Two VHH Antibodies Neutralize Botulinum Neurotoxin E1 by Blocking Its Membrane Translocation in Host Cells


1
Department of Physiology & Biophysics, University of California, Irvine, CA 92617, USA
2
NE-CAT and Department of Chemistry and Chemical Biology, Cornell University, Argonne National Laboratory, Argonne, IL 60439, USA
3
Department of Infectious Disease and Global Health, Cummings School of Veterinary Medicine, Tufts University, North Grafton, MA 01536, USA
*
Author to whom correspondence should be addressed.
Toxins 2020, 12(10), 616; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100616
Submission received: 18 August 2020
/
Revised: 16 September 2020
/
Accepted: 22 September 2020
/
Published: 27 September 2020
(This article belongs to the Special Issue Inhibitors and Countermeasures against Bacterial and Plant Toxins)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Botulinum neurotoxin serotype E (BoNT/E) is one of the major causes of human botulism, which is a life-threatening disease caused by flaccid paralysis of muscles. After receptor-mediated toxin internalization into motor neurons, the translocation domain (HN) of BoNT/E transforms into a protein channel upon vesicle acidification in endosomes and delivers its protease domain (LC) across membrane to enter the neuronal cytosol. It is believed that the rapid onset of BoNT/E intoxication compared to other BoNT serotypes is related to its swift internalization and translocation. We recently identified two neutralizing single-domain camelid antibodies (VHHs) against BoNT/E1 termed JLE-E5 and JLE-E9. Here, we report the crystal structures of these two VHHs bound to the LCHN domain of BoNT/E1. The structures reveal that these VHHs recognize two distinct epitopes that are partially overlapping with the putative transmembrane regions on HN, and therefore could physically block membrane association of BoNT/E1. This is confirmed by our in vitro studies, which show that these VHHs inhibit the structural change of BoNT/E1 at acidic pH and interfere with BoNT/E1 association with lipid vesicles. Therefore, these two VHHs neutralize BoNT/E1 by preventing the transmembrane delivery of LC. Furthermore, structure-based sequence analyses show that the 3-dimensional epitopes of these two VHHs are largely conserved across many BoNT/E subtypes, suggesting a broad-spectrum protection against the BoNT/E family. In summary, this work improves our understanding of the membrane translocation mechanism of BoNT/E and paves the way for developing VHHs as diagnostics or therapeutics for the treatment of BoNT/E intoxication.
Keywords:
botulinum neurotoxin; botulism; single-domain antibody; VHH; neutralizing epitope; antitoxin; membrane translocationKey Contribution: Structures of two single-domain antibodies in complex with BoNT/E1 reveal a novel mechanism by which antibodies neutralize BoNT/E1 by blocking its membrane translocation in host cells.
1. Introduction
Botulinum neurotoxin (BoNT) is one of the most toxic substances known in nature and classified as a Tier 1 select agent by the Centers for Disease Control and Prevention (CDC) in the United States [1,2,3]. There are at least seven immunologically distinct serotypes of BoNT (A–G) that include more than 40 subtypes [4,5,6,7,8]. BoNT/E is one of the four BoNT serotypes (with BoNT/A, BoNT/B, and rarely BoNT/F) known to cause human botulism. There are 12 BoNT/E subtypes (E1–12) identified to date that are produced by Clostridium botulinum and Clostridium butyricum [9]. BoNT/E intoxication is characterized by a rapid rate of onset and a shorter duration of effect when compared to other BoNTs [10,11,12]. While the majority of BoNT/E intoxication cases are foodborne illnesses caused by consumption of contaminated fish or marine mammal products [13], wound and infant botulism have been reported [14,15]. Recently, BoNT/E is being developed in Phase II clinical trial for therapeutic and aesthetic applications, which increases the potential risk of human exposures to this toxin [16].
