TNF Family Cytokines Induce Distinct Cell Death Modalities in the A549 Human Lung Epithelial Cell Line when Administered in Combination with Ricin Toxin

 and
and 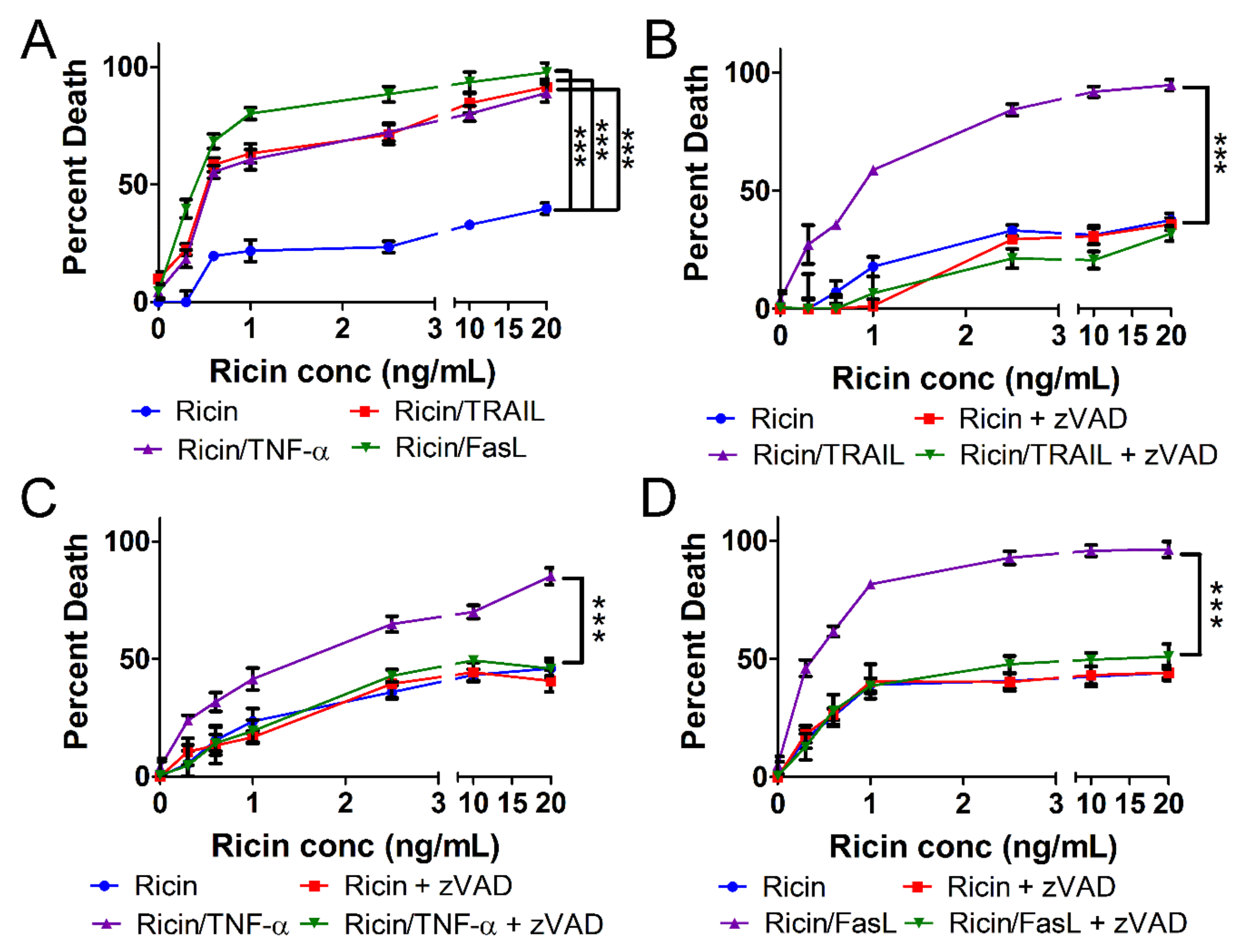{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Ricin-Induced Death Is Primed by TRAIL, TNF-α, and FasL
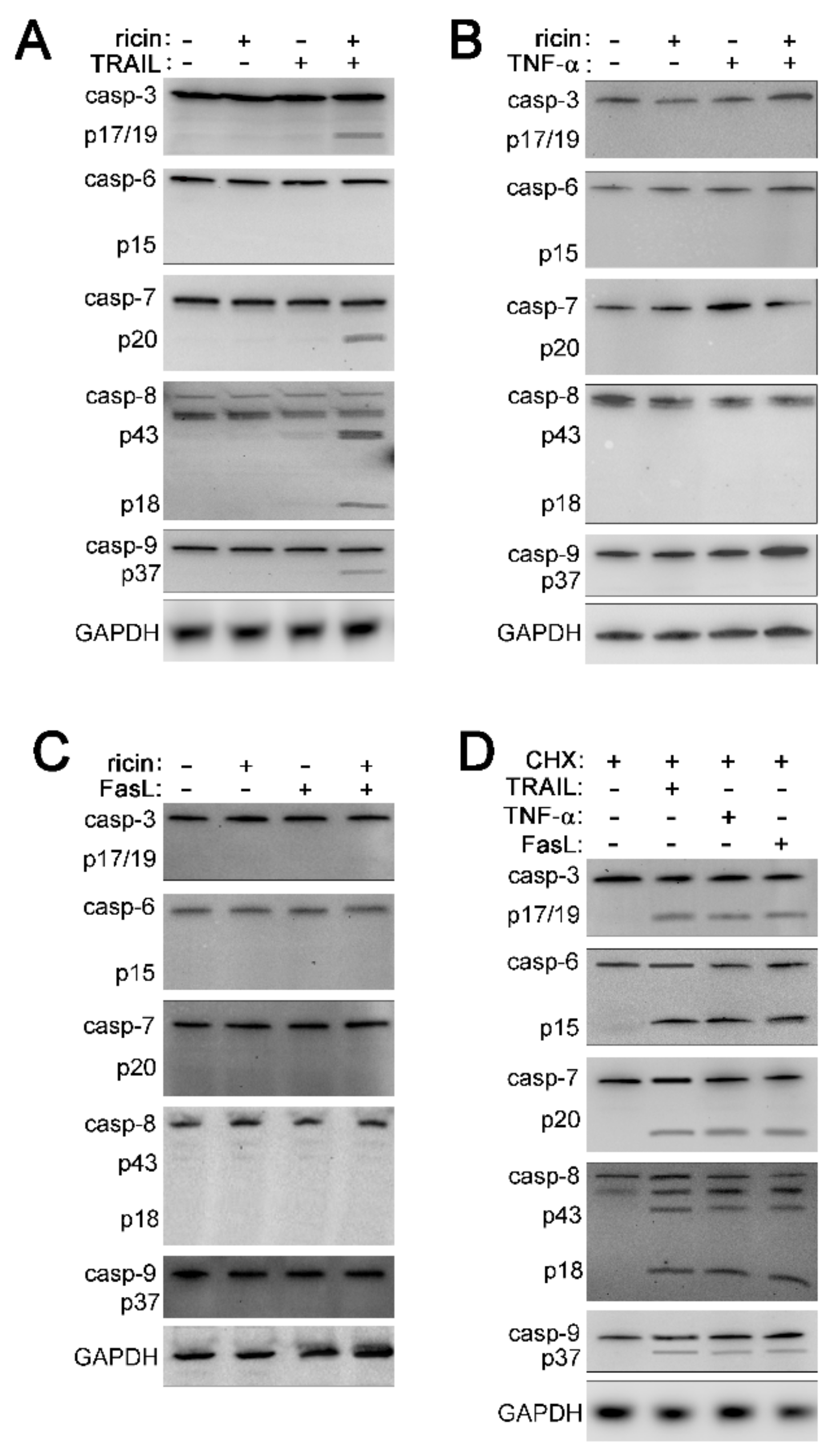
2.2. Cell Death Induced by Ricin/TRAIL Is Associated with Caspase Activation while Death by Ricin/TNF-α and Ricin/FasL Is Not
