Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04)

Abstract
:1. Introduction
2. Results
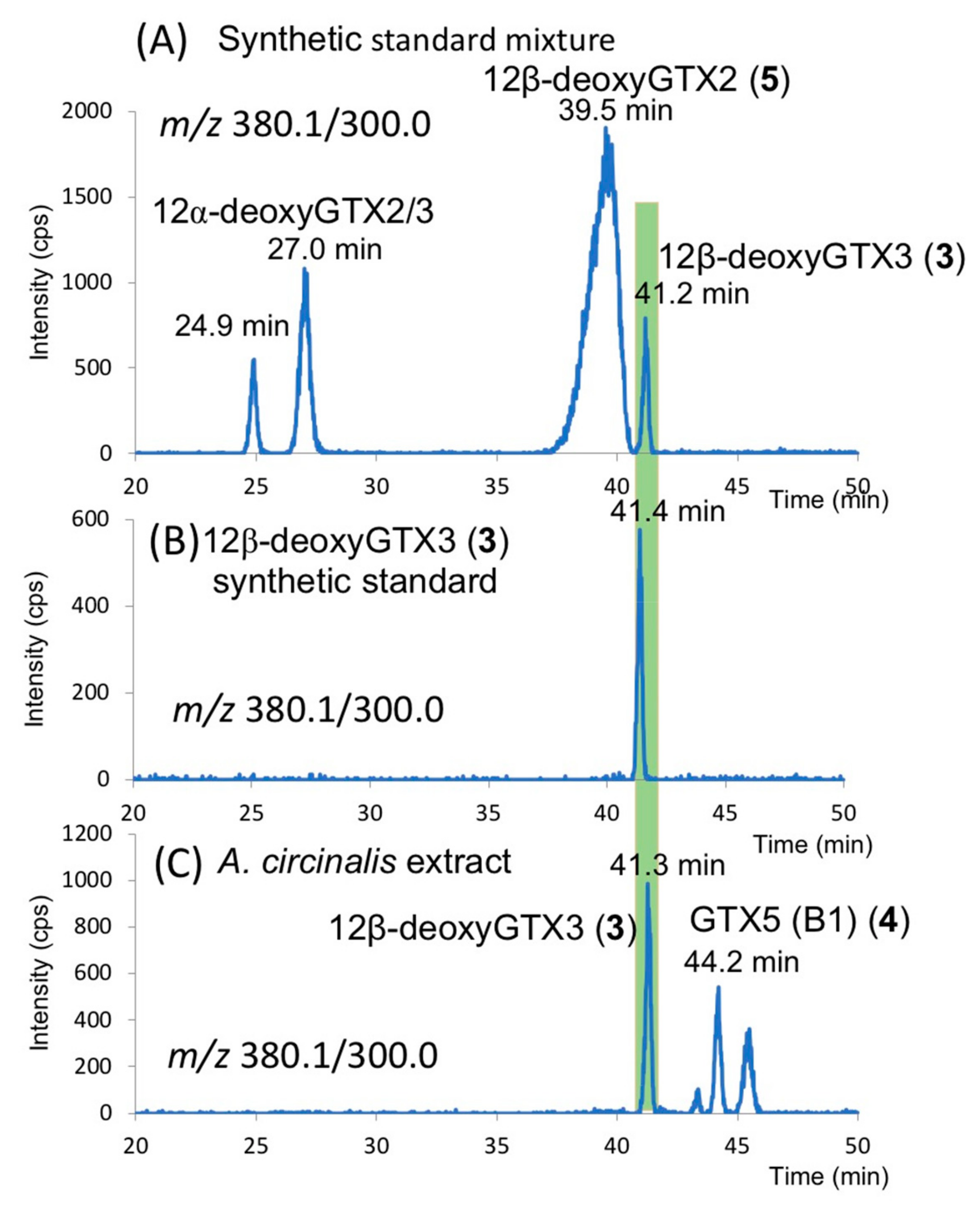
2.1. Screening for Novel PST in A. circinalis (TA04)
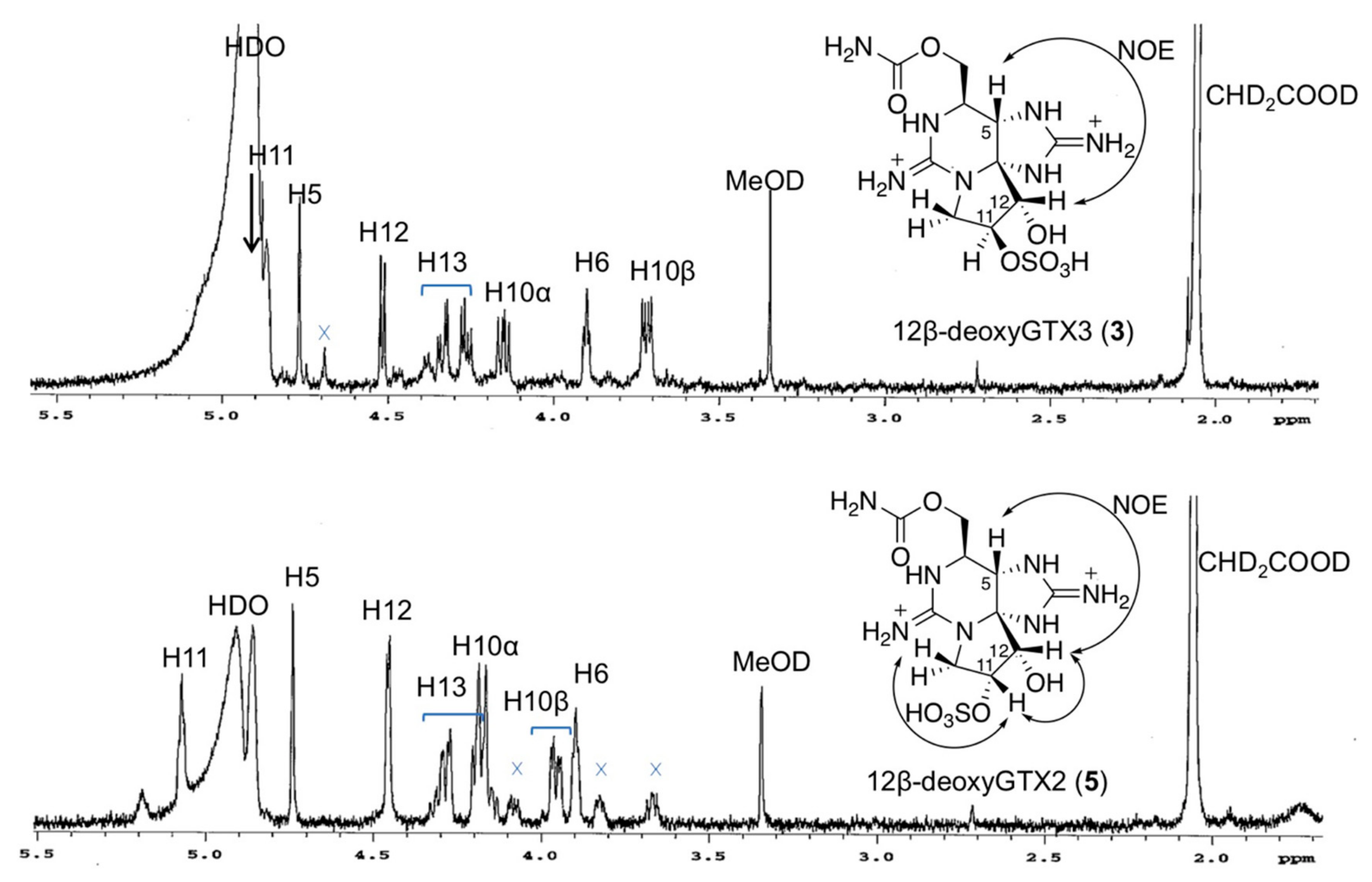
2.2. Preparation and Spectroscopic Identification of 12β-deoxyGTX2 (5) and 12β-deoxyGTX3 (3)
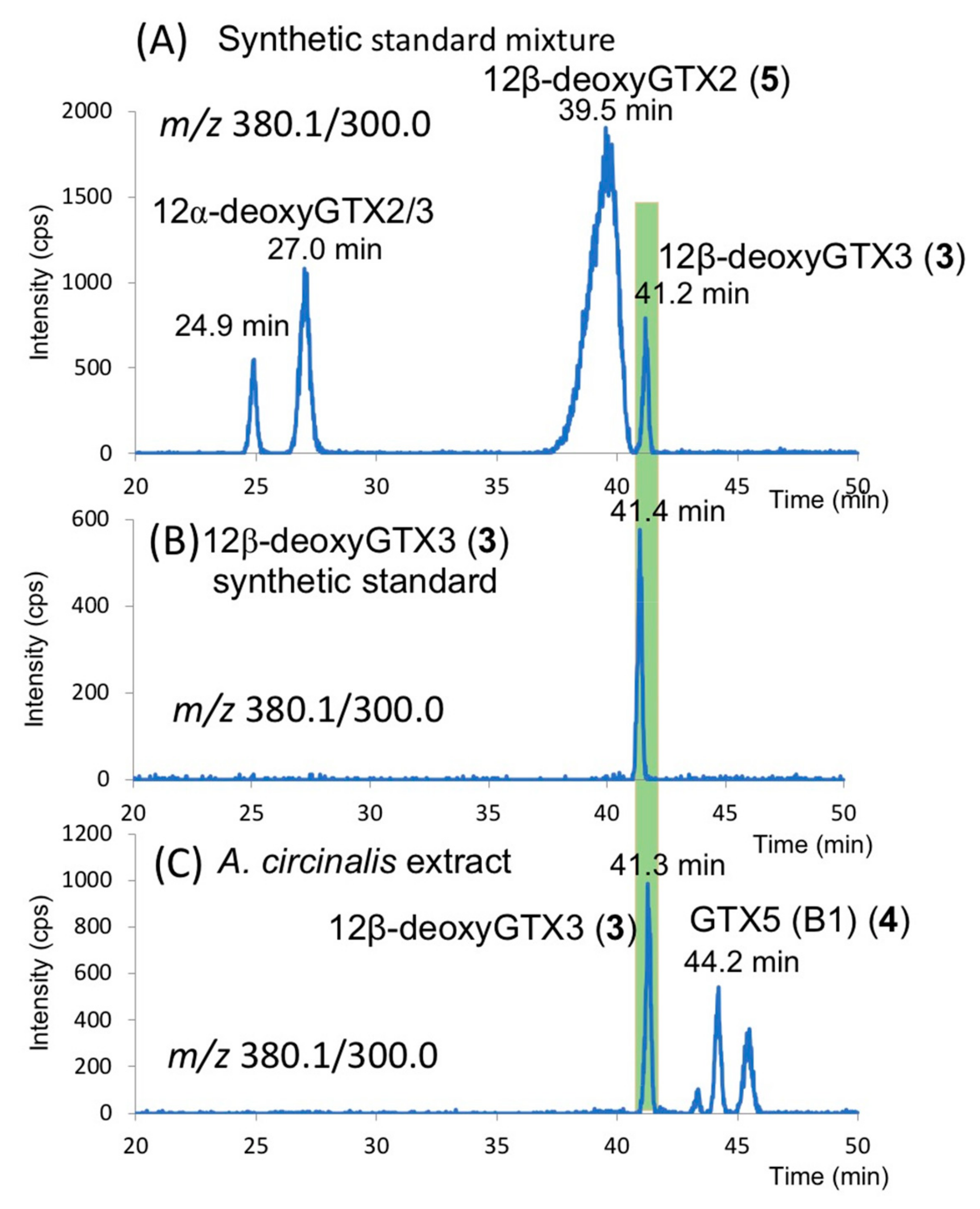
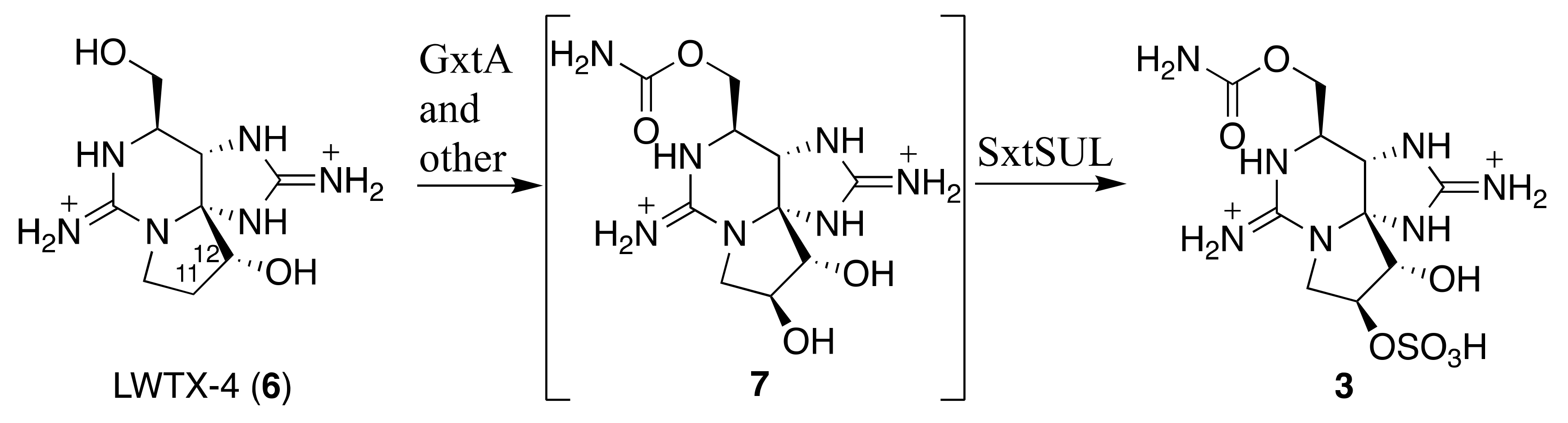
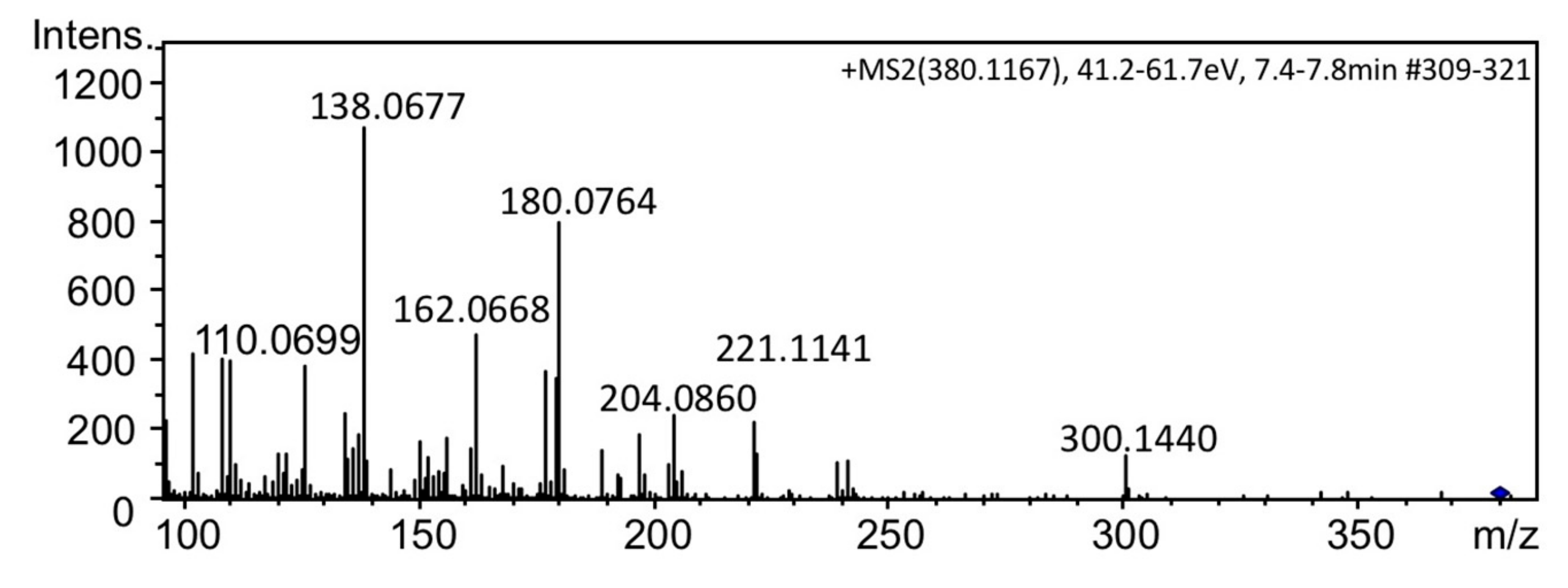
2.3. Identification of 12β-deoxyGTX3 (3) in A. circinalis (TA04)
3. Discussion
4. Methods and Materials
4.1. General Information
4.2. Preparation of 12β-deoxyGTX2 (5) and 12β-deoxyGTX3 (3) from C1/C2
4.3. Preparation of the Mixture of 12α-deoxyGTX2/3 from GTX2/3
4.4. Harvest and Preparation of A. circinalis (TA04) Cell Extract for Screening
4.5. HR–HILIC–LC–MS Conditions for the Screening for Novel PST
4.6. MS Conditions for HR–LC–MS and HR–LC–MS/MS
4.7. Column-Switching LC–MS/MS (MRM) Conditions [28]
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kao, C.Y. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomenon. Pharmacol. Rev. 1966, 18, 997–1049. [Google Scholar] [PubMed]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Thottumkara, A.P.; Parsons, W.H.; Du Bois, J. Saxitoxin. Angew. Chem. Int. Ed. 2014, 53, 5760–5784. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W.; Evans, W.R.; Yin, Q.Q.; Bell, P.; Moczydlowski, E. Evidence for paralytic shellfish poisons in the freshwater cyanobacterium Lyngbya wollei (Farlow ex Gomont) comb. nov. Appl. Environ. Microbiol. 1997, 63, 3104–3110. [Google Scholar] [PubMed]