The current treatment for adult botulism is an equine heptavalent antitoxin (BAT) in limited supply, which has a short serum half-life and can cause adverse effects including serum sickness and asystole [17,18]. Hence, alternative measures for the treatment of botulism are needed. A cocktail of three monoclonal antibodies (mAbs) against multiple BoNT/E subtypes is currently being developed under Phase I clinical trial [19]. In this study, we aim to develop novel alpaca heavy-chain only antibody (VHH)-based neutralizing agents (VNAs) against BoNT/E as next generation antitoxin therapeutics. We have recently developed a structure-based rational design platform to develop VNAs that show high binding affinity and neutralizing potency against BoNT/A and B [20]. Compared to the mAbs, VHHs are advantageous in their small size, high stability, ease of production, and unique capability to target “hidden” cavities or clefts on the surface of proteins [21,22,23,24].
All BoNTs share a tripartite architecture that is composed of a protease domain (LC), a translocation domain (HN), and a receptor-binding domain (HC) [25]. The toxin is expressed as a single polypeptide chain that is proteolytically cleaved between the LC and HN to form a dichain linked by a disulfide bridge. The three domains are structurally separated in BoNT/A and B [26,27]. However, BoNT/E adopts a unique compact and ‘closed-wing’-like conformation, which may enable more facile interactions among the three domains and therefore lead to its fast onset of intoxication (Figure 1a) [28,29]. The BoNT intoxication mechanism involves three major steps [1,25]. HC recognizes dual receptors, a polysialo-ganglioside (PSG) and a transmembrane glycoprotein (either synaptotagmin or synaptic vesicle protein 2, no protein receptor has been identified for BoNT/C), on presynaptic neurons and triggers toxin internalization [30,31,32,33,34,35]. HN then acts as a protein conduit and delivers the LC across the vesicle membrane to the neuronal cytosol [36,37]. In the cytosol, LC cleaves synaptobrevins, SNAP-25, or syntaxin (all three proteins form the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex), and their cleavage blocks acetylcholine release at the neuromuscular junction leading to flaccid paralysis [38,39]. It has been shown that antibodies are able to neutralize BoNT action by inhibiting toxin functions at each of these steps [20,40,41,42].
Upon vesicle acidification, the HN of BoNT/E transforms into a LC-conducting channel that shows properties similar to BoNT/A [36,43,44], suggesting the translocation mechanism is conserved across BoNT serotypes. Although the mechanism of channel formation remains poorly characterized, sequence hydropathy analysis of BoNT/E identified three consecutive segments of HN that are predicted to be important for membrane association [45,46]. These segments include a viral-fusion-peptide-like “BoNT-switch” (Q589–E635) [47], the diphtheria toxin-like channel-forming helices (V563–N588) [48], and an extended amphipathic peptide (F636–L654) (Figure 1a) [49]. Prior studies have shown that some neutralizing mAbs and VHHs target the channel-forming helices and interfere with membrane insertion of BoNT/E or A [20,50], but it is not known whether antibodies can neutralize toxin by binding at other putative transmembrane regions.
Here, we report the structural and functional characterization of two neutralizing VHHs raised against BoNT/E1. We determined the crystal structures of these two VHHs in complex with toxin fragments and demonstrated that their mechanisms of neutralization are to inhibit toxin conformational changes at acidic endosomal pH, thus preventing BoNT/E1 membrane interaction. We further show that the binding epitopes of these VHHs are conserved within known BoNT/E subtypes, suggesting that these VHHs merit further evaluation as therapeutic agents for the treatment of BoNT/E intoxication.
2. Results and Discussion
2.1. The Crystal Structures of the LCHN/E1–JLE-E5 and LCHN/E1–JLE-E9 Complexes
We recently obtained a large panel of unique BoNT/E1-binding VHHs and identified three VHHs, termed JLE-E5, JLE-E9, and JLE-G6, that neutralize BoNT/E1 intoxication through binding at unique epitopes [51]. Among them, JLE-E5 and JLE-E9 interact with the LCHN domain (residues M1–K845) (Figure S1), a fusion protein composed of the LC and the HN domains of BoNT/E1. We found that JLE-E5 and JLE-E9, which were selected from alpaca immunized with the catalytically inactive BoNT/E1 (ciBoNT/E1), neutralized wild type BoNT/E1 with comparable potency in a mouse neuronal cell-based assay and both bound ciBoNT/E1 with apparent affinities of ≈1 nM EC50 in dilution ELISA assays [51]. Recent studies with BoNT/A1 showed that neutralizing VHHs (e.g., ciA-B5 and ciA-H7) targeting LCHN/A could counteract toxin action by blocking membrane interaction and delivery of LC [20]. We thus suspected that these VHHs might neutralize BoNT/E1 by a similar mechanism. Furthermore, since the HN is highly conserved among 12 BoNT/E subtypes (97.6% identity), these VHHs could potentially provide broad-spectrum protection against the diverse BoNT/E family [52].
