Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
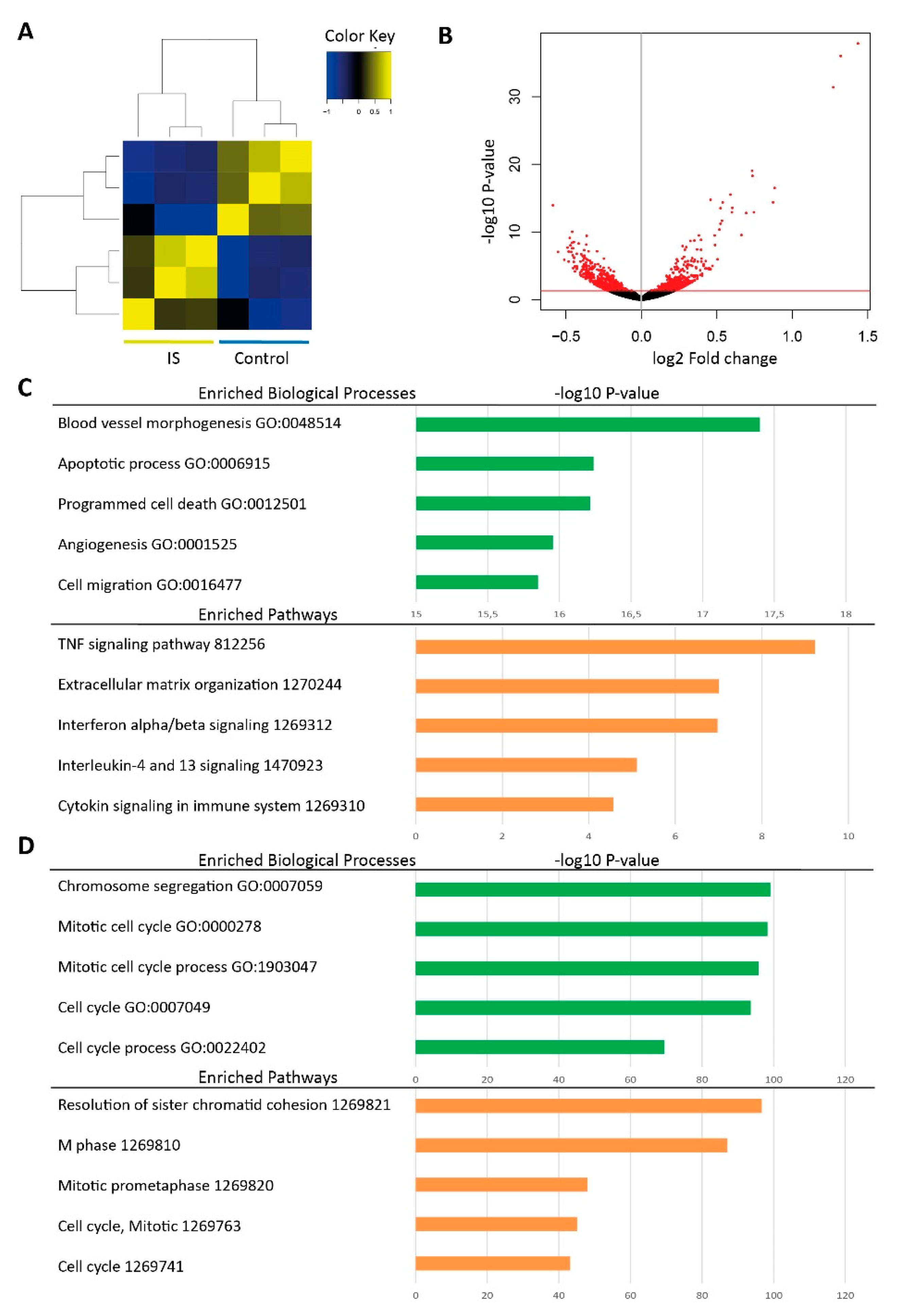
2.1. RNA-seq Reveals Differentially Expressed Genes in IS Treated HUVECs
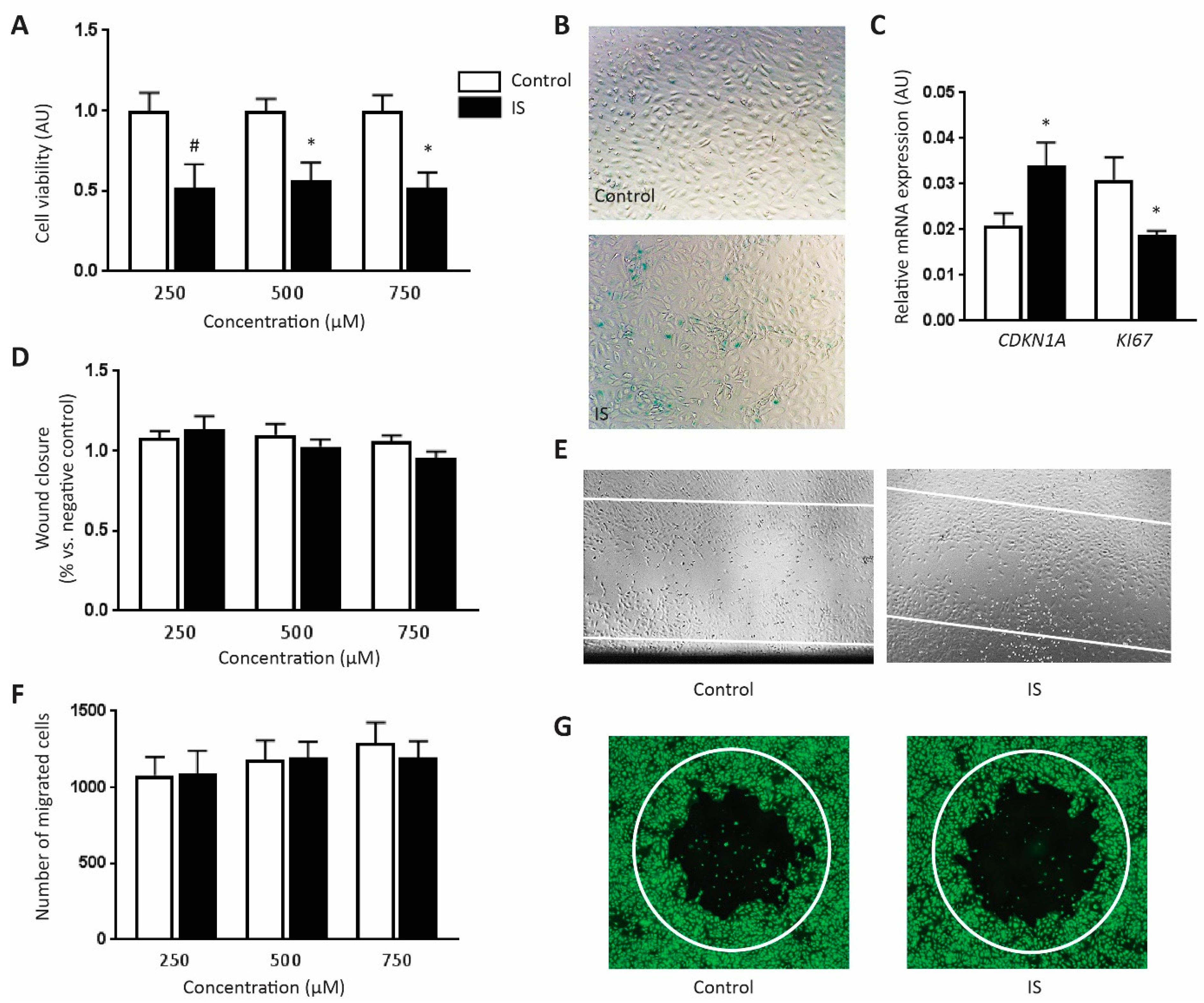
2.2. IS Inhibits Cell Viability at High Concentration
2.3. IS Induces Cell Senescence
2.4. IS Does Not Influence Cell Migration Ability
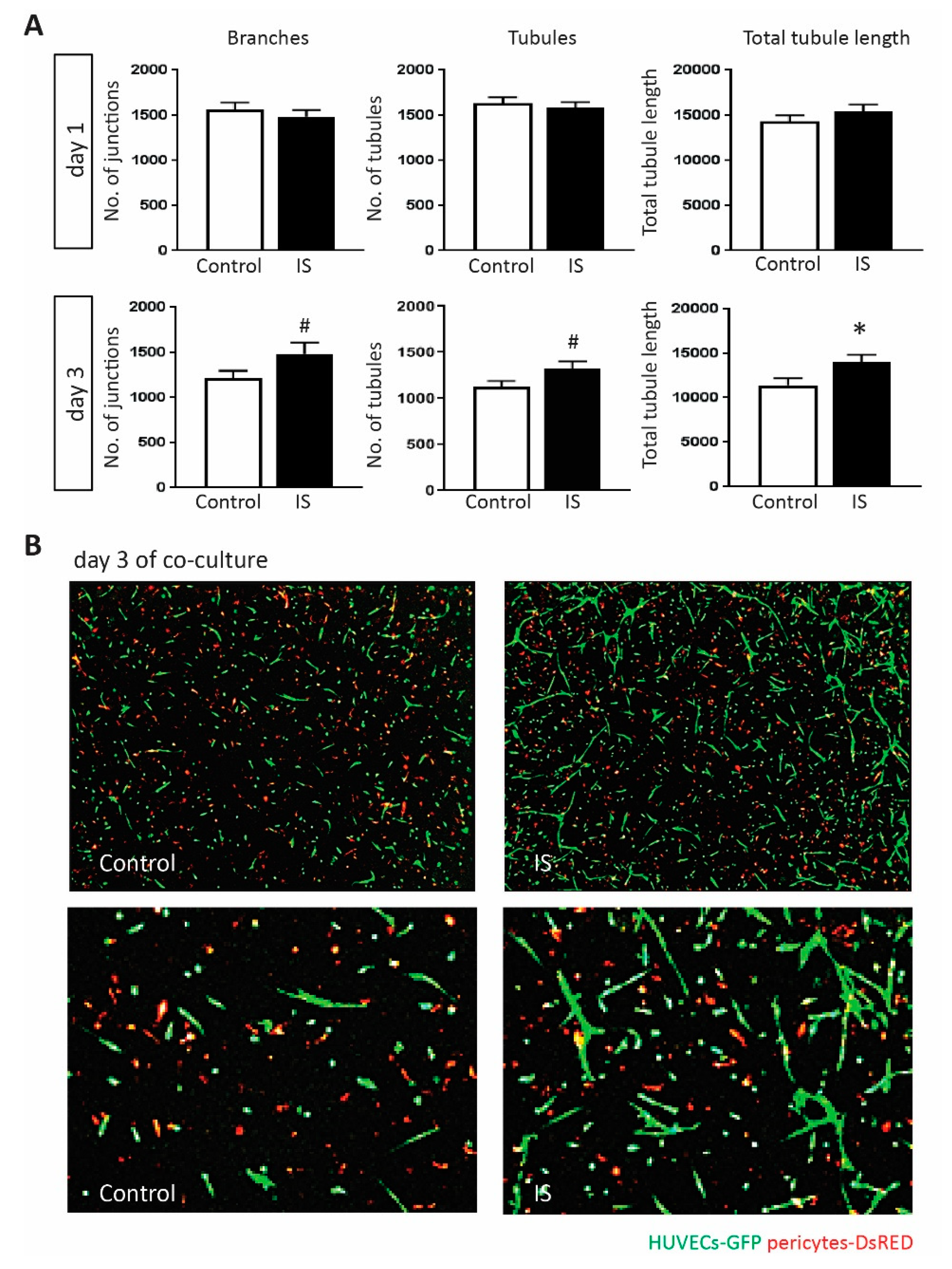
2.5. IS Promotes Angiogenic Response
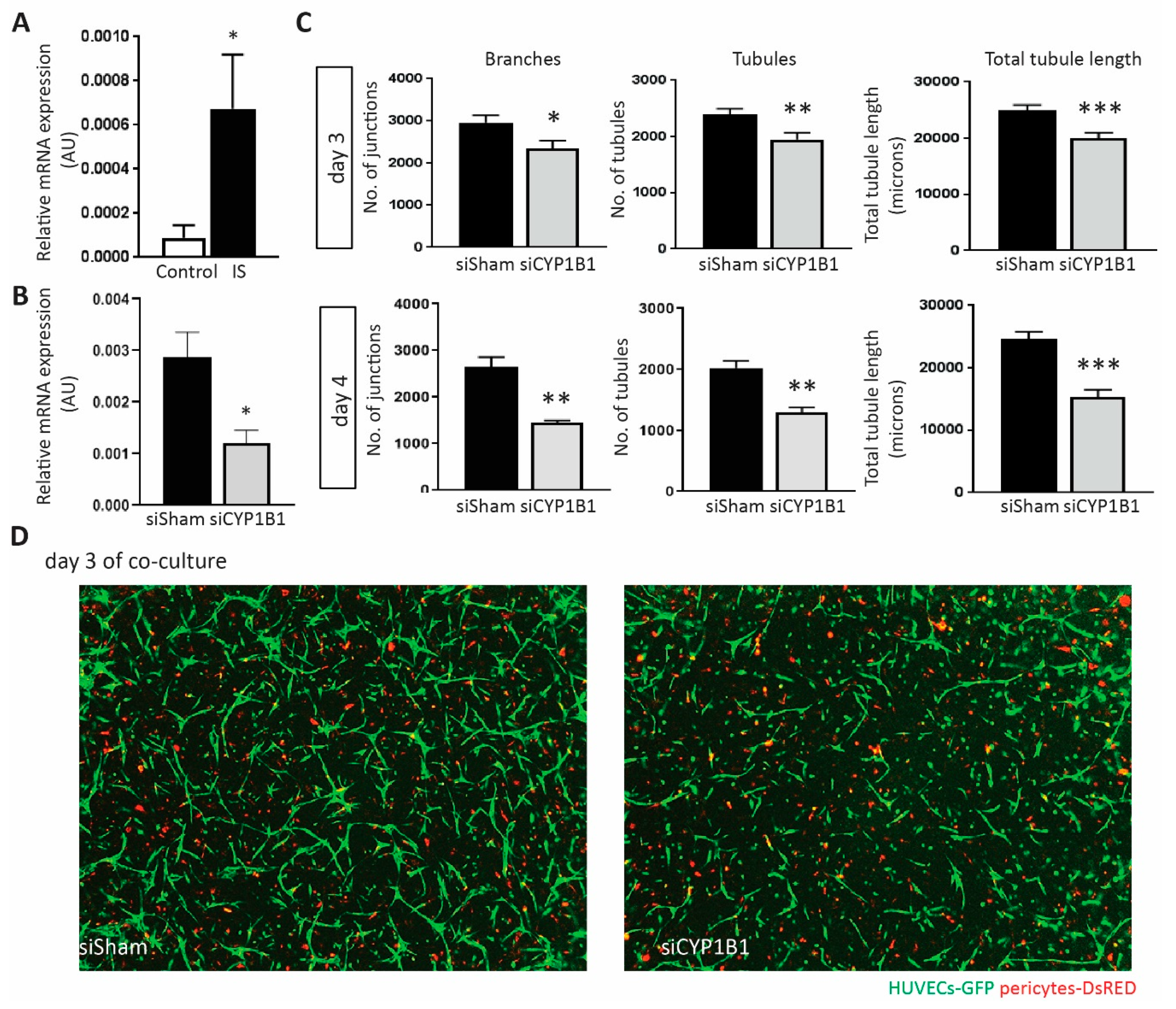
2.6. Depletion of CYP1B1 Inhibits Tubular Formation
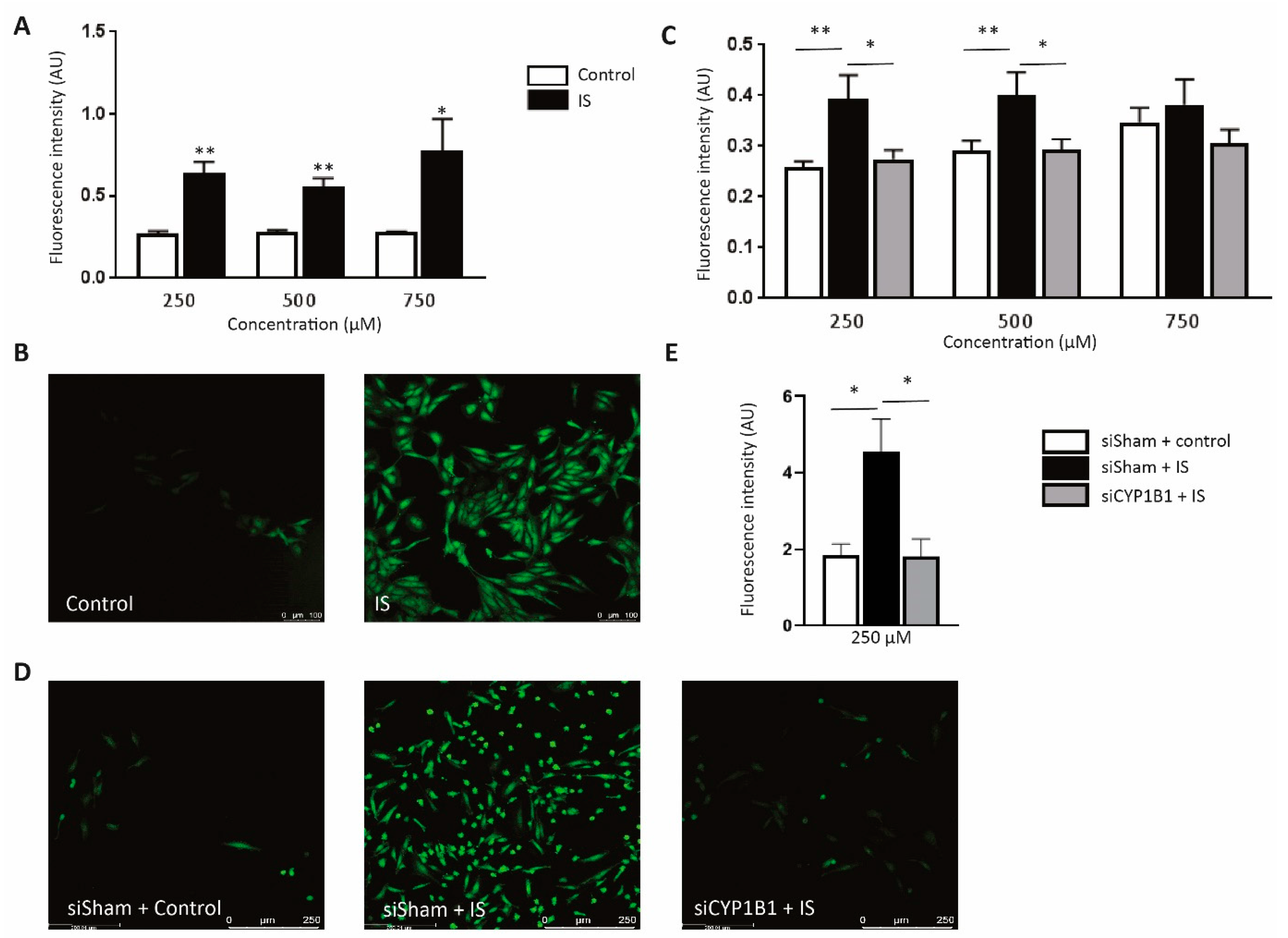
2.7. CYP1B1 Plays an Important Role in IS-Increased ROS Production
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA-seq and Data Analysis
4.3. Gene Enrichment Analysis
4.4. Cell Metabolism Assay
4.5. Senescence-Associated Beta Galactosidase Activity
4.6. Wound Healing Assay
4.7. Cell Migration Plug Assay
4.8. 3D Collagen Co-Culture Assay
4.9. Detection of Intracellular ROS Levels in HUVECs
4.10. Detection of Intracellular ROS Levels in Cardiac Microvascular Endothelial Cells (CMECs)
4.11. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
4.12. Short Interference RNA
4.13. Statistical Analyses
4.14. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ellis, R.J.; Small, D.M.; Vesey, D.A.; Johnson, D.W.; Francis, R.; Vitetta, L.; Gobe, G.C.; Morais, C. Indoxyl sulphate and kidney disease: Causes, consequences and interventions. Nephrology 2016, 21, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.C.; Young, G.H.; Huang, P.H.; Lo, S.C.; Wang, K.C.; Sun, C.Y.; Liang, C.J.; Huang, T.M.; Chen, J.H.; Chang, F.C.; et al. In acute kidney injury, indoxyl sulfate impairs human endothelial progenitor cells: Modulation by statin. Angiogenesis 2013, 16, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Cao, X.; Zou, J.; Shen, B.; Zhang, X.; Liu, Z.; Lv, W.; Teng, J.; Ding, X. Indoxyl sulfate, a valuable biomarker in chronic kidney disease and dialysis. Hemodial Int. 2017, 21, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Inagi, R. Effect of AST-120 in Chronic Kidney Disease Treatment: Still a Controversy? Nephron 2017, 135, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.; Chillon, J.M.; Massy, Z.A.; Boullier, A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bEnd.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-acetylcysteine. Sci. World J. 2015, 2015, 620826. [Google Scholar] [CrossRef]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Cellular Senescence in Human Umbilical Vein Endothelial Cells. J. Atheroscler. Thromb. 2014, 21, 23663. [Google Scholar] [CrossRef]
- Shen, W.C.; Liang, C.J.; Huang, T.M.; Liu, C.W.; Wang, S.H.; Young, G.H.; Tsai, J.S.; Tseng, Y.C.; Peng, Y.S.; Wu, V.C.; et al. Indoxyl sulfate enhances IL-1beta-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFkappaB/AP-1 pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef]
- Chrifi, I.; Hermkens, D.; Brandt, M.M.; Van Dijk, C.G.; Bürgisser, P.E.; Haasdijk, R.; Pei, J.; van de Kamp, E.H.; Zhu, C.; Blonden, L.; et al. Cgnl1, an endothelial junction complex protein, regulates GTPase mediated angiogenesis. Cardiovasc. Res. 2017, 113, 1776–1788. [Google Scholar] [CrossRef] [Green Version]
- Chrifi, I.; Louzao-Martinez, L.; Brandt, M.M.; Van Dijk, C.G.; Bürgisser, P.E.; Zhu, C.; Kros, J.M.; Verhaar, M.C.; Duncker, D.J.; Cheng, C. CMTM4 regulates angiogenesis by promoting cell surface recycling of VE-cadherin to endothelial adherens junctions. Angiogenesis 2018, 22, 75–93. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Nath, K.A.; Norby, S.M. Reactive oxygen species and acute renal failure. Physiol. Med. 2000, 109, 665–678. [Google Scholar] [CrossRef]
