Abdominal Pain. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Filpa, V.; Bistoletti, M.; Caon, I.; Moro, E.; Grimaldi, A.; Moretto, P.; Baj, A.; Giron, M.C.; Karousou, E.; Viola, M.; et al. Changes in hyaluronan deposition in the rat myenteric plexus after experimentally-induced colitis. Sci. Rep. 2017, 7, 17644. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Tytgat, J. Toxins in Drug Discovery and Pharmacology. Toxins 2018, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- González-Cano, R.; Tejada, M.; Artacho-Cordón, A.; Nieto, F.; Entrena, J.; Wood, J.; Cendán, C.; González-Cano, R.; Tejada, M.Á.; Artacho-Cordón, A.; et al. Effects of Tetrodotoxin in Mouse Models of Visceral Pain. Mar. Drugs 2017, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Marcil, J.; Walczak, J.-S.; Guindon, J.; Ngoc, A.H.; Lu, S.; Beaulieu, P. Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br. J. Anaesth. 2006, 96, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, D.M.; de Souza, A.H.; Pereira, E.M.R.; da Silva, J.F.; Rigo, F.K.; Romano-Silva, M.A.; Binda, N.; Castro, C.J.; Cordeiro, M.N.; Ferreira, J.; et al. Effects of the calcium channel blockers Phα1β and ω-conotoxin MVIIA on capsaicin and acetic acid-induced visceral nociception in mice. Pharmacol. Biochem. Behav. 2014, 126, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Harrington, A.M.; Garcia-Caraballo, S.; Maddern, J.; Grundy, L.; Zhang, J.; Page, G.; Miller, P.E.; Craik, D.J.; Adams, D.J.; et al. α-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 2017, 66, 1083–1094. [Google Scholar] [CrossRef]
- Castro, J.; Grundy, L.; Deiteren, A.; Harrington, A.M.; O’Donnell, T.; Maddern, J.; Moore, J.; Garcia-Caraballo, S.; Rychkov, G.Y.; Yu, R.; et al. Cyclic analogues of α-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br. J. Pharmacol. 2018, 175, 2384–2398. [Google Scholar] [CrossRef]
- Sadeghi, M.; Carstens, B.B.; Callaghan, B.P.; Daniel, J.T.; Tae, H.-S.; O’Donnell, T.; Castro, J.; Brierley, S.M.; Adams, D.J.; Craik, D.J.; et al. Structure-Activity Studies Reveal the Molecular Basis for GABAB-Receptor Mediated Inhibition of High Voltage-Activated Calcium Channels by α-Conotoxin Vc1.1. ACS Chem. Biol. 2018, 13, 1577–1587. [Google Scholar] [CrossRef]
- Carstens, B.B.; Berecki, G.; Daniel, J.T.; Lee, H.S.; Jackson, K.A.V.; Tae, H.-S.; Sadeghi, M.; Castro, J.; O’Donnell, T.; Deiteren, A.; et al. Structure-Activity Studies of Cysteine-Rich α-Conotoxins that Inhibit High-Voltage-Activated Calcium Channels via GABA(B) Receptor Activation Reveal a Minimal Functional Motif. Angew. Chem. 2016, 55, 4692–4696. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Korolkova, Y.V.; Dyachenko, I.A.; Bondarenko, D.A.; Skobtsov, D.I.; Murashev, A.N.; Kotova, P.D.; Rogachevskaja, O.A.; Kabanova, N.V.; et al. Polypeptide modulators of TRPV1 produce analgesia without hyperthermia. Mar. Drugs 2013, 11, 5100–5115. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tu, W.; Wen, J.; Zhou, H.; Chen, X.; Zhao, G.; Jiang, Q. The selective ASIC3 inhibitor APETx2 alleviates gastric mucosal lesion in the rat. Die Pharm. 2014, 69, 542–546. [Google Scholar]
- Andreev, Y.; Osmakov, D.; Koshelev, S.; Maleeva, E.; Logashina, Y.; Palikov, V.; Palikova, Y.; Dyachenko, I.; Kozlov, S.; Andreev, Y.A.; et al. Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIC3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds. Mar. Drugs 2018, 16, 500. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Kozlov, S.A.; Andreev, Y.A.; Koshelev, S.G.; Sanamyan, N.P.; Sanamyan, K.E.; Dyachenko, I.A.; Bondarenko, D.A.; Murashev, A.N.; Mineev, K.S.; et al. Sea Anemone Peptide with Uncommon β-Hairpin Structure Inhibits Acid-sensing Ion Channel 3 (ASIC3) and Reveals Analgesic Activity. J. Biol. Chem. 2013, 288, 23116–23127. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.D.; Vrana, K.E.; Ruiz-Velasco, V. The influence of voltage-gated sodium channels on human gastrointestinal nociception. Neurogastroenterol. Motil. 2019, 31, e13460. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355, eaal4326. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4, 207. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, H.A.; Isom, L.L. Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2008, 2, 407–412. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Rocha, H.A.; Dantas, B.P.V.; Rolim, T.L.; Costa, B.A.; de Medeiros, A.C. Main ion channels and receptors associated with visceral hypersensitivity in irritable bowel syndrome. Ann. Gastroenterol. 2014, 27, 200–206. [Google Scholar] [PubMed]
- Wang, J.; Ou, S.-W.; Wang, Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 2003, 23, 8881–8892. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Saab, C.Y.; Klein, J.P.; Craner, M.J.; Waxman, S.G. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J. Neurosci. 2004, 24, 4832–4839. [Google Scholar] [CrossRef]
- Emery, E.C.; Luiz, A.P.; Wood, J.N. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 2016, 20, 975–983. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef]
- Hockley, J.R.F.; Winchester, W.J.; Bulmer, D.C. The voltage-gated sodium channel NaV 1.9 in visceral pain. Neurogastroenterol. Motil. 2016, 28, 316–326. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Chessell, I.P.; Bountra, C.; Chan, C.; Fertleman, C.; Smith, V.; Anand, P. Voltage-gated ion channel Nav1.7 innervation in patients with idiopathic rectal hypersensitivity and paroxysmal extreme pain disorder (familial rectal pain). Neurosci. Lett. 2007, 427, 77–82. [Google Scholar] [CrossRef]