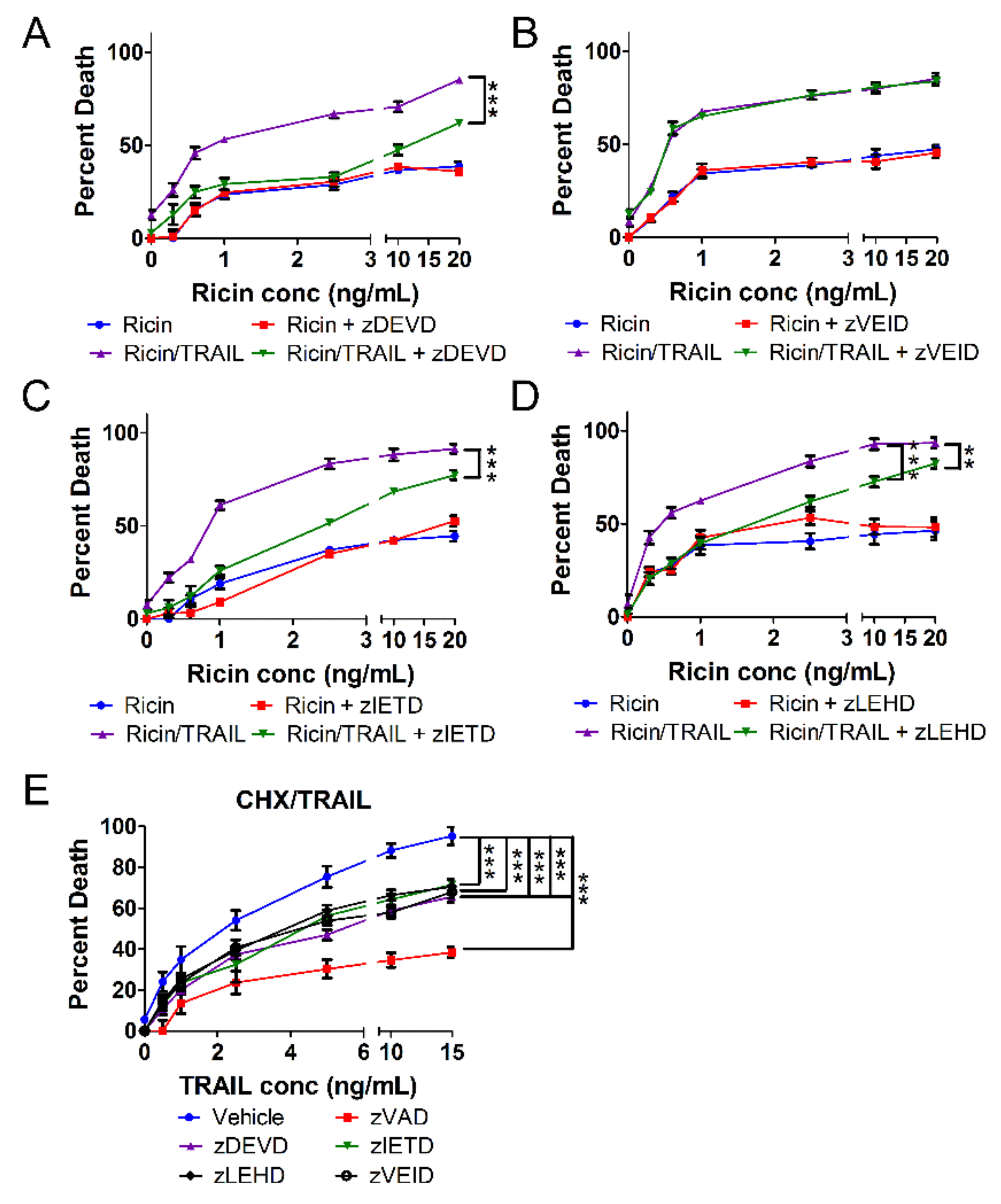
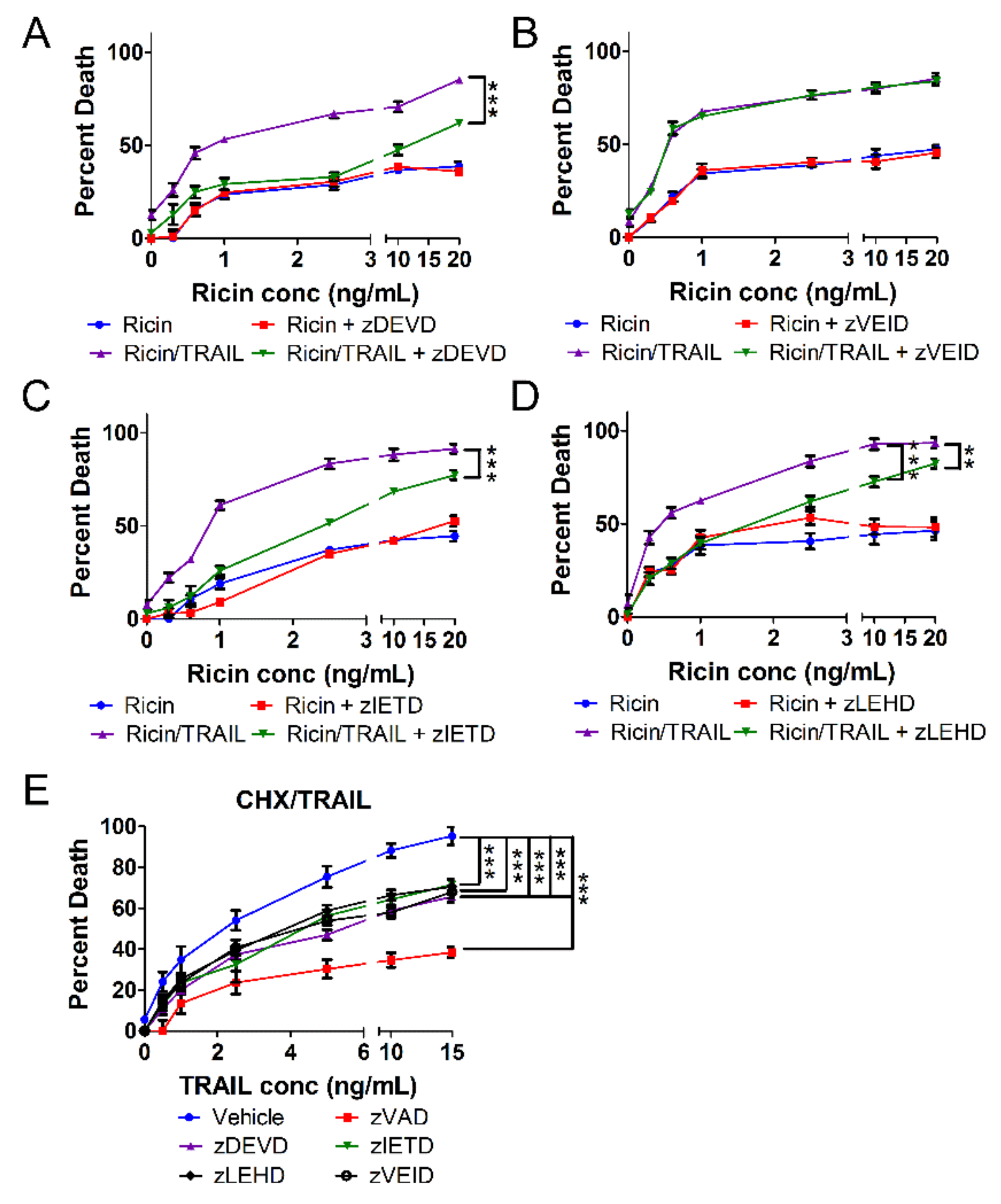
2.3. The Combination of Ricin and TRAIL Induces Caspase-Dependent Apoptosis
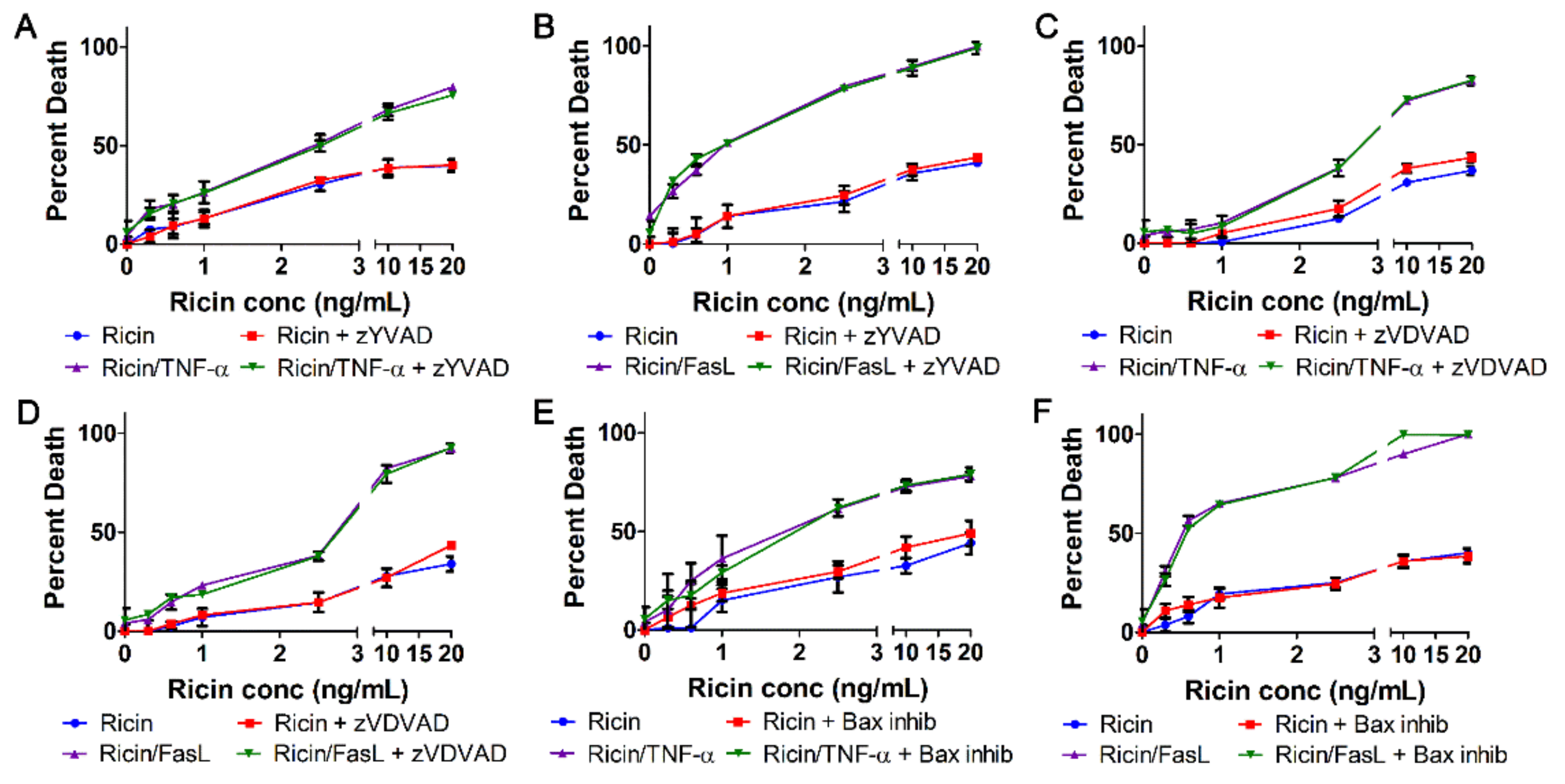
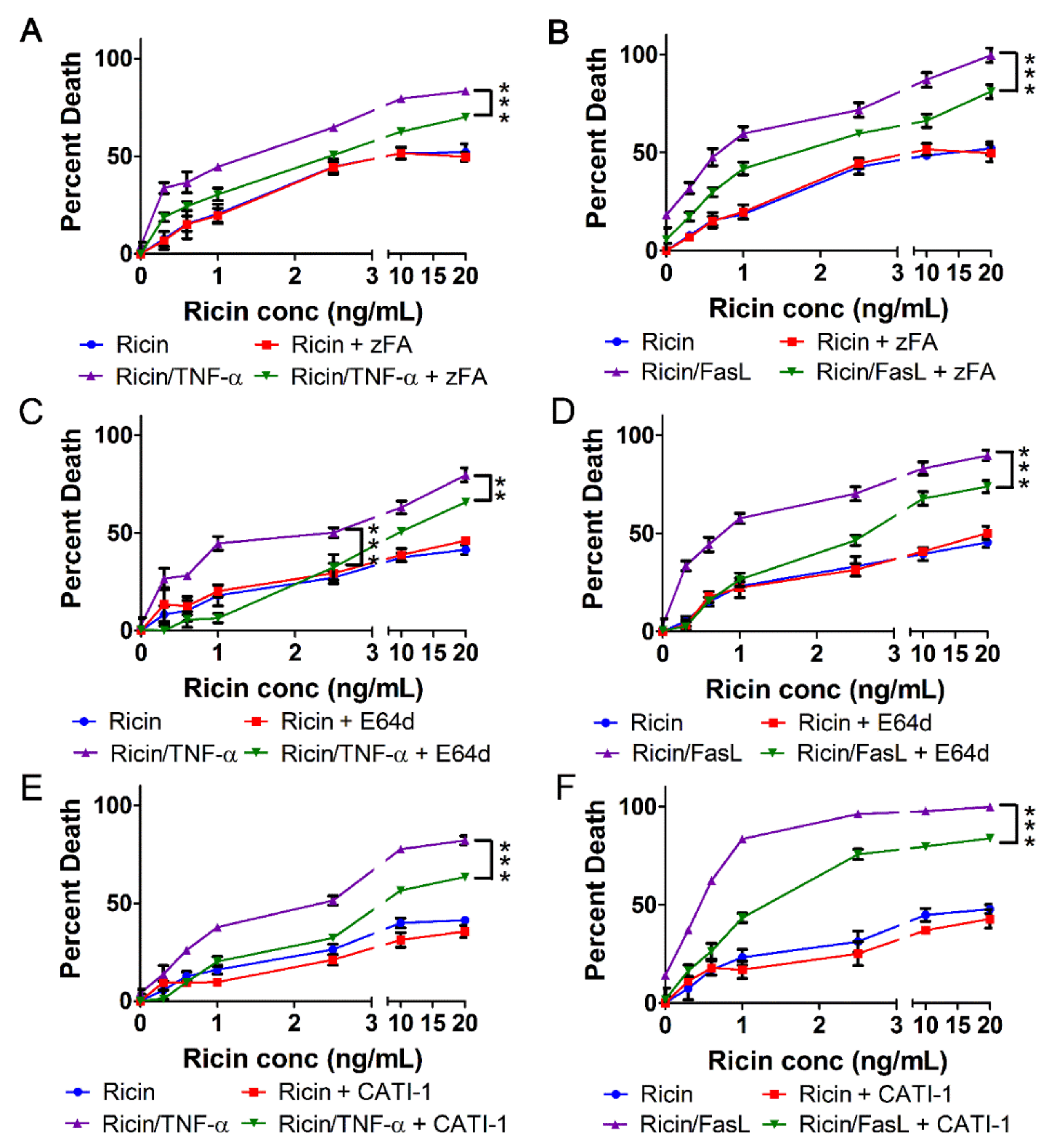
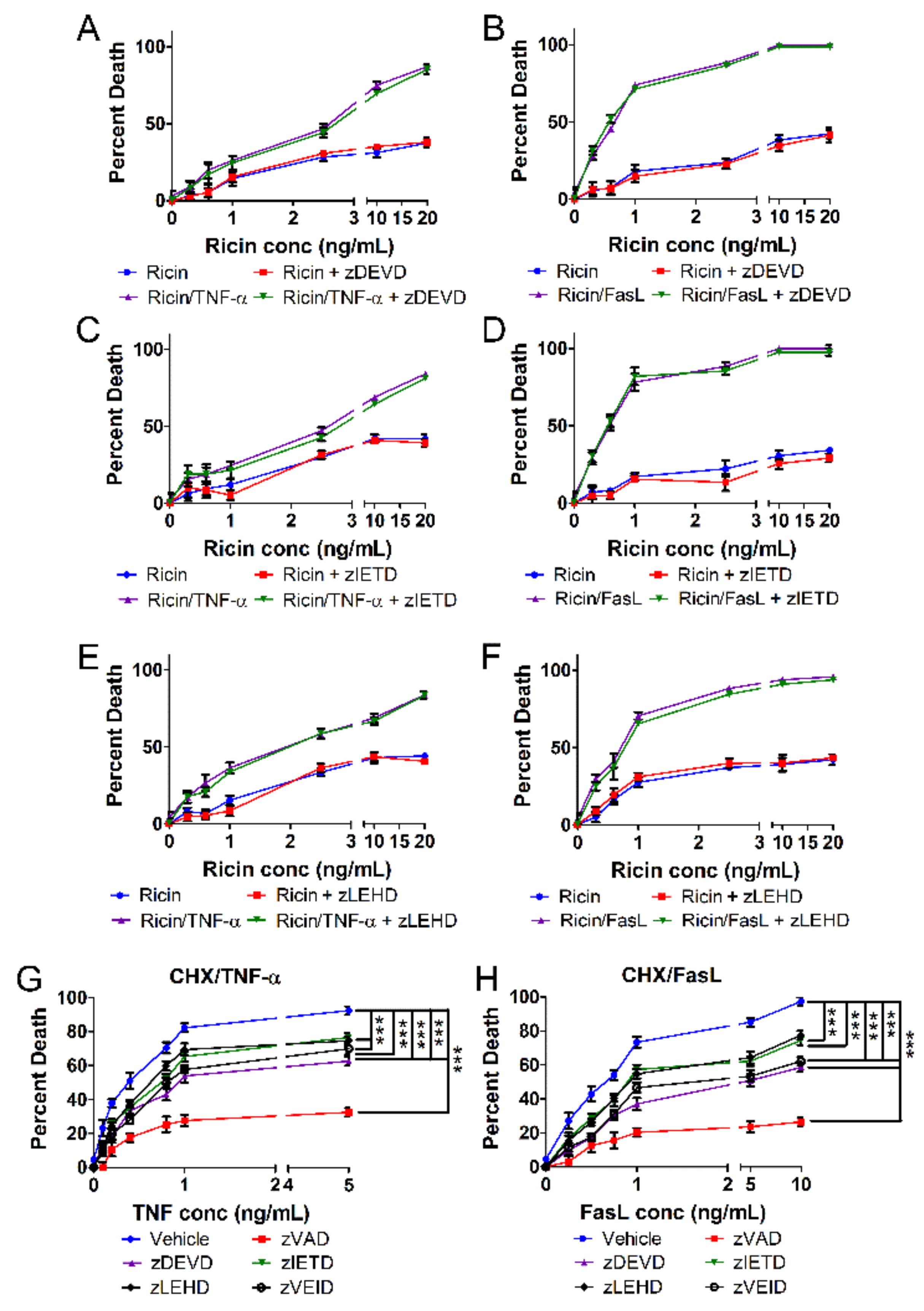
2.4. Ricin Combined with TNF-α or FasL Induces Caspase-Independent Cell Death
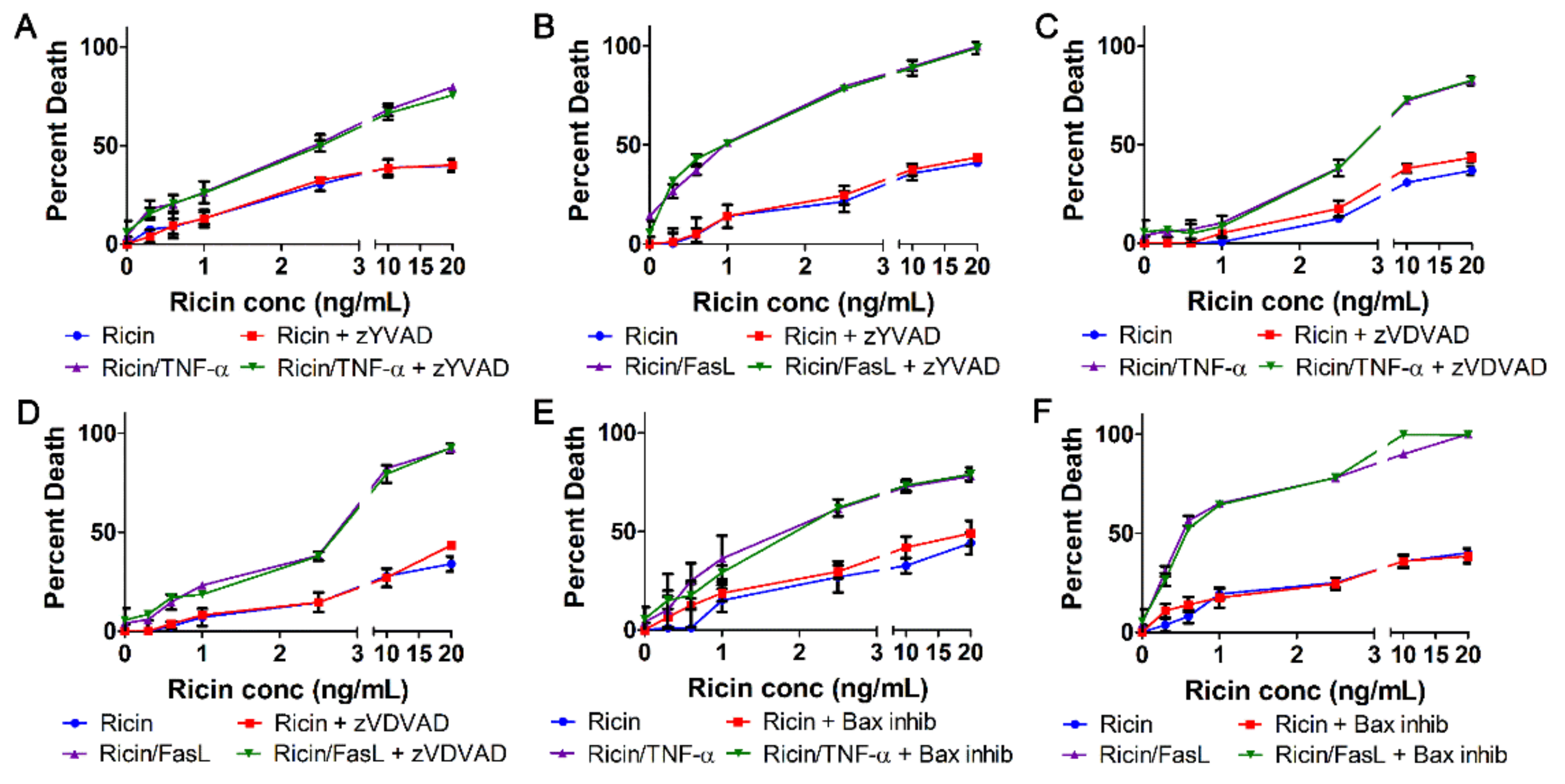
2.5. Cell Death Induced by Ricin/TNF-α and Ricin/FasL Depends on Cathepsins
