Quercetin-3-Rutinoside Blocks the Disassembly of Cholera Toxin by Protein Disulfide Isomerase

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
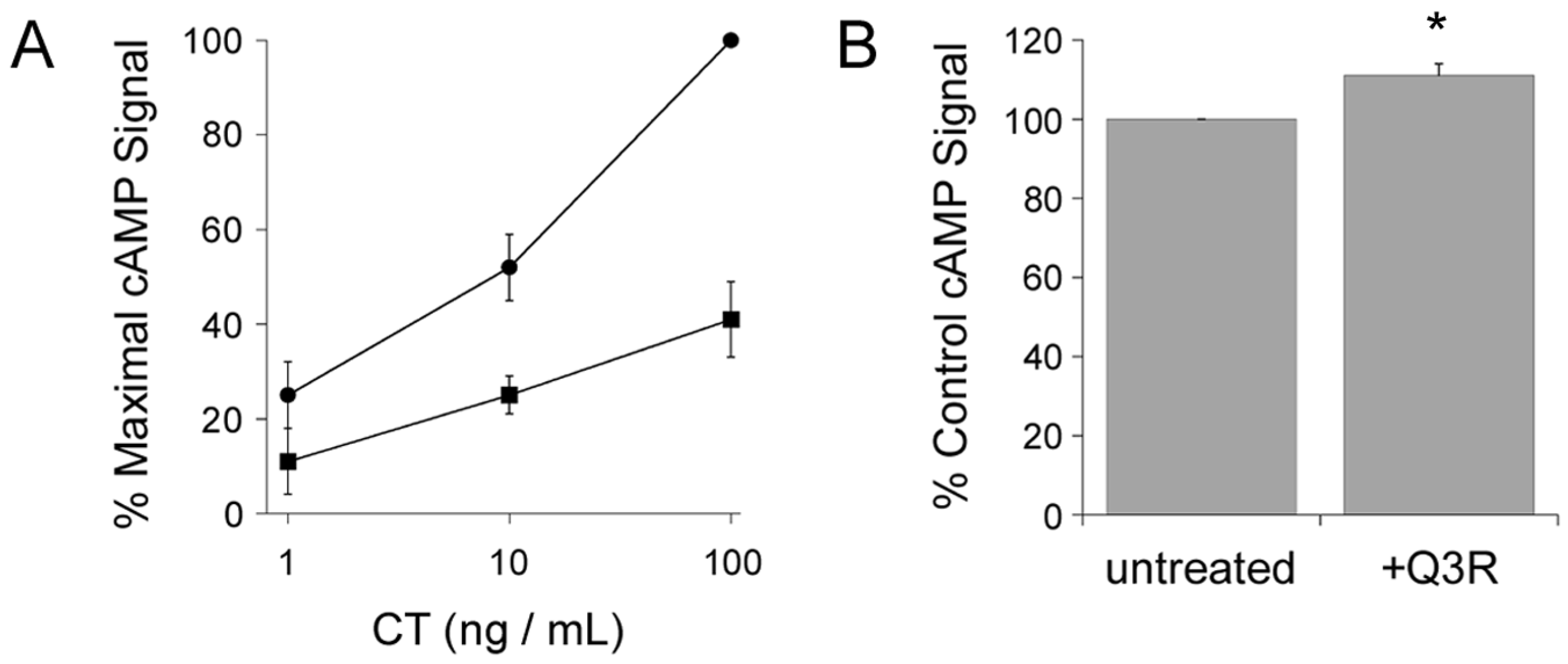
2.1. Q3R Protects Cultured Cells from CT
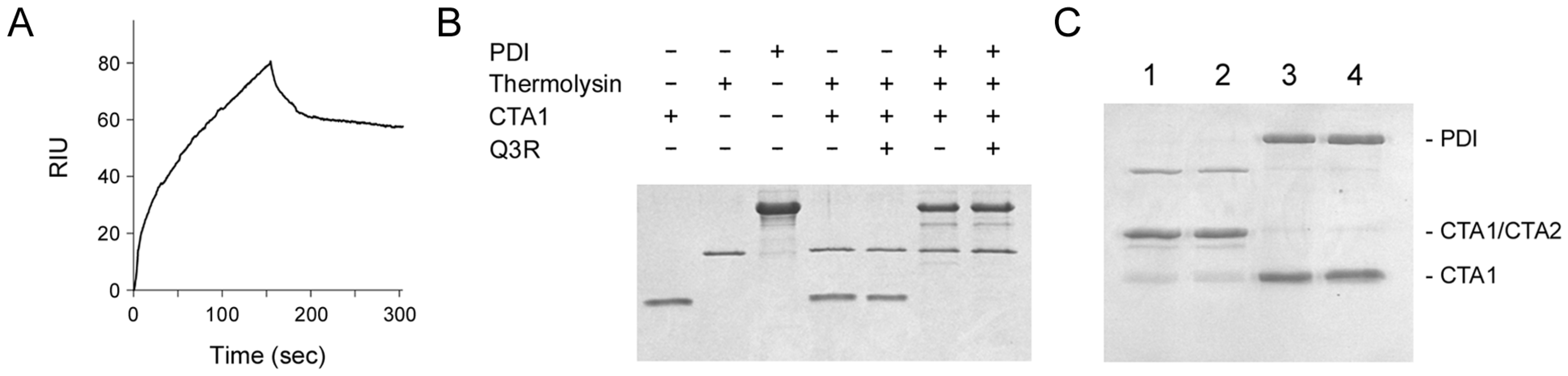
2.2. Q3R Does Not Prevent PDI from Binding or Reducing CTA1
2.3. Q3R Disrupts PDI-Driven Disassembly of the CT Holotoxin
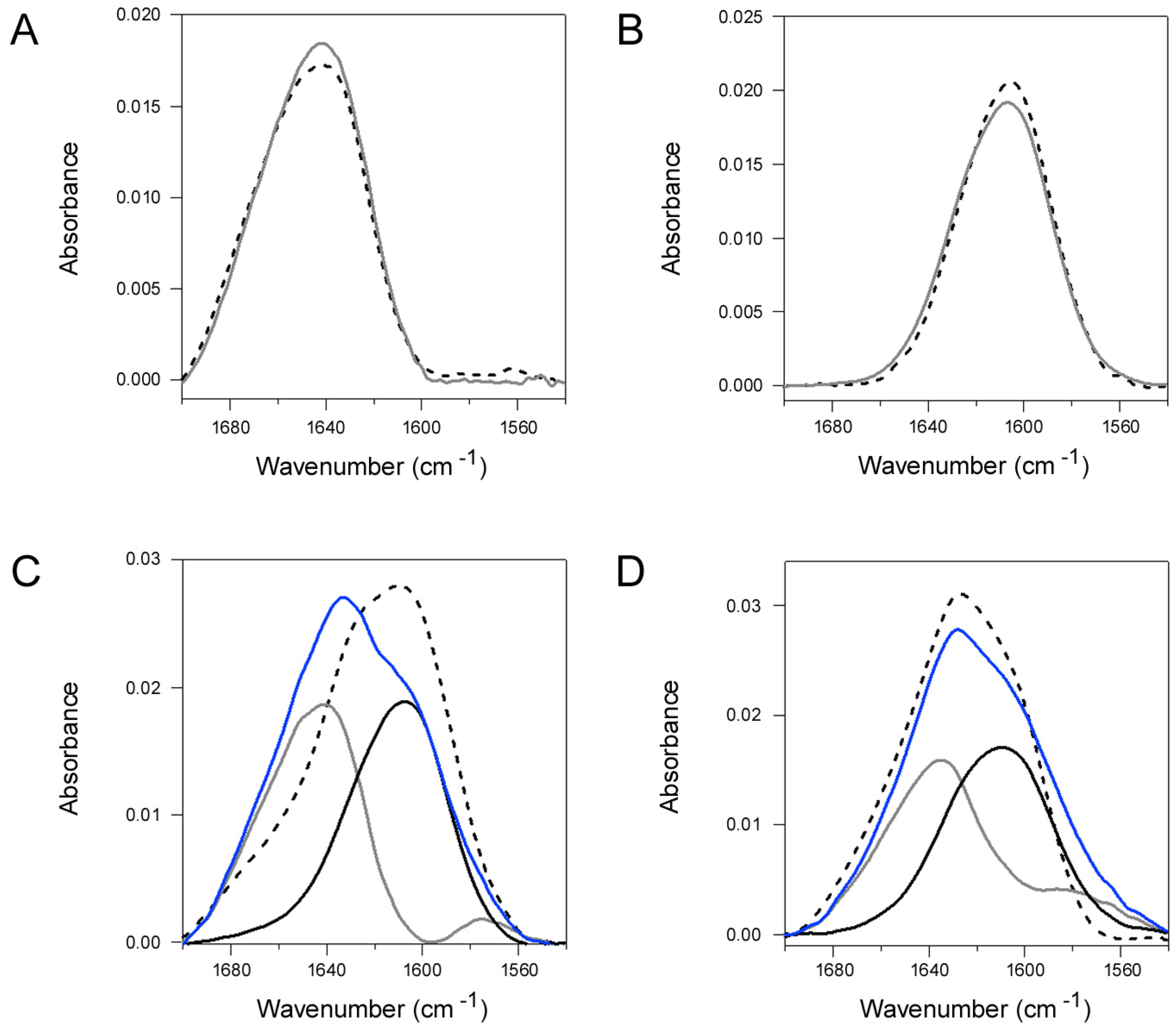
2.4. Q3R Blocks the Conformational Change in PDI that Occurs with Its Binding to CTA1
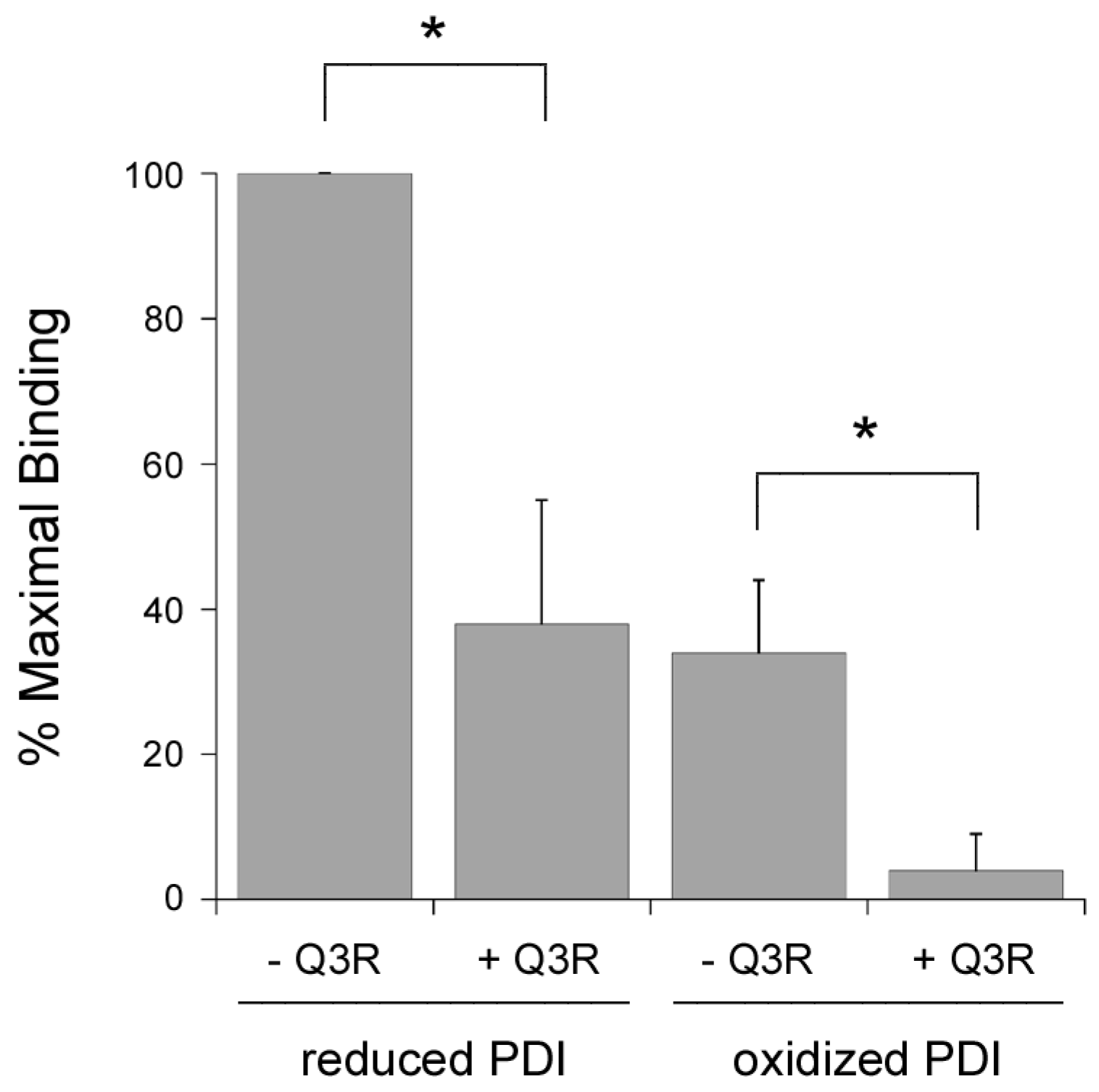
2.5. Q3R Inhibits PDI Binding to the B Chain of Ricin.
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Purification of Recombinant PDI with an N-Terminal His6 Tag
4.3. CT Intoxication Assay
4.4. CTA1 Transfection Intoxication Assay
4.5. SPR
4.6. Toxin Reduction Assay
4.7. CTA1 Protease Sensitivity Assay
4.8. FTIR Spectroscopy
4.9. RTB Binding Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heggelund, J.E.; Bjornestad, V.A.; Krengel, U. Vibrio choleraeand Escherichia coli heat-labile enterotoxins and beyond. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Elsevier: Waltham, MA, USA, 2015; Volume 4, pp. 195–229. [Google Scholar]
- Cho, J.A.; Chinnapen, D.J.; Aamar, E.; Welscher, Y.M.; Lencer, W.I.; Massol, R. Insights on the trafficking and retro-translocation of glycosphingolipid-binding bacterial toxins. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Wernick, N.L.B.; Chinnapen, D.J.-F.; Cho, J.A.; Lencer, W.I. Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef] [PubMed]
- Majoul, I.; Ferrari, D.; Soling, H.D. Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 1997, 401, 104–108. [Google Scholar] [CrossRef]
- Orlandi, P.A. Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem. 1997, 272, 4591–4599. [Google Scholar] [PubMed]
- Taylor, M.; Banerjee, T.; Ray, S.; Tatulian, S.A.; Teter, K. Protein disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem. 2011, 286, 22090–22100. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Rodighiero, C.; Lencer, W.I.; Rapoport, T.A. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 2001, 104, 937–948. [Google Scholar] [CrossRef]