- Zheng, G.; Hong, S.; Hayes, J.M.; Wiley, J.W. Chronic stress and peripheral pain: Evidence for distinct, region-specific changes in visceral and somatosensory pain regulatory pathways. Exp. Neurol. 2015, 273, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, R.; Tao, J.; Wang, Y.; Zhou, Y.; Wu, G.; Xiao, Y.; Hu, C.-Y.; Jiang, X.; Xu, G.-Y.; Neonatal, X.G. Neonatal colonic inflammation sensitizes voltage-gated Na channels via upregulation of cystathionine-synthetase expression in rat primary sensory neurons. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, J.; Zhang, Y.; Xun, X.; Tang, D.; Peng, D.; Yi, J.; Liu, Z.; Shi, X. Synthesis and analgesic effects of μ-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins 2014, 6, 2363–2378. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Melgar, S. Lack of colonic-inflammation-induced acute visceral hypersensitivity to colorectal distension in Na(v)1.9 knockout mice. Eur. J. Pain 2008, 12, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Hillsley, K.; Lin, J.-H.; Stanisz, A.; Grundy, D.; Aerssens, J.; Peeters, P.J.; Moechars, D.; Coulie, B.; Stead, R.H. Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J. Physiol. 2006, 576, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.M.; Martinez-Caro, L.; Garcia-Nicas, E.; Cervero, F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain 2001, 92, 335–342. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Honore, P.; Shieh, C.-C.; Chapman, M.; Joshi, S.; Zhang, X.-F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, T.E.; ter Bekke, R.M.A.; Volders, P.G.A.; Masclee, A.A.M.; Kruimel, J.W. The role of the SCN5A -encoded channelopathy in irritable bowel syndrome and other gastrointestinal disorders. Neurogastroenterol. Motil. 2015, 27, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Beyder, A.; Farrugia, G. Ion channelopathies in functional GI disorders. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, 581–586. [Google Scholar] [CrossRef]
- Beyder, A.; Mazzone, A.; Strege, P.R.; Tester, D.J.; Saito, Y.A.; Bernard, C.E.; Enders, F.T.; Ek, W.E.; Schmidt, P.T.; Dlugosz, A.; et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology 2014, 146, 1659–1668. [Google Scholar] [CrossRef]
- Durán-Riveroll, L.M.; Cembella, A.D. Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels. Mar. Drugs 2017, 15, 303. [Google Scholar] [CrossRef] [PubMed]
- Magarlamov, T.; Melnikova, D.; Chernyshev, A.; Magarlamov, T.Y.; Melnikova, D.I.; Chernyshev, A.V. Tetrodotoxin-Producing Bacteria: Detection, Distribution and Migration of the Toxin in Aquatic Systems. Toxins 2017, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.; Rodríguez, L.; Blanco, L.; Vieites, J.; Cabado, A.; Lago, J.; Rodríguez, L.P.; Blanco, L.; Vieites, J.M.; Cabado, A.G. Tetrodotoxin, an Extremely Potent Marine Neurotoxin: Distribution, Toxicity, Origin and Therapeutical Uses. Mar. Drugs 2015, 13, 6384–6406. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Ebesu, J.S.M. Puffer poisoning: Epidemiology and treatment. J. Toxicol. Toxin Rev. 2001, 20, 1–10. [Google Scholar] [CrossRef]
- Hegyi, B.; Bárándi, L.; Komáromi, I.; Papp, F.; Horváth, B.; Magyar, J.; Bányász, T.; Krasznai, Z.; Szentandrássy, N.; Nánási, P.P. Tetrodotoxin blocks L-type Ca2+ channels in canine ventricular cardiomyocytes. Pflügers Arch. Eur. J. Physiol. 2012, 464, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Swanson, G.T. Recent progress in neuroactive marine natural products. Nat. Prod. Rep. 2014, 31, 273–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.K.; Mikusa, J.P.; Hernandez, G.; Baker, S.; Shieh, C.-C.; Neelands, T.; Zhang, X.-F.; Niforatos, W.; Kage, K.; Han, P.; et al. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 2006, 123, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, D.; Khotimchenko, Y.; Magarlamov, T.; Melnikova, D.I.; Khotimchenko, Y.S.; Magarlamov, T.Y. Addressing the Issue of Tetrodotoxin Targeting. Mar. Drugs 2018, 16, 352. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Takasugi, Y.; Higashino, H.; Ito, H.; Koga, Y.; Nakao, S. Antinociceptive action of carbamazepine on thermal hypersensitive pain at spinal level in a rat model of adjuvant-induced chronic inflammation. J. Anesth. 2011, 25, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Alguacil, L.F.; Pérez-García, C.; Salas, E.; González-Martín, C.; Castillo, C.; Polanco, M.J.; Herradón, G.; Morales, L. Subcutaneous tetrodotoxin and inflammatory pain. Br. J. Anaesth. 2008, 100, 729–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salas, M.M.; McIntyre, M.K.; Petz, L.N.; Korz, W.; Wong, D.; Clifford, J.L. Tetrodotoxin suppresses thermal hyperalgesia and mechanical allodynia in a rat full thickness thermal injury pain model. Neurosci. Lett. 2015, 607, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-J.; Lue, J.-H.; Lin, L.-H.; Huang, C.-T.; Chiang, R.P.-Y.; Chen, C.-L.; Tsai, Y.-J. Effects of pre-emptive drug treatment on astrocyte activation in the cuneate nucleus following rat median nerve injury. Pain 2010, 148, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Strong, J.A.; Meij, J.T.A.; Zhang, J.-M.; Yu, L. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005, 116, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Cantin, L.; Constant, J.; Haller, T.; Blaise, G.; Ong-Lam, M.; du Souich, P.; Korz, W.; Lapointe, B. Tetrodotoxin for Moderate to Severe Cancer-Related Pain: A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel-Design Trial. Pain Res. Manag. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Fisher, K.M.; Lapointe, B.; du Souich, P.; Chary, S.; Moulin, D.; Sellers, E.; Ngoc, A.H.; Canadian Tetrodotoxin Study Group. An Open-Label, Multi-Dose Efficacy and Safety Study of Intramuscular Tetrodotoxin in Patients with Severe Cancer-Related Pain. J. Pain Symptom Manag. 2007, 34, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Lapointe, B.; Ong-Lam, M.; Dubuc, B.; Walde, D.; Gagnon, B.; Love, R.; Goel, R.; Hawley, P.; Ho Ngoc, A.; et al. A multicentre open-label safety and efficacy study of tetrodotoxin for cancer pain. Curr. Oncol. 2011, 18, e109–e116. [Google Scholar] [CrossRef] [PubMed]
- Hockley, J.R.F.; González-Cano, R.; McMurray, S.; Tejada-Giraldez, M.A.; McGuire, C.; Torres, A.; Wilbrey, A.L.; Cibert-Goton, V.; Nieto, F.R.; Pitcher, T.; et al. Visceral and somatic pain modalities reveal NaV 1.7-independent visceral nociceptive pathways. J. Physiol. 2017, 595, 2661–2679. [Google Scholar] [CrossRef]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Moczydlowski, E.G. The molecular mystique of tetrodotoxin. Toxicon 2013, 63, 165–183. [Google Scholar] [CrossRef]