3. Discussion
3.1. Distinct Cell Death Modalities
3.2. Cathepsin-Dependent Cell Death
3.3. Therapeutic Implications
4. Materials and Methods
4.1. Reagents and Inhibitors
4.2. Cell Culture
4.3. Cell Death Assays
4.4. Immunoblots
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bradberry, S.M.; Dickers, K.J.; Rice, P.; Griffiths, G.D.; Vale, J.A. Ricin poisoning. Toxicol. Rev. 2003, 22, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning a comprehensive review. J. Am. Med. Assoc. 2005, 294, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Lopez Nunez, O.F.; Pizon, A.F.; Tamama, K. Ricin Poisoning after Oral Ingestion of Castor Beans: A Case Report and Review of the Literature and Laboratory Testing. J. Emerg. Med. 2017, 53, e67–e71. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.D. Bioterrorism: Toxins as weapons. J. Pharm. Pract. 2012, 25, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnakone, P.; Balali-Mood, M.; Llewellyn, L.; Singh, B.R. Biological Toxins and Bioterrorism; Springer: Dordrecht, The Netherlands, 2015; ISBN 9789400758698. [Google Scholar]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: structure, mode of action, and some current applications. FASEB J. 2018, 8, 201–208. [Google Scholar] [CrossRef]
- Taubenschmid, J.; Stadlmann, J.; Jost, M.; Klokk, T.I.; Rillahan, C.D.; Leibbrandt, A.; Mechtler, K.; Paulson, J.C.; Jude, J.; Zuber, J.; et al. A vital sugar code for ricin toxicity. Cell Res. 2017, 27, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Michael Lord, J. Ricin trafficking in cells. Toxins (Basel) 2015, 7, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.H.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. Nucleic Acids Symp. Ser. 1988, 19, 139–142. [Google Scholar]
- RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130.