- Shimizu, Y.; Norte, M.; Hori, A.; Genenah, A.; Kobayashi, M. Biosynthesis of saxitoxin analogues: The unexpected pathway. J. Am. Chem. Soc. 1984, 106, 6433–6434. [Google Scholar] [CrossRef]
- Kellmann, R.; Mihali, T.K.; Young, J.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef]
- Stüken, A.; Orr, R.J.S.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of nuclear-encoded genes for the neurotoxin saxitoxin in dinoflagellates. PLoS ONE 2011, 6, e20096. [Google Scholar] [CrossRef]
- Verma, A.; Barua, A.; Ruvindy, R.; Savela, H.; Ajani, P.A.; Murray, S.A. The genetic basis of toxin biosynthesis in dinoflagellates. Microorganisms 2019, 7, 222. [Google Scholar] [CrossRef]
- Sako, Y.; Yoshida, T.; Uchida, A.; Arakawa, O.; Noguchi, T.; Ishida, Y. Purification and characterization of a sulfotransferase specific to N-21 of saxitoxin and gonyautoxin 2 + 3 from the toxic dinoflagellate Gymnodinium catenatum (dinophyceae). J. Phycol. 2001, 37, 1044–1051. [Google Scholar] [CrossRef]
- Yoshida, T.; Sako, Y.; Uchida, A.; Kakutani, T.; Arakawa, O.; Noguchi, T.; Ishida, Y. Purification and characterization of sulfotransferase specific to O-22 of 11-hydroxy saxitoxin from the toxic dinoflagellate Gymnodinium catenatum (dinophyceae). Fish. Sci. 2002, 68, 634–642. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Cho, Y.; Konoki, K.; Nagasawa, K.; Oshima, Y.; Yotsu-Yamashita, M. Synthesis and identification of proposed biosynthetic intermediates of saxitoxin in the cyanobacterium Anabaena circinalis (TA04) and the dinoflagellate Alexandrium tamarense (Axat-2). Org. Biomol. Chem. 2014, 12, 3016–3020. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Cho, Y.; Konoki, K.; Nagasawa, K.; Oshima, Y.; Yotsu-Yamashita, M. Synthesis of a tricyclic bisguanidine compound structurally related to saxitoxin and its identification in paralytic shellfish toxin-producing microorganisms. Chem. Eur. J. 2015, 21, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Cho, Y.; Konoki, K.; Nagasawa, K.; Oshima, Y.; Yotsu-Yamashita, M. Biosynthetic route towards saxitoxin and shunt pathway. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Cho, Y.; Yoshioka, R.; Konoki, K.; Nagasawa, K.; Oshima, Y.; Yotsu-Yamashita, M. Synthesis and identification of key biosynthetic intermediates for the formation of the tricyclic skeleton of saxitoxin. Angew. Chem. Int. Ed. 2017, 56, 5327–5331. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.W.; Hinze, M.E.; Skiba, M.A.; Narayan, A.R.H. Chemistry of a unique polyketide-like synthase. J. Am. Chem. Soc. 2018, 140, 2430–2433. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, A.L.; Ellinwood, D.C.; Hinze, M.E.; Deluca, R.J.; Du Bois, J.; Hall, S.; Narayan, A.R.H. C-H Hydroxylation in paralytic shellfish toxin biosynthesis. J. Am. Chem. Soc. 2018, 140, 11863–11869. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, A.L.; Denomme, N.; Hinze, M.E.; Hall, S.; Isom, L.L.; Narayan, A.R.H. Biocatalytic detoxification of paralytic shellfish toxins. ACS Chem. Biol. 2019, 14, 941–948. [Google Scholar] [CrossRef]
- Onodera, H.; Satake, M.; Oshima, Y.; Yasumoto, T.; Carmichael, W.W. New saxitoxin analogues from the freshwater filamentous cyanobacterium Lyngbya wollei. Nat. Toxins 1998, 5, 146–151. [Google Scholar] [CrossRef]
- Hudon, C.; Gagnon, P.; Poirier Larabie, S.; Gagnon, C.; Lajeunesse, A.; Lachapelle, M.; Quilliam, M.A. Spatial and temporal variations of a saxitoxin analogue (LWTX-1) in Lyngbya wollei (Cyanobacteria) mats in the St. Lawrence River (Québec, Canada). Harmful Algae 2016, 57, 69–77. [Google Scholar] [CrossRef]
- D’Agostino, P.M.; Boundy, M.J.; Harwood, T.D.; Carmichael, W.W.; Neilan, B.A.; Wood, S.A. Re-evaluation of paralytic shellfish toxin profiles in cyanobacteria using hydrophilic interaction liquid chromatography-tandem mass spectrometry. Toxicon 2019, 158, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Oshima, Y.; Yasumoto, T. Structures of two paralytic shellfish toxins, gonyautoxins V and VI, isolated from a tropical dinoflagellate, Pyrodinium bahamense var. compressa. Agric. Biol. Chem. 1982, 46, 1861–1864. [Google Scholar]