- Pande, A.H.; Scaglione, P.; Taylor, M.; Nemec, K.N.; Tuthill, S.; Moe, D.; Holmes, R.K.; Tatulian, S.A.; Teter, K. Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 2007, 374, 1114–1128. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.; Banerjee, T.; Pande, A.H.; Taylor, M.; Tatulian, S.A.; Teter, K. Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J. Mol. Biol. 2009, 393, 1083–1096. [Google Scholar] [CrossRef]
- Teter, K.; Holmes, R.K. Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect. Immun. 2002, 70, 6172–6179. [Google Scholar] [CrossRef]
- Ampapathi, R.S.; Creath, A.L.; Lou, D.I.; Craft, J.W., Jr.; Blanke, S.R.; Legge, G.B. Order-disorder-order transitions mediate the activation of cholera toxin. J. Mol. Biol. 2008, 377, 748–760. [Google Scholar] [CrossRef]
- Banerjee, T.; Taylor, M.; Jobling, M.G.; Burress, H.; Yang, Z.; Serrano, A.; Holmes, R.K.; Tatulian, S.A.; Teter, K. ADP-ribosylation factor 6 acts as an allosteric activator for the folded but not disordered cholera toxin A1 polypeptide. Mol. Microbiol. 2014, 94, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burress, H.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Co- and post-translocation roles for Hsp90 in cholera intoxication. J. Biol. Chem. 2014, 289, 33644–33654. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Lipid rafts alter the stability and activity of the cholera toxin A1 subunit. J. Biol. Chem. 2012, 287, 30395–30405. [Google Scholar] [CrossRef] [PubMed]
- Sack, D.A.; Sack, R.B.; Nair, G.B.; Siddique, A.K. Cholera. Lancet 2004, 363, 223–233. [Google Scholar] [CrossRef]
- Muanprasat, C.; Chatsudthipong, V. Cholera: pathophysiology and emerging therapeutic targets. Future Med. Chem. 2013, 5, 781–798. [Google Scholar] [CrossRef] [PubMed]
- Mekalanos, J.J.; Collier, R.J.; Romig, W.R. Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J. Biol. Chem. 1979, 254, 5855–5861. [Google Scholar]