- Sperti, S.; Montanaro, L.; Mattioli, A.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro: 60S ribosomal subunit as the target of the toxin (Short Communication). Biochem. J. 1973, 136, 813–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Khade, P.K.; Sanbonmatsu, K.Y.; Joseph, S. Functional role of the sarcin-ricin loop of the 23s rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 2012, 419, 125–138. [Google Scholar] [CrossRef]
- Griffiths, G.D.; Phillips, G.J.; Holley, J. Inhalation toxicology of ricin preparations: Animal models, prophylactic and therapeutic approaches to protection. Inhal. Toxicol. 2007, 19, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, C.L.; Pitt, M.L.M. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Pincus, S.H.; Bhaskaran, M.; Brey, R.N.; Didier, P.J.; Doyle-Meyers, L.A.; Roy, C.J. Clinical and pathological findings associated with aerosol exposure of macaques to ricin toxin. Toxins (Basel) 2015, 7, 2121–2133. [Google Scholar] [CrossRef]
- Gal, Y.; Mazor, O.; Falach, R.; Sapoznikov, A.; Kronman, C.; Sabo, T. Treatments for pulmonary ricin intoxication: Current aspects and future prospects. Toxins (Basel) 2017, 9, 311. [Google Scholar] [CrossRef]
- Rong, Y.; Westfall, J.; Ehrbar, D.; LaRocca, T.; Mantis, N.J. TRAIL (CD253) Sensitizes Human Airway Epithelial Cells to Toxin-Induced Cell Death. mSphere 2018, 3, e00399-18. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am. J. Pathol. 2007, 170, 1497–1510. [Google Scholar] [CrossRef]
- DaSilva, L.; Cote, D.; Roy, C.; Martinez, M.; Duniho, S.; Pitt, M.L.M.; Downey, T.; Dertzbaugh, M. Pulmonary gene expression profiling of inhaled ricin. Toxicon 2003, 41, 813–822. [Google Scholar] [CrossRef]
- David, J.; Wilkinson, L.J.; Griffiths, G.D. Inflammatory gene expression in response to sub-lethal ricin exposure in Balb/c mice. Toxicology 2009, 264, 119–130. [Google Scholar] [CrossRef]
- Lindauer, M.; Wong, J.; Magun, B. Ricin toxin activates the NALP3 inflammasome. Toxins (Basel) 2010, 2, 1500–1514. [Google Scholar] [CrossRef]
- Wong, J.; Magun, B.E.; Wood, L.J. Lung inflammation caused by inhaled toxicants: A review. Int. J. COPD 2016, 11, 1391–1401. [Google Scholar] [CrossRef]
- Knight, M.J.; Riffkin, C.D.; Muscat, A.M.; Ashley, D.M.; Hawkins, C.J. Analysis of FasL and trail induced apoptosis pathways in glioma cells. Oncogene 2001, 20, 5789–5798. [Google Scholar] [CrossRef]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar]
- Babu, D.J.; Soenen, S.; Raemdonck, K.; Leclercq, G.; De Backer, O.; Motterlini, R.A.; Lefebvre, R. TNF-α/Cycloheximide-Induced Oxidative Stress and Apoptosis in Murine Intestinal Epithelial MODE-K Cells. Curr. Pharm. Des. 2012, 18, 4414–4425. [Google Scholar] [CrossRef]
- Jin, S.; Ray, R.M.; Johnson, L.R. TNF-α/cycloheximide-induced apoptosis in intestinal epithelial cells requires Rac1-regulated reactive oxygen species. Am. J. Physiol. Liver Physiol. 2008, 294, G928–G937. [Google Scholar] [CrossRef]
- Vercammen, D.; Vandenabeele, P.; Beyaert, R.; Declercq, W.; Fiers, W. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 1997, 9, 801–808. [Google Scholar] [CrossRef]
- Lin, Y.; Choksi, S.; Shen, H.M.; Yang, Q.F.; Hur, G.M.; Kim, Y.S.; Tran, J.H.; Nedospasov, S.A.; Liu, Z.G. Tumor Necrosis Factor-induced Nonapoptotic Cell Death Requires Receptor-interacting Protein-mediated Cellular Reactive Oxygen Species Accumulation. J. Biol. Chem. 2004, 279, 10822–10828. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, T.J.; Sosunov, S.A.; Shakerley, N.L.; Ten, V.S.; Ratner, A.J. Hyperglycemic conditions prime cells for RIP1-dependent necroptosis. J. Biol. Chem. 2016, 291, 13753–13761. [Google Scholar] [CrossRef]
- McCaig, W.D.; Patel, P.S.; Sosunov, S.A.; Shakerley, N.L.; Smiraglia, T.A.; Craft, M.M.; Walker, K.M.; Deragon, M.A.; Ten, V.S.; LaRocca, T.J. Hyperglycemia potentiates a shift from apoptosis to RIP1-dependent necroptosis. Cell Death Discov. 2018, 4, 55. [Google Scholar] [CrossRef]
- Mori, T.; Doi, R.; Toyoda, E.; Koizumi, M.; Ito, D.; Kami, K.; Kida, A.; Masui, T.; Kawaguchi, Y.; Fujimoto, K. Regulation of the resistance to TRAIL-induced apoptosis as a new strategy for pancreatic cancer. Surgery 2005, 138, 71–77. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Kohler, B.F.; Lehtivarjo, A.K.; Dussmann, H.; Courtney, M.J.; Prehn, J.H.M.; Rehm, M. Real time analysis of tumor necrosis factor-related apoptosis-inducing ligand/cycloheximide-induced caspase activities during apoptosis initiation. J. Biol. Chem. 2008, 283, 21676–21685. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Bouchier-Hayes, L. The role of caspase-2 in stress-induced apoptosis. J. Cell. Mol. Med. 2010, 14, 1212–1224. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, C.P.; Kadioglu, A.; Yang, A.-L.; Coward, W.R.; Chow, S.C. The Cathepsin B Inhibitor, z-FA-FMK, Inhibits Human T Cell Proliferation In Vitro and Modulates Host Response to Pneumococcal Infection In Vivo. J. Immunol. 2006, 177, 3827–3836. [Google Scholar] [CrossRef] [Green Version]