- Koehn, F.E.; Hall, S.; Fix Wichmann, C.; Schnoes, H.K.; Reichardt, P.B. Dinoflagellate neurotoxins related to saxitoxin: Structure and latent activity of toxins B1 and B2. Tetrahedron Lett. 1982, 23, 2247–2248. [Google Scholar] [CrossRef]
- Dell’Aversano, C.; Hess, P.; Quilliam, M.A. Hydrophilic interaction liquid chromatography-mass spectrometry for the analysis of paralytic shellfish poisoning (PSP) toxins. J. Chromatogr. A 2005, 1081, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Tsuchiya, S.; Omura, T.; Koike, K.; Oikawa, H.; Konoki, K.; Oshima, Y.; Yotsu-Yamashita, M. Metabolomic study of saxitoxin analogues and biosynthetic intermediates in dinoflagellates using 15N-labelled sodium nitrate as a nitrogen source. Sci. Rep. 2019, 9, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Suzuki, T.; Oshima, Y. Preparation of calibration standards of N1-H paralytic shellfish toxin analogues by large-scale culture of cyanobacterium Anabaena circinalis (TA04). Mar. Drugs 2011, 9, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Koehn, F.E.; Ghazarossian, V.E.; Schantz, E.J.; Schnoes, H.K.; Strong, F.M. Derivatives of saxitoxin. Bioorg. Chem. 1981, 10, 412–428. [Google Scholar] [CrossRef]
- Cho, Y.; Tsuchiya, S.; Yoshioka, R.; Omura, T.; Konoki, K.; Oshima, Y.; Yotsu-Yamashita, M. Column switching combined with hydrophilic interaction chromatography-tandem mass spectrometry for the analysis of saxitoxin analogues, and their biosynthetic intermediates in dinoflagellates. J. Chromatogr. A 2016, 1474, 109–120. [Google Scholar] [CrossRef]