- Aksua, U.; Demircic, C.; Incea, C. The Pathogenesis of Acute Kidney Injury and the Toxic Triangle of Oxygen, Reactive Oxygen Species and Nitric Oxide. In Pathophysiology Sepsis Induced Acute Kidney Injury; Karger Publishers: Basel, Switzerland, 2011; Volume 174. [Google Scholar]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Rapsomanikis, K.P.; Dounousi, E. Chronic Kidney Disease and Disproportionally Increased Cardiovascular Damage: Does Oxidative Stress Explain the Burden? Oxid. Med. Cell Longev. 2017, 2017, 9036450. [Google Scholar] [CrossRef]
- Cachofeiro, V.; Goicochea, M.; de Vinuesa, S.G.; Oubina, P.; Lahera, V.; Luno, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. Suppl. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [Green Version]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate-Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, V.; Thijssen, S.; Tao, X.; Fuertinger, D.H.; Kappel, F.; Kotanko, P. In silico comparison of protein-bound uremic toxin removal by hemodialysis, hemodiafiltration, membrane adsorption, and binding competition. Sci. Rep. 2019, 9, 909. [Google Scholar] [CrossRef]
- Wang, W.; Liu, X.; Li, J.; Li, Y.; Li, L.; Wang, S.; Zhang, J.; Zhang, Y.; Huang, H. The Effects of Indoxyl Sulfate on Human Umbilical Cord-Derived Mesenchymal Stem Cells In Vitro. Cell. Physiol. Biochem. 2016, 38, 401–414. [Google Scholar] [CrossRef]