- Schantz, E.J.; Ghazarossian, V.E.; Schnoes, H.K.; Strong, F.M.; Springer, J.P.; Pezzanite, J.O.; Clardy, J. Structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 1238–1239. [Google Scholar] [CrossRef]
- Cirés, S.; Ballot, A. A review of the phylogeny, ecology and toxin production of bloom-forming Aphanizomenon spp. and related species within the Nostocales (cyanobacteria). Harmful Algae 2016, 54, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.H. Effects of “paralytic shellfish poison” on frog nerve and muscle. Br. J. Pharmacol. Chemother. 1964, 22, 478. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Nishiyama, A. Actions of saxitoxin on peripheral neuromuscular systems. J. Physiol. 1965, 180, 50–66. [Google Scholar] [PubMed]
- Wang, J.; Salata, J.J.; Bennett, P.B. Saxitoxin Is a Gating Modifier of hERG K+ Channels. J. Gen. Physiol. 2003, 121, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Sheets, M.; Ishida, H.; Li, F.; Barry, W.H. Saxitoxin blocks L-type ICa. J. Pharmacol. Exp. Ther. 2004, 308, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.J.; Blair, M.R.; Takman, B.H. The local anesthetic activity of saxitoxin alone and with vasoconstrictor and local anesthetic agents. Arch. Int. Pharmacodyn. Ther. 1976, 224, 275–282. [Google Scholar]
- Chorny, M.; Levy, R.J. Site-specific analgesia with sustained release liposomes. Proc. Natl. Acad. Sci. USA 2009, 106, 6891–6892. [Google Scholar] [CrossRef] [Green Version]
- Shankarappa, S.A.; Tsui, J.H.; Kim, K.N.; Reznor, G.; Dohlman, J.C.; Langer, R.; Kohane, D.S. Prolonged nerve blockade delays the onset of neuropathic pain. Proc. Natl. Acad. Sci. USA 2012, 109, 17555–1756. [Google Scholar] [CrossRef]
- Llewellyn, L.; Negri, A.; Robertson, A. Paralytic shellfish toxins in tropical oceans. Toxin Rev. 2006, 25, 159–196. [Google Scholar] [CrossRef]
- Garrido, R.; Lagos, N.; Lagos, M.; Rodríguez-Navarro, A.J.; Garcia, C.; Truan, D.; Henriquez, A. Treatment of chronic anal fissure by gonyautoxin. Colorectal Dis. 2007, 9, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hinzpeter, J.; Barrientos, C.; Zamorano, Á.; Martinez, Á.; Palet, M.; Wulf, R.; Barahona, M.; Sepúlveda, J.M.; Guerra, M.; Bustamante, T.; et al. Gonyautoxins: First evidence in pain management in total knee arthroplasty. Toxicon 2016, 119, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Zepeda, R.; Candiracci, M.; Lobos, N.; Lux, S.; Miranda, H.; Zepeda, R.J.; Candiracci, M.; Lobos, N.; Lux, S.; Miranda, H.F. Chronic Toxicity Study of Neosaxitoxin in Rats. Mar. Drugs 2014, 12, 5055–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Navarro, A.J.; Lagos, N.; Lagos, M.; Braghetto, I.; Csendes, A.; Hamilton, J.; Figueroa, C.; Truan, D.; Garcia, C.; Rojas, A.; et al. Neosaxitoxin as a Local Anesthetic. Anesthesiology 2007, 106, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Lobo, K.; Donado, C.; Cornelissen, L.; Kim, J.; Ortiz, R.; Peake, R.W.A.; Kellogg, M.; Alexander, M.E.; Zurakowski, D.; Kurgansky, K.E.; et al. A Phase 1, Dose-escalation, Double-blind, Block-randomized, Controlled Trial of Safety and Efficacy of Neosaxitoxin Alone and in Combination with 0.2% Bupivacaine, with and without Epinephrine, for Cutaneous Anesthesia. Anesthesiology 2015, 123, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manríquez, V.; Castro Caperan, D.; Guzmán, R.; Naser, M.; Iglesia, V.; Lagos, N. First evidence of neosaxitoxin as a long-acting pain blocker in bladder pain syndrome. Int. Urogynecol. J. 2015, 26, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navaro, A.J.; Lagos, N.; Lagos, M.; Braghetto, I.; Csendes, A.; Hamilton, J.; Berger, Z.; Wiedmaier, G.; Henriquez, A. Intrasphincteric Neosaxitoxin Injection: Evidence of Lower Esophageal Sphincter Relaxation in Achalasia. Am. J. Gastroenterol. 2006, 101, 2667–2668. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harbor Perspect. Biol. 2011, 3, a003947–a003947. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Gandini, M.A.; Snutch, T.P. Voltage-Gated Calcium Channels: Molecular Targets for Treating Chronic Pain. In The Oxford Handbook of the Neurobiology of Pain; Wood, J.N., Ed.; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Hofmann, F.; Biel, M.; Flockerzi, V. Molecular Basis for CA2+ Channel Diversity. Annu. Rev. Neurosci. 1994, 17, 399–418. [Google Scholar] [CrossRef]
- McKay, B.E.; McRory, J.E.; Molineux, M.L.; Hamid, J.; Snutch, T.P.; Zamponi, G.W.; Turner, R.W. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur. J. Neurosci. 2006, 24, 2581–2594. [Google Scholar] [CrossRef]
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, A.; Dickenson, A.H. Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain 1997, 69, 93–100. [Google Scholar] [CrossRef]
- Murakami, M.; Nakagawasai, O.; Fujii, S.; Hosono, M.; Hozumi, S.; Esashi, A.; Taniguchi, R.; Okamura, T.; Suzuki, T.; Sasano, H.; et al. Antinociceptive effect of cilnidipine, a novel N-type calcium channel antagonist. Brain Res. 2000, 868, 123–127. [Google Scholar] [CrossRef]
- Gohil, K.; Bell, J.R.; Ramachandran, J.; Miljanich, G.P. Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist, SNX-230 (ω-conopeptide MVIIC). Brain Res. 1994, 653, 258–266. [Google Scholar] [CrossRef]
- Cizkova, D.; Marsala, J.; Lukacova, N.; Marsala, M.; Jergova, S.; Orendacova, J.; Yaksh, T. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 2002, 147, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.A.; Tramontana, M.; Cecconi, R.; Santicioli, P. Neurochemical evidence for the involvement of N-type calcium channels in transmitter secretion from peripheral endings of sensory nerves in guinea pigs. Neurosci. Lett. 1990, 114, 203–206. [Google Scholar] [CrossRef]
- Needham, K.; Bron, R.; Hunne, B.; Nguyen, T.V.; Turner, K.; Nash, M.; Furness, J.B. Identification of subunits of voltage-gated calcium channels and actions of pregabalin on intrinsic primary afferent neurons in the guinea-pig ileum. Neurogastroenterol. Motil. 2010, 22, e301–e308. [Google Scholar] [CrossRef]
- Qian, A.; Song, D.; Li, Y.; Liu, X.; Tang, D.; Yao, W.; Yuan, Y. Role of Voltage Gated Ca2+ Channels in Rat Visceral Hypersensitivity Change Induced by 2, 4, 6-Trinitrobenzene Sulfonic Acid. Mol. Pain 2013, 9, 15. [Google Scholar] [CrossRef]