- Cho, Y.; Tsuchiya, S.; Yoshioka, R.; Omura, T.; Konoki, K.; Oshima, Y.; Yotsu-Yamashita, M. The presence of 12β-deoxydecarbamoylsaxitoxin in the Japanese toxic dinoflagellate Alexandrium determined by simultaneous analysis for paralytic shellfish toxins using HILIC-LC-MS/MS. Harmful Algae 2015, 49, 58–67. [Google Scholar] [CrossRef]
- Lim, P.T.; Sato, S.; Van Thuoc, C.; Tu, P.T.; Huyen, N.T.M.; Takata, Y.; Yoshida, M.; Kobiyama, A.; Koike, K.; Ogata, T. Toxic Alexandrium minutum (Dinophyceae) from Vietnam with new gonyautoxin analogue. Harmful Algae 2007, 6, 321–331. [Google Scholar] [CrossRef]
- Mihali, T.K.; Carmichael, W.W.; Neilan, B.A. A putative gene cluster from a Lyngbya wollei bloom that encodes paralytic shellfish toxin biosynthesis. PLoS ONE 2011, 6, e14657. [Google Scholar] [CrossRef] [PubMed]
- Cullen, A.; D’Agostino, P.M.; Mazmouz, R.; Pickford, R.; Wood, S.; Neilan, B.A. Insertions within the saxitoxin biosynthetic gene cluster result in differential toxin profiles. ACS Chem. Biol. 2018, 13, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.P.; Jones, G.; Blackburn, S.I.; Oshima, Y.; Onodara, H. Effect of culture and bloom development and of sample storage on paralytic shellfish poisons in the cyanobacterum Anabaena circinalis. J. Phycol. 1997, 33, 26–35. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 12β-deoxyGTX2 (5) | 12β-deoxyGTX3 (3) | LWTX-1 (2) | ||||