- Kharait, S.; Haddad, D.J.; Springer, M.L. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem. Biophys. Res. Commun. 2011, 409, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Edamatsu, T.; Fujieda, A.; Itoh, Y. Phenyl sulfate, indoxyl sulfate and p-cresyl sulfate decrease glutathione level to render cells vulnerable to oxidative stress in renal tubular cells. PLoS ONE 2018, 13, e0193342. [Google Scholar] [CrossRef]
- Edamatsu, T.; Fujieda, A.; Ezawa, A.; Itoh, Y. Classification of Five Uremic Solutes According to Their Effects on Renal Tubular Cells. Int. J. Nephrol. 2014, 2014, 512178. [Google Scholar] [CrossRef]
- Saito, S.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Enomoto, A.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor is involved in expression of TGF-beta1 and alpha-smooth muscle actin in proximal tubular cells. Endocrinology 2014, 155, 1899–1907. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.F.V. Oxidative stress: The role of cytochromes P450 in oxygen activation. J. Chem. Technol. Biotechnol. 2002, 77, 1095–1100. [Google Scholar] [CrossRef]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and function of the cytochrome P450 peroxygenase enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.I.; Melvin, W.T.; Greenlee, W.F.; Burke, M.D. Regulation, Function, and Tissue-specific Expression of Cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. 2001, 41, 297–316. [Google Scholar] [CrossRef]
- Stengel, B. Chronic kidney disease and cancer: A troubling connection. J. Nephrol. 2010, 23, 253. [Google Scholar]
- McFadyen, M.C.; Melvin, W.T.; Murray, G.I. Cytochrome P450 CYP1B1 activity in renal cell carcinoma. Br. J. Cancer. 2004, 91, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef]