- Wegener, J.W.; Schulla, V.; Koller, A.; Klugbauer, N.; Feil, R.; Hofmann, F. Control of intestinal motility by the Ca(v)1.2 L-type calcium channel in mice. FASEB J. 2006, 20, 1260–1262. [Google Scholar] [CrossRef]
- Choudhury, B.K.; Shi, X.-Z.; Sarna, S.K. Gene plasticity in colonic circular smooth muscle cells underlies motility dysfunction in a model of postinfective IBS. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G632–G642. [Google Scholar] [CrossRef] [PubMed]
- Marger, F.; Gelot, A.; Alloui, A.; Matricon, J.; Ferrer, J.F.S.; Barrère, C.; Pizzoccaro, A.; Muller, E.; Nargeot, J.; Snutch, T.P.; et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 11268–11273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Luo, Z.D. Calcium channel functions in pain processing. Channels 2010, 4, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, A.; Schüetter, N.; Laffray, S.; Sanguesa, J.; Pizzoccaro, A.; Dubel, S.; Mantilleri, A.; Nargeot, J.; Noël, J.; Wood, J.N.; et al. The Low-Threshold Calcium Channel Cav3.2 Determines Low-Threshold Mechanoreceptor Function. Cell Rep. 2015, 10, 370–382. [Google Scholar] [CrossRef] [PubMed]
- García-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The Deubiquitinating Enzyme USP5 Modulates Neuropathic and Inflammatory Pain by Enhancing Cav3.2 Channel Activity. Neuron 2014, 83, 1144–1158. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.-F.; Tsai, M.-L.; Chen, C.-C.; Yen, C.-T. Involvement of the Cav3.2 T-Type Calcium Channel in Thalamic Neuron Discharge Patterns. Mol. Pain 2011, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, D.; Choi, S.; Lee, S.; Sun, M.; Kim, C.; Shin, H.-S. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science 2003, 302, 117–119. [Google Scholar] [CrossRef]
- Layer, R.T.; McIntosh, J.M. Conotoxins: Therapeutic Potential and Application. Mar. Drugs 2006, 4, 119. [Google Scholar] [CrossRef]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Zhang, J.-F.; Horne, W.A.; Tsien, R.W. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature 1994, 372, 272–275. [Google Scholar] [CrossRef]
- Wang, Y.X.; Gao, D.; Pettus, M.; Phillips, C.; Bowersox, S.S. Interactions of intrathecally administered ziconotide, a selective blocker of neuronal N-type voltage-sensitive calcium channels, with morphine on nociception in rats. Pain 2000, 84, 271–281. [Google Scholar] [CrossRef]
- Bowersox, S.S.; Gadbois, T.; Singh, T.; Pettus, M.; Wang, Y.X.; Luther, R.R. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J. Pharmacol. Exp. Ther. 1996, 279, 1243–1249. [Google Scholar] [PubMed]
- Xiao, W.H.; Bennett, G.J. Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J. Pharmacol. Exp. Ther. 1995, 274, 666–672. [Google Scholar] [PubMed]
- Safavi-Hemami, H.; Brogan, S.E.; Olivera, B.M. Pain therapeutics from cone snail venoms: From Ziconotide to novel non-opioid pathways. J. Proteom. 2019, 190, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Eastman, M.J.; Johnson, S.P. Ziconotide Combination Intrathecal Therapy. Available online: https://www.practicalpainmanagement.com/treatments/interventional/pumps/ziconotide-combination-intrathecal-therapy (accessed on 30 June 2019).
- Brookes, M.E.; Eldabe, S.; Batterham, A. Ziconotide Monotherapy: A Systematic Review of Randomised Controlled Trials. Curr. Neuropharmacol. 2017, 15, 217–231. [Google Scholar] [CrossRef]
- Webster, L.R. The Relationship Between the Mechanisms of Action and Safety Profiles of Intrathecal Morphine and Ziconotide: A Review of the Literature. Pain Med. 2015, 16, 1265–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauck, R.L.; Wallace, M.S.; Burton, A.W.; Kapural, L.; North, J.M. Intrathecal Ziconotide for Neuropathic Pain: A Review. Pain Pract. 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Deer, T.; Rauck, R.L.; Kim, P.; Saulino, M.F.; Wallace, M.; Grigsby, E.J.; Huang, I.-Z.; Mori, F.; Vanhove, G.F.; McDowell, G.C. Effectiveness and Safety of Intrathecal Ziconotide: Interim Analysis of the Patient Registry of Intrathecal Ziconotide Management (PRIZM). Pain Pract. 2018, 18, 230–238. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Palecek, J.; Paleckova, V.; Sorkin, L.S.; Willis, W.D. The role of NMDA and non-NMDA excitatory amino acid receptors in the excitation of primate spinothalamic tract neurons by mechanical, chemical, thermal, and electrical stimuli. J. Neurosci. 1992, 12, 3025–3041. [Google Scholar] [CrossRef]
- Filpa, V.; Moro, E.; Protasoni, M.; Crema, F.; Frigo, G.; Giaroni, C. Role of glutamatergic neurotransmission in the enteric nervous system and brain-gut axis in health and disease. Neuropharmacology 2016, 111, 14–33. [Google Scholar] [CrossRef]
- Carpanese, E.; Moretto, P.; Filpa, V.; Marchet, S.; Moro, E.; Crema, F.; Frigo, G.; Giaroni, C. Antagonism of ionotropic glutamate receptors attenuates chemical ischemia-induced injury in rat primary cultured myenteric ganglia. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.E.; Robertson, A.D.; Whorlow, S.L.; Angus, J.A. Cardiovascular and autonomic effects of ω-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br. J. Pharmacol. 2000, 131, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Kolosov, A.; Goodchild, C.S.; Cooke, I. CNSB004 (Leconotide) causes antihyperalgesia without side effects when given intravenously: A comparison with ziconotide in a rat model of diabetic neuropathic pain. Pain Med. 2010, 11, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Smith, A.B.; Schroeder, C.I.; Yasuda, T.; Lewis, R.J. ω-Conotoxin CVID Inhibits a Pharmacologically Distinct Voltage-sensitive Calcium Channel Associated with Transmitter Release from Preganglionic Nerve Terminals. J. Biol. Chem. 2003, 278, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Cabot, P.J.; Ross, F.B.; Robertson, A.D.; Lewis, R.J. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002, 96, 119–127. [Google Scholar] [CrossRef]