To gain insight into the neutralization mechanisms of JLE-E5 and JLE-E9, we copurified these two VHHs with either ciBoNT/E1 or LCHN/E1 in different combinations and carried out crystallization screens. The protein crystals of the ciBoNT/E1–JLE-E5–JLE-E9 and the LCHN/E1–JLE-E5–JLE-E9 complexes poorly diffracted X-ray and were not pursued further. After extensive crystallization screening and optimization, we successfully obtained high-quality crystals and determined the structures of the LCHN/E1–JLE-E5 and the LCHN/E1–JLE-E9 complexes at 2.5 and 3.6 Å resolutions, respectively (Table S1).
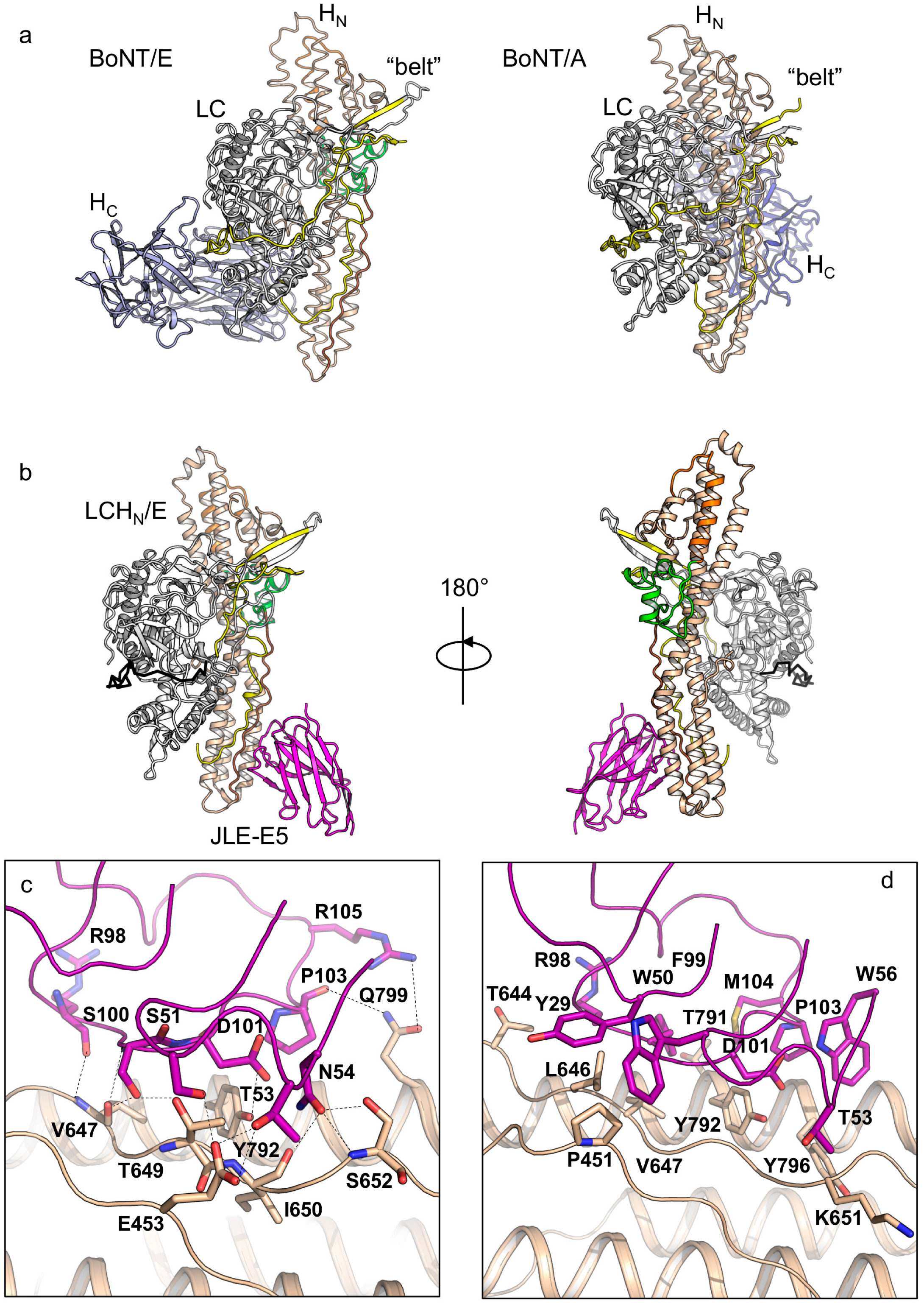
Both of these VHHs bind LCHN in a 1:1 ratio, and there are one and four protein complex molecules in one asymmetric unit (ASU) in the crystals of the LCHN/E1–JLE-E5 and the LCHN/E1–JLE-E9 complexes, respectively (Figure 1b and Figure 2a). Each VHH shows a typical immunoglobulin fold that comprises four framework regions (FR) and three complementary-determining regions (CDR). The absence of HC or the VHH binding does not induce pronounced structural changes in LCHN/E1, as the structures of the VHH-bound LCHN/E1 domain are highly similar to that of the apo holotoxin (root-mean-square-deviation r.m.s.d = 1.25 Å over 731 Cα atoms, PDB code 3FFZ) [28].
Analysis of the electron density map of LCHN/E1 in both structures reveals that the C-terminal loop (residues D832–K845) and the N-terminal “belt” of HN (residues V458–K500) are missing (Figure 1b and Figure S2), indicative of local structural flexibility. However, the inability to detect the 43 amino acids of the belt is unusual because the belt acts as a pseudo-substrate that wraps around LC in all known BoNT structures including BoNT/E1 holotoxin [26,27,53]. Comparing our structures with the holotoxin suggests that the “missing” belt on LCHN/E1 could be due to the deletion of HC, which interacts with the belt in the context of BoNT/E1 holotoxin [28] and stabilizes the belt and protects LC. This is unique to BoNT/E that adopts a compact closed-wing-like conformation, while all other BoNTs with known structures show an open-wing-like conformation and their HC does not interact with the belt (Figure 1a). Since HN is suggested to be responsible for the rapid onset of BoNT/E intoxication [10], we hypothesize that the exceptional flexibility of the belt may contribute to the speedy translocation of BoNT/E1 by lowering the energy requirement for protein unfolding and LC delivery across the HN channel. Therefore, testing the physiological role of the belt in facilitating BoNT/E translocation is of high interest in future studies.