- Tang, Y.; Scheef, E.A.; Wang, S.; Sorenson, C.M.; Marcus, C.B.; Jefcoate, C.R.; Sheibani, N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 2009, 113, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Palenski, T.L.; Gurel, Z.; Sorenson, C.M.; Hankenson, K.D.; Sheibani, N. Cyp1B1 expression promotes angiogenesis by suppressing NF-kappaB activity. Am. J. Physiol. Cell. Physiol. 2013, 305, C1170–C1184. [Google Scholar] [CrossRef]
- Futrakul, N.; Butthep, P.; Laohareungpanya, N.; Chaisuriya, P.; Ratanabanangkoon, K. A defective angiogenesis in chronic kidney disease. Ren. Fail. 2008, 30, 215–217. [Google Scholar] [CrossRef]
- Anderson, C.E.; Hamm, L.L.; Batuman, G.; Kumbala, D.R.; Chen, C.S.; Kallu, S.G.; Siriki, R.; Gadde, S.; Kleinpeter, M.A.; Krane, N.K.; et al. The association of angiogenic factors and chronic kidney disease. BMC Nephrol. 2018, 19, 117. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia and Dysregulated Angiogenesis in Kidney Disease. Kidney Dis. 2015, 1, 80–89. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Huang, H.L.; Lin, C.C.; Tsai, T.H.; Wang, C.H.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Indoxyl sulfate suppresses endothelial progenitor cell-mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Moore, D.B.; Woodard, W.; Fenoglio, A.; Yoder, M.C. Vessel wall–derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Hemost. Thromb. Vasc. Biol. 2005, 105. [Google Scholar] [CrossRef]
- Patan, S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004, 117, 3–32. [Google Scholar]
- Schroeder, J.C.; DiNatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef]