- Taylor, M.; Burress, H.; Banerjee, T.; Ray, S.; Curtis, D.; Tatulian, S.A.; Teter, K. Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathog. 2014, 10, e1003925. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, C.C. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef]
- Song, J.L.; Wang, C.C. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur. J. Biochem. 1995, 231, 312–316. [Google Scholar] [CrossRef]
- Quan, H.; Fan, G.; Wang, C.C. Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site. J. Biol. Chem. 1995, 270, 17078–17080. [Google Scholar] [CrossRef]
- Hatahet, F.; Ruddock, L.W. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007, 274, 5223–5234. [Google Scholar] [CrossRef] [PubMed]
- Klappa, P.; Ruddock, L.W.; Darby, N.J.; Freedman, R.B. The b’ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 1998, 17, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.Y.; Churchich, J.E. Recognition of protein substrates by protein-disulfide isomerase. A sequence of the b’ domain responds to substrate binding. J. Biol. Chem. 1999, 274, 32757–32761. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.B.; Desmond, J.L.; Byrne, L.J.; Heal, J.W.; Howard, M.J.; Sanghera, N.; Walker, K.L.; Wallis, A.K.; Wells, S.A.; Williamson, R.A.; et al. ‘Something in the way she moves’: The functional significance of flexibility in the multiple roles of protein disulfide isomerase (PDI). Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.J.; Sidhu, A.; Wallis, A.K.; Ruddock, L.W.; Freedman, R.B.; Howard, M.J.; Williamson, R.A. Mapping of the ligand-binding site on the b’ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 2009, 423, 209–217. [Google Scholar] [CrossRef]
- Inagaki, K.; Satoh, T.; Yagi-Utsumi, M.; Le Gulluche, A.C.; Anzai, T.; Uekusa, Y.; Kamiya, Y.; Kato, K. Redox-coupled structural changes of the catalytic a’ domain of protein disulfide isomerase. FEBS Lett. 2015, 589, 2690–2694. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Wang, X.; Wang, L.; Wallis, A.K.; Freedman, R.B.; Wang, C.C. Plasticity of human protein disulfide isomerase: evidence for mobility around the X-linker region and its functional significance. J. Biol. Chem. 2010, 285, 26788–26797. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase: a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2. [Google Scholar] [CrossRef]