Further analysis of the crystal packing shows that the LCHN/E1 forms a symmetrical homodimer via LC–LC interactions (Figure S2). Prior study also revealed that LC/E forms homodimers in solution as well as in crystal lattice, and the binding mode of LC/E homodimer is nearly identical to that of LCHN/E1 homodimer [54]. The holotoxin is not able to form such a homodimer because the HC domain clashes with the interacting partner and sterically blocks the interaction. We observed such LC/E–LC/E interactions in crystals that grew under three different crystallization conditions with two different BoNT/E1 constructs. These findings suggest that LC/E dimerization may represent a natural occurrence rather than a crystallization artifact, but its functional role remains to be clarified.
VHH JLE-E5 binds to the C-terminal boundary of the rod-like HN domain and buries a molecular surface of ≈733.5 Å2 per molecule (calculated by PDBePISA v1.51) (Figure 1b) [55]. JLE-E5 binds HNE primarily through CDR2 and CDR3 and directly interacts with the belt, the amphipathic segment, and the C-terminal helix of HNE (Figure 1c,d, Tables S2 and S3). In CDR2 of JLE-E5, residues S51 and T53 electrostatically interact with E453, while W50 forms an aromatic-proline interaction with P451 of the belt. Residues N54 of CDR2 and R98/S100 of CDR3 form multiple hydrogen bonds with V647, T649, I650, and S652 in the amphipathic segment of HNE. In addition, D101, P103, and R105 of CDR3 interact with Y792 and Q799 of the C-terminal helix to stabilize complex formation. Prior studies showed that, upon acidification, the C-terminus of HN interacts with membrane when the toxin engages the PSG receptor (Figure 1a) [28,56]. As a result, the conserved amphipathic peptide transforms from an extended conformation into a transmembrane helix with ion-conducive properties upon membrane insertion [49]. These structural findings suggest JLE-E5 might inhibit the channel formation of BoNT/E1 because it stabilizes HNE at its neutral conformation and thus prevents the conformational change of the amphipathic peptide and blocks channel formation.