- Kolosov, A.; Aurini, L.; Williams, E.D.; Cooke, I.; Goodchild, C.S. Intravenous Injection of Leconotide, an Omega Conotoxin: Synergistic Antihyperalgesic Effects with Morphine in a Rat Model of Bone Cancer Pain. Pain Med. 2011, 12, 923–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berecki, G.; Motin, L.; Haythornthwaite, A.; Vink, S.; Bansal, P.; Drinkwater, R.; Wang, C.I.; Moretta, M.; Lewis, R.J.; Alewood, P.F.; et al. Analgesic (omega)-conotoxins CVIE and CVIF selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol. Pharmacol. 2010, 77, 139–148. [Google Scholar] [CrossRef]
- Lee, S.; Kim, Y.; Back, S.K.; Choi, H.-W.; Lee, J.Y.; Jung, H.H.; Ryu, J.H.; Suh, H.-W.; Na, H.S.; Kim, H.J.; et al. Analgesic Effect of Highly Reversible ω-Conotoxin FVIA on N Type Ca 2+ Channels. Mol. Pain 2010, 6, 97. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Adams, D.J.; Berecki, G. Mechanisms of conotoxin inhibition of N-type (Ca(v)2.2) calcium channels. Biochim. Biophys. Acta 2013, 1828, 1619–1628. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Craik, D.J. Therapeutic potential of conopeptides. Future Med. Chem. 2012, 4, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Novère, N.; Corringer, P.-J.; Changeux, J.-P. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 2002, 53, 447–456. [Google Scholar] [CrossRef]
- Hone, A.J.; Meyer, E.L.; McIntyre, M.; McIntosh, J.M. Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J. 2012, 26, 917–926. [Google Scholar] [CrossRef]
- Hone, A.J.; Servent, D.; McIntosh, J.M. α9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 2018, 175, 1915–1927. [Google Scholar] [CrossRef]
- Pacini, A.; Micheli, L.; Maresca, M.; Branca, J.J.V.; McIntosh, J.M.; Ghelardini, C.; Di Cesare Mannelli, L. The α9α10 nicotinic receptor antagonist α-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp. Neurol. 2016, 282, 37–48. [Google Scholar] [CrossRef]
- Bagdas, D.; Meade, J.A.; Alkhlaif, Y.; Muldoon, P.P.; Carroll, F.I.; Damaj, M.I. Effect of nicotine and alpha-7 nicotinic modulators on visceral pain-induced conditioned place aversion in mice. Eur. J. Pain 2018, 22, 1419–1427. [Google Scholar] [CrossRef]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on α7 nAChRs. Curr. Neuropharmacol. 2018, 16, 415–425. [Google Scholar] [CrossRef]
- Jain, K.K. Modulators of nicotinic acetylcholine receptors as analgesics. Curr. Opin. Investig. Drugs 2004, 5, 76–81. [Google Scholar]
- Grau, V.; Richter, K.; Hone, A.J.; McIntosh, J.M. Conopeptides [V11L;V16D]ArIB and RgIA4: Powerful Tools for the Identification of Novel Nicotinic Acetylcholine Receptors in Monocytes. Front. Pharmacol. 2018, 9, 1499. [Google Scholar] [CrossRef] [PubMed]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef]
- Vincler, M.; McIntosh, J.M. Targeting the α9α10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin. Ther. Targets 2007, 11, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Sandall, D.W.; Satkunanathan, N.; Keays, D.A.; Polidano, M.A.; Liping, X.; Pham, V.; Down, J.G.; Khalil, Z.; Livett, B.G.; Gayler, K.R. A Novel α-Conotoxin Identified by Gene Sequencing Is Active in Suppressing the Vascular Response to Selective Stimulation of Sensory Nerves in Vivo. Biochemistry 2003, 42, 6904–6911. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, J.A.; Keays, D.A.; Kelley, W.P.; Sandall, D.W.; Bingham, J.-P.; Livett, B.G.; Gayler, K.R.; Sweedler, J. V Determining sequences and post-translational modifications of novel conotoxins in Conus victoriae using cDNA sequencing and mass spectrometry. J. Mass Spectrom. 2004, 39, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B.; Hone, A.J.; Roux, I.; Kniazeff, J.; Pin, J.-P.; Upert, G.; Servent, D.; Glowatzki, E.; McIntosh, J.M. RgIA4 Potently Blocks Mouse α9α10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia. Front. Cell. Neurosci. 2017, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Nevin, S.T.; Clark, R.J.; Klimis, H.; Christie, M.J.; Craik, D.J.; Adams, D.J. Are α9α10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol. Pharmacol. 2007, 72, 1406–1410. [Google Scholar] [CrossRef]
- Napier, I.A.; Klimis, H.; Rycroft, B.K.; Jin, A.H.; Alewood, P.F.; Motin, L.; Adams, D.J.; Christie, M.J. Intrathecal α-conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 2012, 62, 2202–2207. [Google Scholar] [CrossRef]
- Klimis, H.; Adams, D.J.; Callaghan, B.; Nevin, S.; Alewood, P.F.; Vaughan, C.W.; Mozar, C.A.; Christie, M.J. A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011, 152, 259–266. [Google Scholar] [CrossRef]
- Smith, N.J.; Hone, A.J.; Memon, T.; Bossi, S.; Smith, T.E.; McIntosh, J.M.; Olivera, B.M.; Teichert, R.W. Comparative functional expression of nAChR subtypes in rodent DRG neurons. Front. Cell. Neurosci. 2013, 7, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.; Haythornthwaite, A.; Berecki, G.; Clark, R.J.; Craik, D.J.; Adams, D.J. Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 2008, 28, 10943–10951. [Google Scholar] [CrossRef] [PubMed]
- Brusberg, M.; Ravnefjord, A.; Martinsson, R.; Larsson, H.; Martinez, V.; Lindström, E. The GABA(B) receptor agonist, baclofen, and the positive allosteric modulator, CGP7930, inhibit visceral pain-related responses to colorectal distension in rats. Neuropharmacology 2009, 56, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, J.N.; Medda, B.K.; Shaker, R. Effect of GABA(B) receptor agonist on distension-sensitive pelvic nerve afferent fibers innervating rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1343–G1351. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.; Brusberg, M.; Ravnefjord, A.; Kakol-Palm, D.; Påhlman, I.; Novén, A.; Larsson, H.; Martinez, V. Oral baclofen reduces visceral pain-related pseudo-affective responses to colorectal distension in rats: Relation between plasma exposure and efficacy. Scand. J. Gastroenterol. 2011, 46, 652–662. [Google Scholar] [CrossRef]
- ASX Announcement ASX Code: MBP Metabolic’s Pain Drug ACV1 Enters Second Phase 2 Human Clinical Trial; Commencement of Second ACV1 Phase 2A Trial in Patients with Neuropathic Pain; Metabolic Pharmaceuticals Limited: Victoria, Australia, 2007.
- Metabolic Discontinues Clinical Trial Programme for Neuropathic Pain Drug, ACV1; ASX: Melbourne, Australia, 2007.