- Turano, C.; Coppari, S.; Altieri, F.; Ferraro, A. Proteins of the PDI family: unpredicted non-ER locations and functions. J. Cell. Physiol. 2002, 193, 154–163. [Google Scholar] [CrossRef]
- Conway, M.E.; Harris, M. S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions. Front. Chem. 2015, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Halloran, M.; Parakh, S.; Atkin, J.D. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 2013, 797914. [Google Scholar] [CrossRef] [PubMed]
- Chuiu, J.; Passam, F.; Butera, D.; Hogg, P.J. Protein Disulfide Isomerase in Thrombosis. Semin. Thromb. Hemost. 2015, 41, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Bendapudi, P.; Sharda, A.; Chen, V.; Bellido-Martin, L.; Jasuja, R.; Furie, B.C.; Flaumenhaft, R.; Furie, B. Extracellular thiol isomerases and their role in thrombus formation. Antioxid. Redox Signal. 2016, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cho, J. Protein disulfide isomerase in thrombosis and vascular inflammation. J. Thromb. Haemost. 2013, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xue, G.; Song, M.; Xu, P.; Chen, D.; Yuan, C.; Lin, L.; Flaumenhaft, R.; Li, J.; Huang, M. Molecular basis of rutin inhibition of protein disulfide isomerase (PDI) by combined in silico and experimental methods. RSC Adv. 2018, 8, 18480–18491. [Google Scholar] [CrossRef]
- Lin, L.; Gopal, S.; Sharda, A.; Passam, F.; Bowley, S.R.; Stopa, J.; Xue, G.; Yuan, C.; Furie, B.C.; Flaumenhaft, R.; et al. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J. Biol. Chem. 2015, 290, 23543–23552. [Google Scholar] [CrossRef]
- Jasuja, R.; Passam, F.H.; Kennedy, D.R.; Kim, S.H.; van Hessem, L.; Lin, L.; Bowley, S.R.; Joshi, S.S.; Dilks, J.R.; Furie, B.; et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Investig. 2012, 122, 2104–2113. [Google Scholar] [CrossRef]