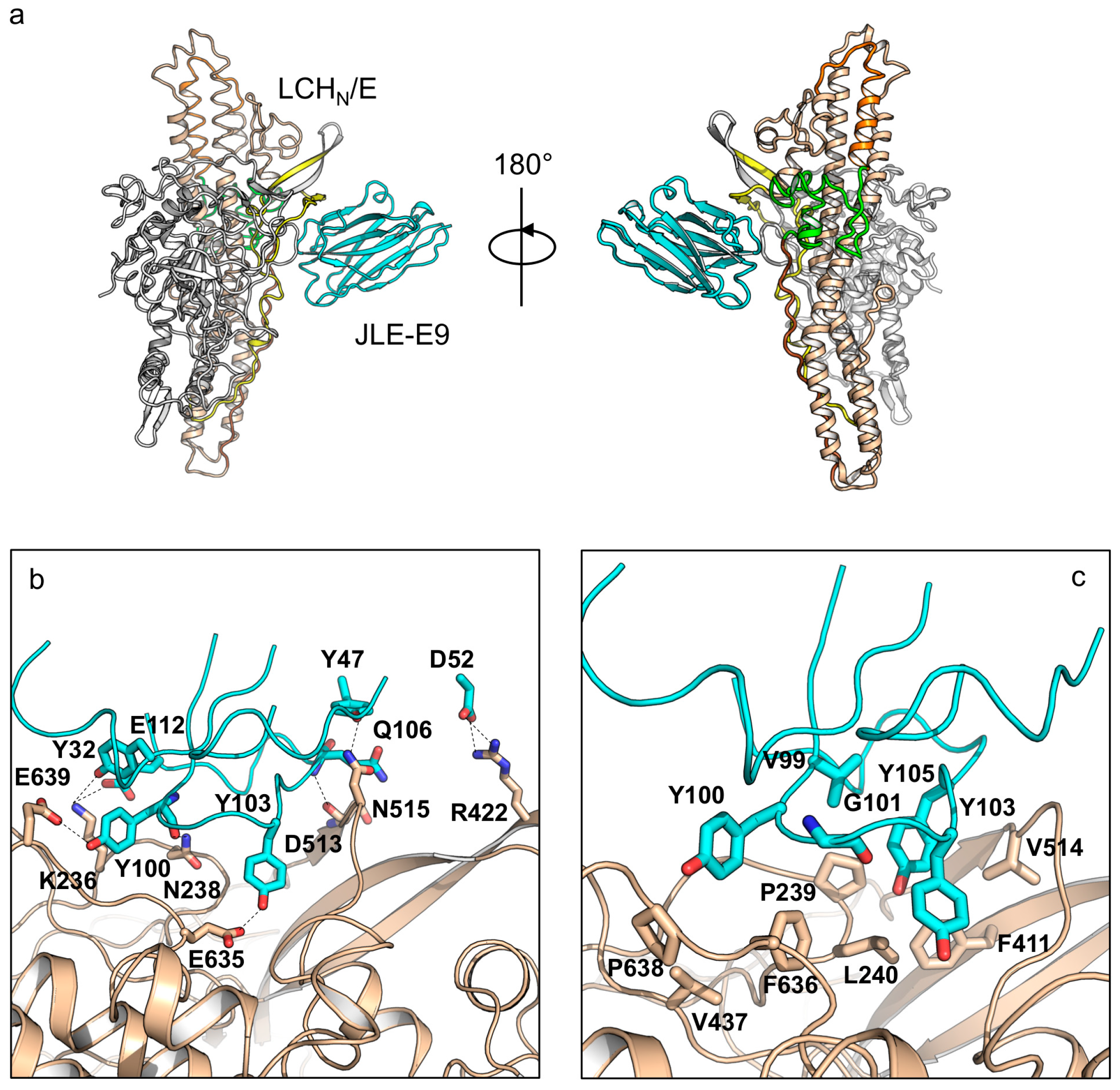
Structure analysis of JLE-E9 binding to LCHN/E1 shows that it uniquely recognizes a composite epitope involving both the LC and the HN domains, which buries an interface area of ≈860 Å2 (Figure 2a). A detailed structural analysis revealed that the CDR1, FR2, and CDR3 of JLE-E9 interact with LCHN/E1 involving the 230-loop of the LC, the belt, and the molecular switch of HN (Figure 2b,c). Specifically, residues Y32 of CDR1, Y100, and E112 of CDR3 electrostatically interact with K236 and N238 of LC. D52 of FR2 forms two salt bridges with R422 of the belt, while Y47 of FR2 and Q106 of CDR3 form hydrogen bonds with D513 and N515 of the belt. Another interesting finding is that residues Y100 and Y103 of CDR3 interact with E635 of the BoNT-switch and E639 of the amphipathic peptide, two conserved carboxylates in HN that are important for pH sensing (Tables S4 and S5) [37,57]. The LCHN/E1–JLE-E9 binding is further strengthened by hydrophobic interactions between residues V99, Y100, G101, Y103, and Y105 of CDR3 with a hydrophobic ridge on BoNT/E1 that is composed of residues P239, L240, F411, V437, V514, F636, and P638. It is believed that the LC and the belt are partially unfolded prior to their entry into the cytosol by translocation