- Halai, R.; Clark, R.J.; Nevin, S.T.; Jensen, J.E.; Adams, D.J.; Craik, D.J. Scanning mutagenesis of alpha-conotoxin Vc1.1 reveals residues crucial for activity at the alpha9alpha10 nicotinic acetylcholine receptor. J. Biol. Chem. 2009, 284, 20275–20284. [Google Scholar] [CrossRef]
- Lovelace, E.S.; Gunasekera, S.; Alvarmo, C.; Clark, R.J.; Nevin, S.T.; Grishin, A.A.; Adams, D.J.; Craik, D.J.; Daly, N.L. Stabilization of α-conotoxin AuIB: Influences of disulfide connectivity and backbone cyclization. Antioxid. Redox Signal. 2011, 14, 87–95. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. 2010, 49, 6545–6548. [Google Scholar] [CrossRef]
- Holzer, P.; Hassan, A.M.; Jain, P.; Reichmann, F.; Farzi, A. Neuroimmune pharmacological approaches. Curr. Opin. Pharmacol. 2015, 25, 13–22. [Google Scholar] [CrossRef]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Sub-Cell. Biochem. 2018, 87, 141–165. [Google Scholar]
- Latorre, R.; Zaelzer, C.; Brauchi, S. Structure-functional intimacies of transient receptor potential channels. Q. Rev. Biophys. 2009, 42, 201–246. [Google Scholar] [CrossRef] [PubMed]
- Blackshaw, L.A. Transient receptor potential cation channels in visceral sensory pathways. Br. J. Pharmacol. 2014, 171, 2528–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, E.S.; Fernandes, M.A.; Keeble, J.E. The functions of TRPA1 and TRPV1: Moving away from sensory nerves. Br. J. Pharmacol. 2012, 166, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.J.; Aziz, Q.; Facer, P.; Davis, J.B.; Thompson, D.G.; Anand, P. Increased capsaicin receptor TRPV1 nerve fibres in the inflamed human oesophagus. Eur. J. Gastroenterol. Hepatol. 2004, 16, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Adam, B.; Liebregts, T.; Gschossmann, J.M.; Krippner, C.; Scholl, F.; Ruwe, M.; Holtmann, G. Severity of mucosal inflammation as a predictor for alterations of visceral sensory function in a rat model. Pain 2006, 123, 179–186. [Google Scholar] [CrossRef]
- Szallasi, A.; Cortright, D.N.; Blum, C.A.; Eid, S.R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 2007, 6, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 Ion Channels in Inflammatory Bowel Diseases: Potential Therapeutic Targets? Pharmaceuticals 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Facer, P.; Davis, J.; Smith, G.; Egerton, J.; Bountra, C.; Williams, N.; Anand, P. Sensory fibres expressing capsaicin receptor TRPV1 in patients with rectal hypersensitivity and faecal urgency. Lancet 2003, 361, 385–391. [Google Scholar] [CrossRef]
- Ji, R.-R.; Samad, T.A.; Jin, S.-X.; Schmoll, R.; Woolf, C.J. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 2002, 36, 57–68. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Dyer, N.H.; Chan, C.L.; Knowles, C.; Williams, N.S.; Anand, P. Vanilloid receptor 1 immunoreactivity in inflamed human bowel. Lancet 2001, 357, 1338–1339. [Google Scholar] [CrossRef]
- Premkumar, L.S.; Ahern, G.P. Induction of vanilloid receptor channel activity by protein kinase C. Nature 2000, 408, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Jara-Oseguera, A.; Simon, S.A.; Rosenbaum, T. TRPV1: On the road to pain relief. Curr. Mol. Pharmacol. 2008, 1, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Duggan, A.; Kumar, G.; García-Añoveros, J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 2005, 25, 4052–4061. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.P.; Pelayo, J.C.; Cattaruzza, F.; Kuo, Y.; Gai, G.; Chiu, J.V.; Bron, R.; Furness, J.B.; Grady, E.F.; Bunnett, N.W. Transient Receptor Potential Ankyrin 1 Is Expressed by Inhibitory Motoneurons of the Mouse Intestine. Gastroenterology 2011, 141, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.S.; La, J.-H.; Scheff, N.N.; Davis, B.M.; Albers, K.M.; Gebhart, G.F. TRPV1 and TRPA1 Antagonists Prevent the Transition of Acute to Chronic Inflammation and Pain in Chronic Pancreatitis. J. Neurosci. 2013, 33, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, F.; Spreadbury, I.; Miranda-Morales, M.; Grady, E.F.; Vanner, S.; Bunnett, N.W. Transient receptor potential ankyrin-1 has a major role in mediating visceral pain in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G81–G91. [Google Scholar] [CrossRef] [Green Version]
- Brierley, S.M.; Hughes, P.A.; Page, A.J.; Kwan, K.Y.; Martin, C.M.; O’Donnell, T.A.; Cooper, N.J.; Harrington, A.M.; Adam, B.; Liebregts, T.; et al. The Ion Channel TRPA1 Is Required for Normal Mechanosensation and Is Modulated by Algesic Stimuli. Gastroenterology 2009, 137, 2084–2095.e3. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.; Yiangou, Y.; Facer, P.; Brydon, W.G.; Walters, J.R.F.; Anand, P.; Ghosh, S. Expression of the TRPV1 receptor differs in quiescent inflammatory bowel disease with or without abdominal pain. Gut 2010, 59, 767–774. [Google Scholar] [CrossRef]
- Winston, J.; Shenoy, M.; Medley, D.; Naniwadekar, A.; Pasricha, P.J. The vanilloid receptor initiates and maintains colonic hypersensitivity induced by neonatal colon irritation in rats. Gastroenterology 2007, 132, 615–627. [Google Scholar] [CrossRef]
- Spicarova, D.; Adamek, P.; Kalynovska, N.; Mrozkova, P.; Palecek, J. TRPV1 receptor inhibition decreases CCL2-induced hyperalgesia. Neuropharmacology 2014, 81, 75–84. [Google Scholar] [CrossRef]
- Kimball, E.S.; Wallace, N.H.; Schneider, C.R.; D’Andrea, M.R.; Hornby, P.J. Vanilloid receptor 1 antagonists attenuate disease severity in dextran sulphate sodium-induced colitis in mice. Neurogastroenterol. Motil. 2004, 16, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.A.; Harrington, A.M.; Castro, J.; Liebregts, T.; Adam, B.; Grasby, D.J.; Isaacs, N.J.; Maldeniya, L.; Martin, C.M.; Persson, J.; et al. Sensory neuro-immune interactions differ between irritable bowel syndrome subtypes. Gut 2013, 62, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea Anemones: Quiet Achievers in the Field of Peptide Toxins. Toxins 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.V.; Dorofeeva, N.A.; Komarova, M.S.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Grishin, E.V.; Tikhonov, D.B.; Kozlov, S.A. TRPV1 activation power can switch an action mode for its polypeptide ligands. PLoS ONE 2017, 12, e0177077. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.A.; Andreev, Y.A.; Murashev, A.N.; Skobtsov, D.I.; D’yachenko, I.A.; Grishin, E.V. New polypeptide components from the Heteractis crispa sea anemone with analgesic activity. Russ. J. Bioorg. Chem. 2009, 35, 711–719. [Google Scholar] [CrossRef]