- Galinski, C.N.; Zwicker, J.I.; Kennedy, D.R. Revisiting the mechanistic basis of the French Paradox: red wine inhibits the activity of protein disulfide isomerase in vitro. Thromb. Res. 2016, 137, 169–173. [Google Scholar] [CrossRef]
- Teter, K.; Jobling, M.G.; Holmes, R.K. Vesicular transport is not required for the cytoplasmic pool of cholera toxin to interact with the stimulatory alpha subunit of the heterotrimeric G protein. Infect. Immun. 2004, 72, 6826–6835. [Google Scholar] [CrossRef]
- Cherubin, P.; Guyette, J.; Taylor, M.; O’Donnell, M.; Herndon, L.; Burress, H.; Riad, A.; Tatulian, S.A.; Teter, K. Protein disulfide isomerase does not act as an unfoldase in the disassembly of cholera toxin. Biosci. Rep. 2018, 38, BSR20181320. [Google Scholar] [CrossRef] [PubMed]
- Bekendam, R.H.; Bendapudi, P.K.; Lin, L.; Nag, P.P.; Pu, J.; Kennedy, D.R.; Feldenzer, A.; Chiu, J.; Cook, K.M.; Furie, B.; et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 2016, 7, 12579. [Google Scholar] [CrossRef] [PubMed]
- Tatulian, S.A. Structural analysis of proteins by isotope-edited FTIR spectroscopy. Spectrosc. Int. J. 2010, 24, 37–43. [Google Scholar] [CrossRef]
- Tatulian, S.A. FTIR analysis of proteins and protein-membrane interactions. Methods Mol. Biol. 2019, 2003, 281–325. [Google Scholar] [PubMed]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosen, A.; Solda, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharm. 2004, 67, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.M.; Mantis, N.J. Neutralizing monoclonal antibodies against ricin’s enzymatic subunit interfere with protein disulfide isomerase-mediated reduction of ricin holotoxin in vitro. J. Immunol. Methods 2013, 395, 71–78. [Google Scholar] [CrossRef]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Rasooly, R.; He, X.; Friedman, M. Milk inhibits the biological activity of ricin. J. Biol. Chem. 2012, 287, 27924–27929. [Google Scholar] [CrossRef]
- Cherubin, P.; Garcia, M.C.; Curtis, D.; Britt, C.B.T.; Craft, J.W., Jr.; Burress, H.; Berndt, C.; Reddy, S.; Guyette, J.; Zheng, T.; et al. Inhibition of cholera toxin and other AB toxins by polyphenolic compounds. PLoS ONE 2016, 11, e0166477. [Google Scholar] [CrossRef]