through the HN channel (≈15 Å diameter) [36,58]. Therefore, the binding of JLE-E9 might interfere with LCHN unfolding in endosomes and prevent LC translocation. Furthermore, JLE-E9 directly interacts with the pH sensing residues in the BoNT-switch and the amphipathic peptide. These interactions might desensitize the pH-sensing of HN and inhibit its interaction with membrane since the hydrophobic BoNT-switch of HN is believed to be released upon acidification to engage endosomal membrane [47].
2.2. JLE-E5 and JLE-E9 Inhibit the Conformational Change of ciBoNT/E1 at Acidic pH
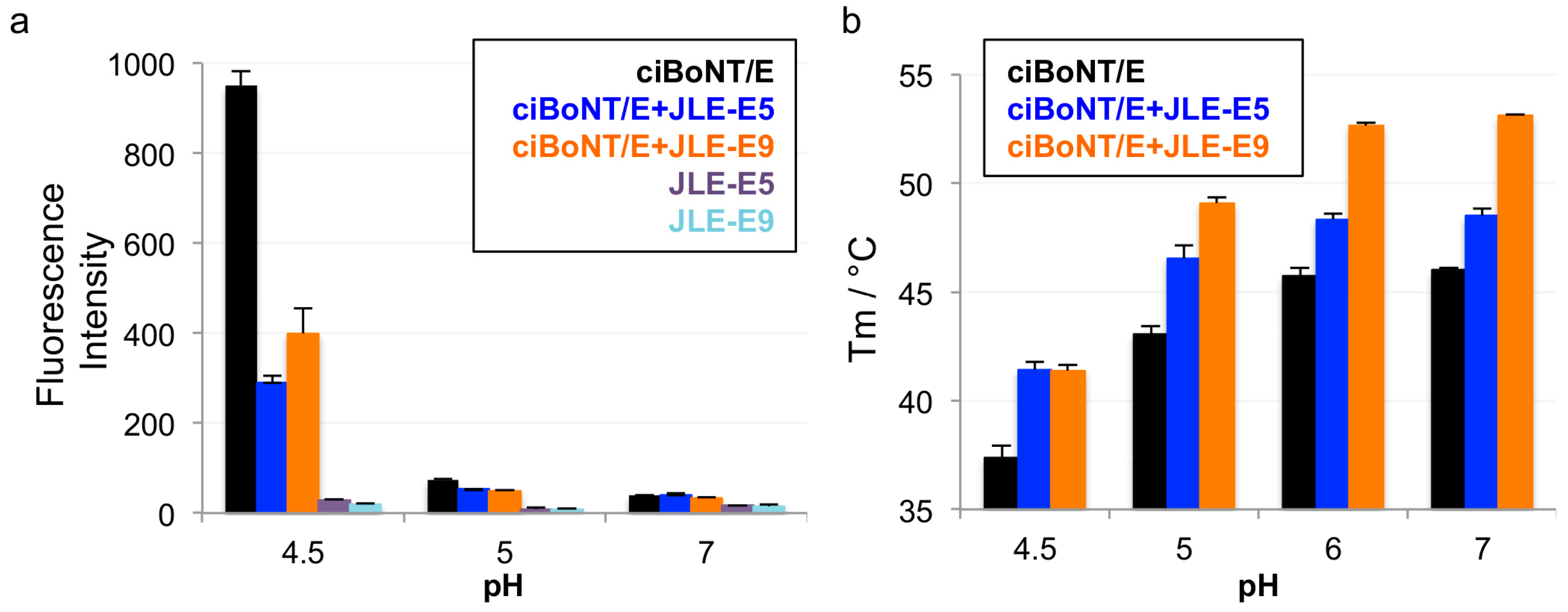
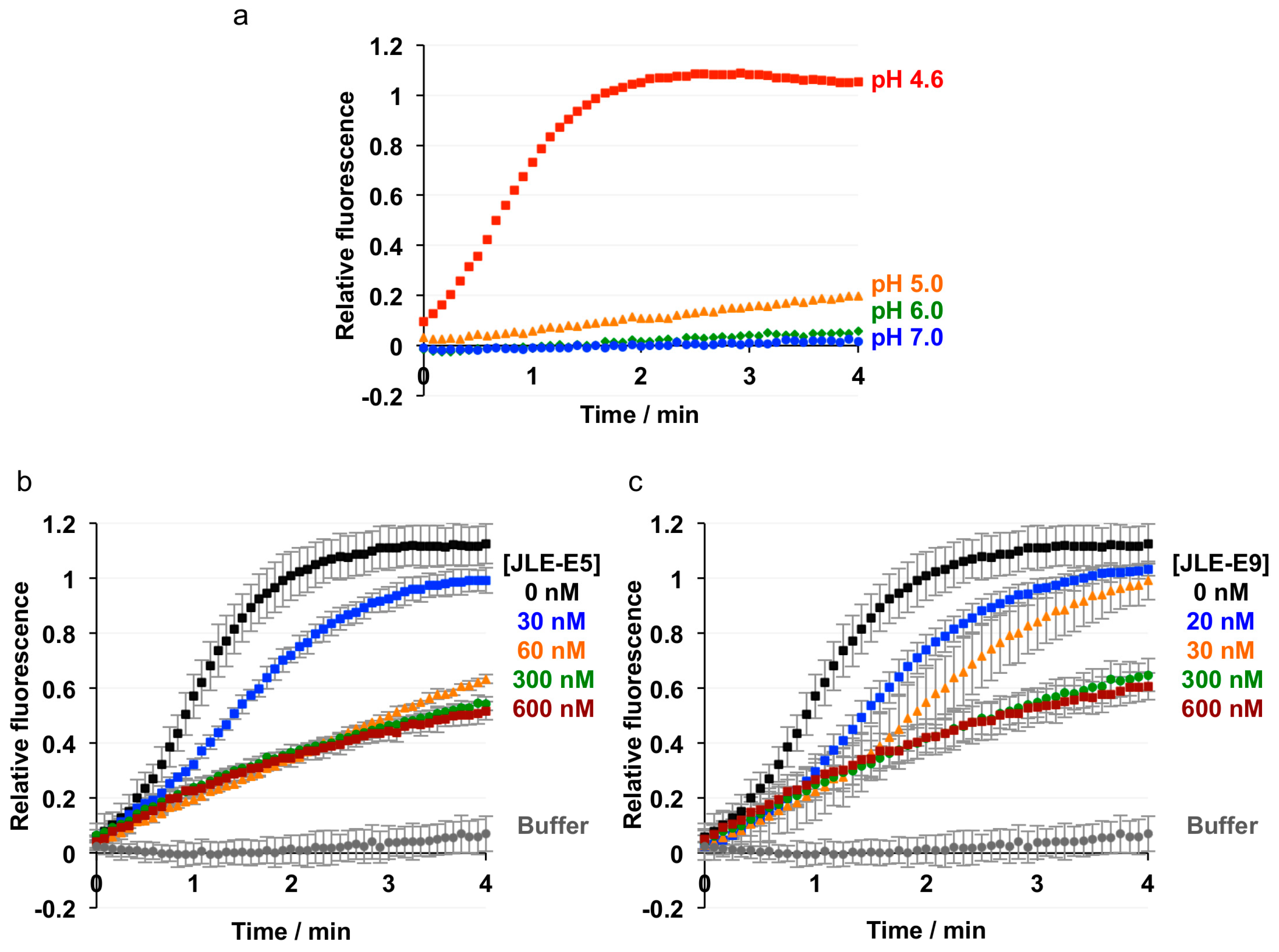
To test our hypothesis that both JLE-E5 and JLE-E9 inhibit the partial unfolding of LC and/or HN at endosomal pH, we examined the unfolding of ciBoNT/E1 using a hydrophobic fluorescence dye ANS [20,47,59]. As shown in Figure 3a, the fluorescence of ANS substantially increased when incubated with ciBoNT/E1 at pH 4.5, but not at pH 7.0 or pH 5.0, indicating ciBoNT/E1 became partially unfolded at pH < 5.0 and agreeing with similar previous studies [59]. The presence of either JLE-E5 or JLE-E9 caused a significant reduction in the ANS fluorescence intensity by ≈60% and ≈72 %, respectively, demonstrating that these VHHs