- Philyppov, I.B.; Paduraru, O.N.; Andreev, Y.A.; Grishin, E.V.; Shuba, Y.M. Modulation of TRPV1-dependent contractility of normal and diabetic bladder smooth muscle by analgesic toxins from sea anemone Heteractis crispa. Life Sci. 2012, 91, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Solstad, R.G.; Mineev, K.S.; Korolkova, Y.V.; Mosharova, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Arseniev, A.S.; et al. New Disulfide-Stabilized Fold Provides Sea Anemone Peptide to Exhibit Both Antimicrobial and TRPA1 Potentiating Properties. Toxins 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvåg, K.; et al. Peptide from Sea Anemone Metridium senile Affects Transient Receptor Potential Ankyrin-repeat 1 (TRPA1) Function and Produces Analgesic Effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofori-Armstrong, B.; Rash, L.D. Acid-sensing ion channel (ASIC) structure and function: Insights from spider, snake and sea anemone venoms. Neuropharmacology 2017, 127, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, E.B.; Kawate, T.; Gouaux, E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 2009, 460, 599–604. [Google Scholar] [CrossRef]
- Baron, A.; Lingueglia, E. Pharmacology of acid-sensing ion channels—Physiological and therapeutical perspectives. Neuropharmacology 2015, 94, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.-S.; Debayle, D.; Friend, V.; Alloui, A.; Lazdunski, M.; et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Alvarez de la Rosa, D.; Zhang, P.; Shao, D.; White, F.; Canessa, C.M. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc. Natl. Acad. Sci. USA 2002, 99, 2326–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Kolker, S.J.; Burnes, L.A.; Walder, R.Y.; Sluka, K.A. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain 2008, 137, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, P. Acid-sensing ion channels in gastrointestinal function. Neuropharmacology 2015, 94, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, R.; Bassilana, F.; de Weille, J.; Champigny, G.; Heurteaux, C.; Lazdunski, M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J. Biol. Chem. 1997, 272, 20975–20978. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Noël, J.; Lay, N.; Alloui, A.; Diochot, S.; Friend, V.; Jodar, M.; Lazdunski, M.; Lingueglia, E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008, 27, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Zimmer, A.; Sun, W.-H.; Hall, J.; Brownstein, M.J.; Zimmer, A. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc. Natl. Acad. Sci. USA 2002, 99, 8992–8997. [Google Scholar] [CrossRef] [Green Version]
- Schicho, R.; Florian, W.; Liebmann, I.; Holzer, P.; Lippe, I.T. Increased expression of TRPV1 receptor in dorsal root ganglia by acid insult of the rat gastric mucosa. Eur. J. Neurosci. 2004, 19, 1811–1818. [Google Scholar] [CrossRef]
- Hughes, P.A.; Brierley, S.M.; Young, R.L.; Blackshaw, L.A. Localization and comparative analysis of acid-sensing ion channel (ASIC1, 2, and 3) mRNA expression in mouse colonic sensory neurons within thoracolumbar dorsal root ganglia. J. Comp. Neurol. 2007, 500, 863–875. [Google Scholar] [CrossRef]
- Deval, E.; Gasull, X.; Noël, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-sensing ion channels (ASICs): Pharmacology and implication in pain. Pharmacol. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Wultsch, T.; Painsipp, E.; Shahbazian, A.; Mitrovic, M.; Edelsbrunner, M.; Lazdunski, M.; Waldmann, R.; Holzer, P. Deletion of the acid-sensing ion channel ASIC3 prevents gastritis-induced acid hyperresponsiveness of the stomach–brainstem axis. Pain 2008, 134, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Brierley, S.M.; Martin, C.M.; Price, M.P.; Symonds, E.; Butler, R.; Wemmie, J.A.; Blackshaw, L.A. Different contributions of ASIC channels 1a, 2, and 3 in gastrointestinal mechanosensory function. Gut 2005, 54, 1408–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.C.W.; Xu, L.; Gebhart, G.F. The Mechanosensitivity of Mouse Colon Afferent Fibers and Their Sensitization by Inflammatory Mediators Require Transient Receptor Potential Vanilloid 1 and Acid-Sensing Ion Channel 3. J. Neurosci. 2005, 25, 10981–10989. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.C.W.; Otsuka, E.; Wagstrom, E.; Jensen, C.S.; Price, M.P.; Gebhart, G.F. Short-Term Sensitization of Colon Mechanoreceptors Is Associated With Long-Term Hypersensitivity to Colon Distention in the Mouse. Gastroenterology 2007, 133, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Gelot, A.; Etienne, M.; Lazdunski, M.; Muller, E.; Ardid, D. Spinal cord plasticity and acid-sensing ion channels involvement in a rodent model of irritable bowel syndrome. Eur. J. Pain 2011, 15, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Muller, E.; Accarie, A.; Meleine, M.; Etienne, M.; Voilley, N.; Busserolles, J.; Eschalier, A.; Lazdunski, M.; Bourdu, S.; et al. Peripheral contribution of NGF and ASIC1a to colonic hypersensitivity in a rat model of irritable bowel syndrome. Neurogastroenterol. Motil. 2013, 25, e740–e754. [Google Scholar] [CrossRef] [PubMed]
- Delafoy, L.; Gelot, A.; Ardid, D.; Eschalier, A.; Bertrand, C.; Doherty, A.M.; Diop, L. Interactive involvement of brain derived neurotrophic factor, nerve growth factor, and calcitonin gene related peptide in colonic hypersensitivity in the rat. Gut 2006, 55, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Stanzel, R.D.P.; Lourenssen, S.; Blennerhassett, M.G. Inflammation causes expression of NGF in epithelial cells of the rat colon. Exp. Neurol. 2008, 211, 203–213. [Google Scholar] [CrossRef]
- Torres, A.M.; Kuchel, P.W. The β-defensin-fold family of polypeptides. Toxicon 2004, 44, 581–588. [Google Scholar] [CrossRef]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Rasmussen, L.A.; Rainier, J.D.; Light, A.R.; Wemmie, J.A.; Sluka, K.A. ASIC1 and ASIC3 Play Different Roles in the Development of Hyperalgesia Following Inflammatory Muscle Injury. J. Pain 2010, 11, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharmacol. 2010, 161, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.P.; McIlwrath, S.L.; Xie, J.; Cheng, C.; Qiao, J.; Tarr, D.E.; Sluka, K.A.; Brennan, T.J.; Lewin, G.R.; Welsh, M.J. The DRASIC Cation Channel Contributes to the Detection of Cutaneous Touch and Acid Stimuli in Mice. Neuron 2001, 32, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Izumi, M.; Ikeuchi, M.; Ji, Q.; Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J. Biomed. Sci. 2012, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, M.G.; Rash, L.D.; Kellenberger, S. Inhibition of voltage-gated Na+ currents in sensory neurones by the sea anemone toxin APETx2. Br. J. Pharmacol. 2012, 165, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Cristofori-Armstrong, B.; Anangi, R.; Rosengren, K.J.; Lau, C.H.Y.; Mobli, M.; Brust, A.; Alewood, P.F.; King, G.F.; Rash, L.D. Understanding the Molecular Basis of Toxin Promiscuity: The Analgesic Sea Anemone Peptide APETx2 Interacts with Acid-Sensing Ion Channel 3 and hERG Channels via Overlapping Pharmacophores. J. Med. Chem. 2014, 57, 9195–9203. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Acid sensing by visceral afferent neurones. Acta Physiol. 2011, 201, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Compound/Target | Dose/Concentration (Administration route) | Model | Parameter Evaluated | Effect | REF |