|---|---|---|---|---|---|---|
| No. | δH | Multiplicity (J in Hz) | δH | Multiplicity (J in Hz) | δH | (J in Hz) *** |
| 5 | 4.74 | s | 4.77 | s | 4.74 | s |
| 6 | 3.90 | t 4.3 | 3.90 | t 4.3 | 3.93 | 4.8 |
| 10α | 4.17 | d 12.3 | 4.15 | dd 11.5, 11.9 | 4.16 | 8.1, 11.6 |
| 10β | 3.96 | dd 11.5, 4.4 | 3.72 | dd 11.5, 4.3 | 3.67 | 5.4, 11.6 |
| 11 | 5.07 | t 3.1 | 4.91 * | n.d. ** | 4.90 | 5.4, 7.2, 8.1 |
| 12 | 4.45 | d 4.0 | 4.52 | d 7.0 | 4.49 | 7.2 |
| 13a | 3.96 | m | 4.27 | dd 13.9, 5.6 | 4.29 | 4.8 |
| 13b | 4.29 | dd 11.5, 4.4 | 4.33 | dd 13.9, 4.4 | 4.29 | 4.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minowa, T.; Cho, Y.; Oshima, Y.; Konoki, K.; Yotsu-Yamashita, M. Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04). Toxins 2019, 11, 539. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090539
Minowa T, Cho Y, Oshima Y, Konoki K, Yotsu-Yamashita M. Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04). Toxins. 2019; 11(9):539. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090539
Chicago/Turabian StyleMinowa, Takashi, Yuko Cho, Yasukatsu Oshima, Keiichi Konoki, and Mari Yotsu-Yamashita. 2019. "Identification of a Novel Saxitoxin Analogue, 12β-Deoxygonyautoxin 3, in the Cyanobacterium, Anabaena circinalis (TA04)" Toxins 11, no. 9: 539. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090539