inhibited the structural change of ciBoNT/E1 at acidic pH. We employed an alternative method to measure the effect of these two VHHs on the thermostability of ciBoNT/E1 using a fluorescence-based thermo shift assay (Figure 3b). The melting temperature (TM) of ciBoNT/E1 dropped dramatically from 53.1 °C at pH 7 to 37.4 °C at pH 4.5, which indicates ciBoNT/E1 is partially unfolded at low pH (Figure 3b). JLE-E5 and JLE-E9 increased the TM of ciBoNT/E1 at pH 5–7 by 2.5–3.5 °C and 6.0–7.1 °C, respectively. At pH 4.5, both VHHs increased the TM of the holotoxin by ≈4.0 °C. These data consistently demonstrated that these two BoNT/E-neutralizing VHHs stabilize the native conformation of ciBoNT/E1 and thus inhibit the pH-dependent LCHN/E1 unfolding, which is an obligatory step for transmembrane delivery of LC/E to the cytosol.
2.3. JLE-E5 and JLE-E9 Inhibit the Membrane Interaction of ciBoNT/E
We next investigated the effect of these two neutralizing VHHs on membrane interaction of BoNT/E using a membrane depolarization assay. Similar experiments were successfully employed to study the binding of HNA to membrane in recent reports [20,47]. The anionic liposome was prepared containing ganglioside GT1b, the coreceptor for the membrane association of BoNT/E [12]. We found that