|---|---|---|---|---|---|
| TTX/Nav | 1, 3, 6 g/kg (s.c.) | Intracolonic instillation of capsaicin in WT (C57BL/6 background) and Nav 1.7 conditional KO | -Pain-related behaviors (licking of the abdomen, stretching of abdomen, abdominal retractions); -Referred mechanical hyperalgesia (response to abdominal stimulation with von Frey electrodes) | -Similar dose-dependent reduction in WT and KO -Similar dose-dependent reduction in WT and KO | [55] |
| 3, 6 g/kg (s.c.) | Intracolonic instillation of mustard oil in WT and Nav 1.7 conditional KO mice | Pain-related behaviors (licking of the abdomen, stretching of abdomen, abdominal retractions) | Dose-dependent reduction, similar in WT and KO | [55] | |
| 0.3, 1, 3, 6 g/kg (s.c.) | Intraperitoneal acetic-acid induced writhing test in Swiss Webster mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Dose-dependent reduction | [56] | |
| ω-conotoxin MVIIA/Cav 2.2 | 1, 10, 30, 100 pmol/site (i.t.) | Intraperitoneal acetic-acid induced writhing test in Swiss mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Dose-dependent reduction | [57] |
| 1, 10, 30, 100 pmol/site (i.t.) | Intracolonic instillation of capsaicin in Swiss mice | Pain-related behaviors (licking of the abdomen, stretching of abdomen, abdominal retractions). | Dose-dependent reduction | [57] | |
| 30 pmol/site (i.t.) | Intraperitoneal acetic-acid induced writhing test in Swiss mice | Measurement of glutamate levels in the CSF | Reduction of nociceptive stimulus-induced increase of glutamate levels in the CSF | [57] | |
| 30 pmol/site (i.t.) | Intracolonic instillation of capsaicin in Swiss mice | Measurement of glutamate levels in the CSF | Reduction of nociceptive stimulus-induced increase of glutamate levels in the CSF | [57] | |
| Vc1.1/GABAB | 1 M (in vitro) | Human thoracolumbar DRG | Whole-cell patch clamp recordings | Inhibition of a selective population of DRG neurons | [58] |
| 1, 10, 100, 1000 nM (in vitro) | CVH mouse model induced by intrarectal TNBS administration | Ex vivo single fiber recordings of primary afferents splanchnic colonic and pelvic colorectum afferents | Concentration-dependent inhibition of mechanosensitivity (the effect was higher in CVH animals) | [58] | |
| 1, 10, 100, 1000 nM (in vitro) | CVH mouse model induced by intrarectal TNBS administration | Ex vivo single fiber recordings of splanchnic colonic primary afferents. | Concentration-dependent inhibition of mechanosensitivity (the effect was higher in CVH animals) | [59] | |
| 10 nM (in vitro) | CVH mouse model induced by intrarectal TNBS administration | Whole-cell patch clamp recordings on colonic extrinsic primary afferents | Significant inhibition of the excitability of colonic control DRG which was higher in CVH animals | [59] | |
| 1 M (intrarectal enema) | CVH mouse model induced by intrarectal TNBS administration followed by noxious distension of the colorectum | VMR to colorectal distension by electromyography assessment | Significant reduction of VMR in CVH mice to noxious distension pressures | [60] | |
| [Ser3] Vc1.1(1-8)/GABAB | 100 pM, 30 nM, 1 M (in vitro) | Mouse DRG | Whole-cell patch clamp recordings | Inhibition of VGCC | [61] |
| 1, 10, 100 and 1000 nM (ex vivo, applied on the colonic surface) | CVH mouse model induced by intrarectal TNBS administration | In vitro single-unit extracellular recordings of action potential discharge from splanchnic colonic afferents. | Concentration-dependent inhibition of mechanosensitivity of splanchnic colonic primary afferents | [61] | |
| 1 M (intrarectal enema) | Noxious distension of the mouse colorectum | VMR to colorectal distension by electromyography assessment | Significant reduction of VMR to colorectal distension vs vehicle treated animals | [61] | |
| APCH1/TRPV1 | 0.5 mg/kg (i.v.) | Intraperitoneal acetic-acid induced writhing test in CD1 mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Reduction | [62] |
| APCH3/TRPV1 | 0.1, 0.5 mg/kg (i.v.) | Intraperitoneal acetic-acid induced writhing test in CD1 mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Reduction | [62] |
| APETx2/ASIC3 | 25 g/kg | Acute gastric mucosal damage induced by WIRS in Wistar rats | -intragastric pH -gastric histopathological changes -UI -ASIC3 expression in thoracic DRG neurons projecting to the stomach | Significant reduction of WIRS-induced: -gastric mucosal injury, UI score, gastric acidity -ASIC3 expression in DRG | [63] |
| 0.002, 0.02, 0.2, 1 mg/kg (i.m.) | Intraperitoneal acetic-acid induced writhing test in CD1 mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Bell-shaped reduction of abdominal contractile responses | [64] | |
| Ugr9-1/ASIC3 | −0.002, 0.02, 0.2, 1 mg/kg (i.m.); −0.5, 0.1, 0.01 mg/kg (i.v.) | Intraperitoneal acetic-acid induced writhing test in CD1 mice | Number of complete abdominal contractions accompanied with stretching of hind limbs | Dose-dependent reduction of abdominal contractile responses | [64,65] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baj, A.; Bistoletti, M.; Bosi, A.; Moro, E.; Giaroni, C.; Crema, F. Marine Toxins and Nociception: Potential Therapeutic Use in the Treatment of Visceral Pain Associated with Gastrointestinal Disorders. Toxins 2019, 11, 449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080449
Baj A, Bistoletti M, Bosi A, Moro E, Giaroni C, Crema F. Marine Toxins and Nociception: Potential Therapeutic Use in the Treatment of Visceral Pain Associated with Gastrointestinal Disorders. Toxins. 2019; 11(8):449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080449
Chicago/Turabian StyleBaj, Andreina, Michela Bistoletti, Annalisa Bosi, Elisabetta Moro, Cristina Giaroni, and Francesca Crema. 2019. "Marine Toxins and Nociception: Potential Therapeutic Use in the Treatment of Visceral Pain Associated with Gastrointestinal Disorders" Toxins 11, no. 8: 449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080449