- Moriguchi, T.; Motohashi, H.; Hosoya, T.; Nakajima, O.; Takahashi, S.; Ohsako, S.; Aoki, Y.; Nishimura, N.; Tohyama, C.; Fujii-Kuriyama, Y.; et al. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Nakamura, M.; Tsutsumi, Y.; Suenaga, A.; Otagiri, M. Pharmacokinetics and tissue distribution of uraemic indoxyl sulphate in rats. Biopharm. Drug Dispos. 2003, 24, 345–355. [Google Scholar] [CrossRef]
- Sindhu, R.K.; Vaziri, N.D. Upregulation of cytochrome P450 1A2 in chronic renal failure: Does oxidized tryptophan play a role? Adv. Exp. Med. Biol. 2003, 527, 401–407. [Google Scholar]
- Patel, R.D.; Murray, I.A.; Flaveny, C.A.; Kusnadi, A.; Perdew, G.H. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab. Investig. 2009, 89, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Ikuta, T.; Kawajiri, K. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp. Cell Res. 2006, 312, 3585–3594. [Google Scholar] [CrossRef]
- Hollingshead, B.D.; Beischlag, T.V.; Dinatale, B.C.; Ramadoss, P.; Perdew, G.H. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008, 68, 3609–3617. [Google Scholar] [CrossRef]
- Patel, R.D.; Kim, D.J.; Peters, J.M.; Perdew, G.H. The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin. Toxicol. Sci. 2006, 89, 75–82. [Google Scholar] [CrossRef]
- Van Dijk, C.G.; Oosterhuis, N.R.; Xu, Y.J.; Brandt, M.; Paulus, W.J.; Van Heerebeek, L.; Duncker, D.J.; Verhaar, M.C.; Fontoura, D.; Lourenço, A.P.; et al. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat With Cardiorenal Metabolic Syndrome. Circ. Heart Fail. 2016, 9, e002760. [Google Scholar] [CrossRef]
- Brandt, M.M.; Meddens, C.A.; Louzao-Martinez, L.; Van Den Dungen, N.A.; Lansu, N.R.; Nieuwenhuis, E.E.; Duncker, D.J.; Verhaar, M.C.; Joles, J.A.; Mokry, M.; et al. Chromatin Conformation Links Distal Target Genes to CKD Loci. J. Am. Soc. Nephrol. 2018, 29, 462–476. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.D.; Goebel, M.; Helmes, M.; van der Velden, J.; Musters, R.; Koolwijk, P.; Paulus, W.; van Hinsbergh, V. Loss of Cardiac Microvascular Endothelial Cell-Cardiomyocyte Interaction by Inflammatory Activation is Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, in press. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Ensembl ID | Gene Symbol | Gene Name | Angiogenic Function † | Fold Change (log2) | p-Value |
|---|---|---|---|---|---|---|
| Up-regulation | ENSG00000138061 | CYP1B1 | Cytochrome P450 Family 1 Subfamily B Member 1 | Promote angiogenesis | 1.434 | 1.390 × 10−38 |
| ENSG00000114812 | VIPR1 | Vasoactive Intestinal Peptide Receptor 1 | Not known | 1.321 | 1.072 × 10−36 | |
| ENSG00000137809 | ITGA11 | Integrin Subunit Alpha 11 | Not known | 1.272 | 4.190 × 10−32 | |
| ENSG00000178695 | KCTD12 | Potassium Channel Tetramerization Domain Containing 12 | Not known | 0.883 | 3.247 × 10−17 | |
| ENSG00000063438 | AHRR | Aryl-Hydrocarbon Receptor Repressor | Not known | 0.872 | 4.144 × 10−15 | |
| ENSG00000007908 | SELE | Selectin E | Not known | 0.746 | 1.240 × 10−13 | |
| ENSG00000137331 | IER3 | Immediate Early Response 3 | Not known | 0.736 | 5.396 × 10−19 | |
| ENSG00000163659 | TIPARP | TCDD Inducible Poly(ADP-Ribose) Polymerase | Not known | 0.734 | 8.865 × 10−20 | |
| ENSG00000144476 | ACKR3 | Atypical Chemokine Receptor 3 | Promote angiogenesis | 0.695 | 1.596 × 10−13 | |
| ENSG00000144802 | NFKBIZ | NFKB Inhibitor Zeta | Not known | 0.663 | 3.022 × 10−10 | |
| Down-regulation | ENSG00000117724 | CENPF | Centromere Protein F | Not known | −0.586 | 1.114 × 10−14 |
| ENSG00000143476 | DTL | Denticleless E3 Ubiquitin Protein Ligase Homolog | Not known | −0.554 | 8.619 × 10−8 | |