- Rozman-Pungerčar, J.; Kopitar-Jerala, N.; Bogyo, M.; Turk, D.; Vasiljeva, O.; Stefe, I.; Vandenabeele, P.; Brömme, D.; Puizdar, V.; Fonović, M.; et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: When reaction mechanism is more important than specificity. Cell Death Differ. 2003, 10, 881–888. [Google Scholar] [CrossRef]
- Lopez-Hernandez, F.J.; Ortiz, M.A.; Bayon, Y.; Piedrafita, F.J. Z-FA-fmk inhibits effector caspases but not initiator caspases 8 and 10, and demonstrates that novel anticancer retinoid-related molecules induce apoptosis via the intrinsic pathway. Mol. Cancer Ther. 2003, 2, 255–263. [Google Scholar]
- Ahmed, N.K.; Martin, L.A.; Watts, L.M.; Palmer, J.; Thornburg, L.; Prior, J.; Esser, R.E. Peptidyl fluoromethyl ketones as inhibitors of cathepsin B. Implication for treatment of rheumatoid arthritis. Biochem. Pharmacol. 1992, 44, 1201–1207. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mizoue, K.; Kitamura, K.; Tse, W.C.; Huber, C.P.; Ishida, T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Biopolym. Pept. Sci. Sect. 1999, 51, 99–107. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef]
- Demuth, H.U.; Schierhorn, A.; Bryan, P.; Höfke, R.; Kirschke, H.; Brömme, D. N-peptidyl, O-acyl hydroxamates: Comparison of the selective inhibition of serine and cysteine proteinases. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 1996, 1295, 179–186. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Cooper, J.R.; Abdullatif, M.B.; Burnett, E.C.; Kempsell, K.E.; Conforti, F.; Tolley, H.; Collins, J.E.; Davies, D.E. Long term culture of the a549 cancer cell line promotes multilamellar body formation and differentiation towards an alveolar type II Pneumocyte phenotype. PLoS ONE 2016, 11, e0164438. [Google Scholar] [CrossRef]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Li, X.P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar] [CrossRef]
- Jin, Z.; El-Deiry, W.S. Distinct Signaling Pathways in TRAIL- versus Tumor Necrosis Factor-Induced Apoptosis. Mol. Cell. Biol. 2006, 26, 8136–8148. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.; Jeong, E.J.; Kim, I.K.; Jung, Y.K.; Kim, K.S. Fas- and tumor necrosis factor-mediated apoptosis uses the same binding surface of FADD to trigger signal transduction: A typical model for convergent signal transduction. J. Biol. Chem. 2000, 275, 36217–36222. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Lavrik, I. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Sikriwal, D.; Batra, J.K. Ribosome inactivating proteins and apoptosis. Plant Cell Monogr. 2010, 18, 167–189. [Google Scholar]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Suntres, Z.E.; Stone, W.L.; Smith, M.G. Ricin—Induced Toxicity: The Role of Oxidative Stress. J. Med. CBR Def. 2005, 3, 1–21. [Google Scholar]
- Oda, T.; Iwaoka, J.; Komatsu, N.; Tsuyoshi, M. Involvement of N-acetylcysteine-sensitive pathways in ricin-induced apoptotic cell death in U937 cells. Biosci. Biotechnol. Biochem. 1999, 63, 341–348. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Ling, C. Cytotoxic genes from traditional Chinese medicine inhibit tumor growth both in vitro and in vivo. J. Integr. Med. 2014, 12, 483–494. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodges, A.L.; Kempen, C.G.; McCaig, W.D.; Parker, C.A.; Mantis, N.J.; LaRocca, T.J. TNF Family Cytokines Induce Distinct Cell Death Modalities in the A549 Human Lung Epithelial Cell Line when Administered in Combination with Ricin Toxin. Toxins 2019, 11, 450. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080450
Hodges AL, Kempen CG, McCaig WD, Parker CA, Mantis NJ, LaRocca TJ. TNF Family Cytokines Induce Distinct Cell Death Modalities in the A549 Human Lung Epithelial Cell Line when Administered in Combination with Ricin Toxin. Toxins. 2019; 11(8):450. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080450
Chicago/Turabian StyleHodges, Alexa L., Cody G. Kempen, William D. McCaig, Cory A. Parker, Nicholas J. Mantis, and Timothy J. LaRocca. 2019. "TNF Family Cytokines Induce Distinct Cell Death Modalities in the A549 Human Lung Epithelial Cell Line when Administered in Combination with Ricin Toxin" Toxins 11, no. 8: 450. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080450