- Friedman, M.; Rasooly, R. Review of the inhibition of biological activities of food-related selected toxins by natural compounds. Toxins 2013, 5, 743–775. [Google Scholar] [CrossRef]
- Reddy, S.; Taylor, M.; Zhao, M.; Cherubin, P.; Geden, S.; Ray, S.; Francis, D.; Teter, K. Grape extracts inhibit multiple events in the cell biology of cholera intoxication. PLoS ONE 2013, 8, e73390. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Furie, B.; Zwicker, J.I. Therapeutic implications of protein disulfide isomerase inhibition in thrombotic disease. Arter. Thromb. Vasc. Biol. 2015, 35, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.; Burress, H.; Taylor, M.; Nemec, K.N.; Ray, S.; Haslam, D.B.; Teter, K. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect. Immun. 2011, 79, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Twiddy, E.M. Characterization of monoclonal antibodies that react with unique and cross-reacting determinants of cholera enterotoxin and its subunits. Infect. Immun. 1983, 42, 914–923. [Google Scholar] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guyette, J.; Cherubin, P.; Serrano, A.; Taylor, M.; Abedin, F.; O’Donnell, M.; Burress, H.; Tatulian, S.A.; Teter, K. Quercetin-3-Rutinoside Blocks the Disassembly of Cholera Toxin by Protein Disulfide Isomerase. Toxins 2019, 11, 458. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080458
Guyette J, Cherubin P, Serrano A, Taylor M, Abedin F, O’Donnell M, Burress H, Tatulian SA, Teter K. Quercetin-3-Rutinoside Blocks the Disassembly of Cholera Toxin by Protein Disulfide Isomerase. Toxins. 2019; 11(8):458. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080458
Chicago/Turabian StyleGuyette, Jessica, Patrick Cherubin, Albert Serrano, Michael Taylor, Faisal Abedin, Morgan O’Donnell, Helen Burress, Suren A. Tatulian, and Ken Teter. 2019. "Quercetin-3-Rutinoside Blocks the Disassembly of Cholera Toxin by Protein Disulfide Isomerase" Toxins 11, no. 8: 458. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11080458