| ENSG00000163808 | KIF15 | Kinesin Family Member 15 | Not known | −0.510 | 1.195 × 10−6 | |
| ENSG00000138778 | CENPE | Centromere Protein E | Not known | −0.490 | 1.912 × 10−8 | |
| ENSG00000137812 | CASC5 | Kinetochore Scaffold 1 | Not known | −0.483 | 8.198 × 10−8 | |
| ENSG00000184661 | CDCA2 | Cell Division Cycle Associated 2 | Not known | −0.482 | 2.314 × 10−6 | |
| ENSG00000066279 | ASPM | Abnormal Spindle Microtubule Assembly | Not known | −0.473 | 7.741 × 10−10 | |
| ENSG00000156802 | ATAD2 | ATPase Family AAA Domain Containing 2 | Inhibit angiogenesis | −0.470 | 9.314 × 10−9 | |
| ENSG00000196549 | MME | Membrane Metalloendopeptidase | Not known | −0.465 | 9.572 × 10−8 | |
| ENSG00000132646 | PCNA | Proliferating Cell Nuclear Antigen | Not known | −0.461 | 3.809 × 10−9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, J.; Juni, R.; Harakalova, M.; Duncker, D.J.; Asselbergs, F.W.; Koolwijk, P.; van Hinsbergh, V.; Verhaar, M.C.; Mokry, M.; Cheng, C. Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1. Toxins 2019, 11, 454. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080454
Pei J, Juni R, Harakalova M, Duncker DJ, Asselbergs FW, Koolwijk P, van Hinsbergh V, Verhaar MC, Mokry M, Cheng C. Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1. Toxins. 2019; 11(8):454. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080454
Chicago/Turabian StylePei, Jiayi, Rio Juni, Magdalena Harakalova, Dirk J. Duncker, Folkert W. Asselbergs, Pieter Koolwijk, Victor van Hinsbergh, Marianne C. Verhaar, Michal Mokry, and Caroline Cheng. 2019. "Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1" Toxins 11, no. 8: 454. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080454