ciBoNT/E1 triggered an immediate dissipation of valinomycin-induced membrane potential at pH < 5, suggesting that ciBoNT/E1 associated with the artificial lipid bilayer at acidic pH in a way similar to BoNT/A1 [47] (Figure 4a). Notably, the presence of either JLE-E5 or JLE-E9 potently inhibited the ability of ciBoNT/E to depolarize membrane in a VHH concentration-dependent manner (Figure 4b,c). These data collectively suggest that both of these VHHs inhibit the conformational change of BoNT/E1 at endosomal pH and thereby block its channel formation. Recent studies with BoNT/A1 found that a VHH (ciA-B5) that binds in the N-terminal region of HNA neutralizes the toxin by inhibiting membrane insertion of HNA and blocking channel formation. However, unlike JLE-E5 and JLE-E9, ciA-B5 does not affect the low-pH triggered structural rearrangement of BoNT/A. These antibodies thus likely trap the toxins in different intermediate states through the course of channel formation of HN. VHHs have been previously applied to study the dynamic conformations of membrane proteins [60]. Our work thus provides valuable tools for future mechanistic studies of channel formation mechanisms for BoNTs.
2.4. The JLE-E5- and JLE-E9-Binding Epitopes are Conserved Across Several BoNT/E Subtypes
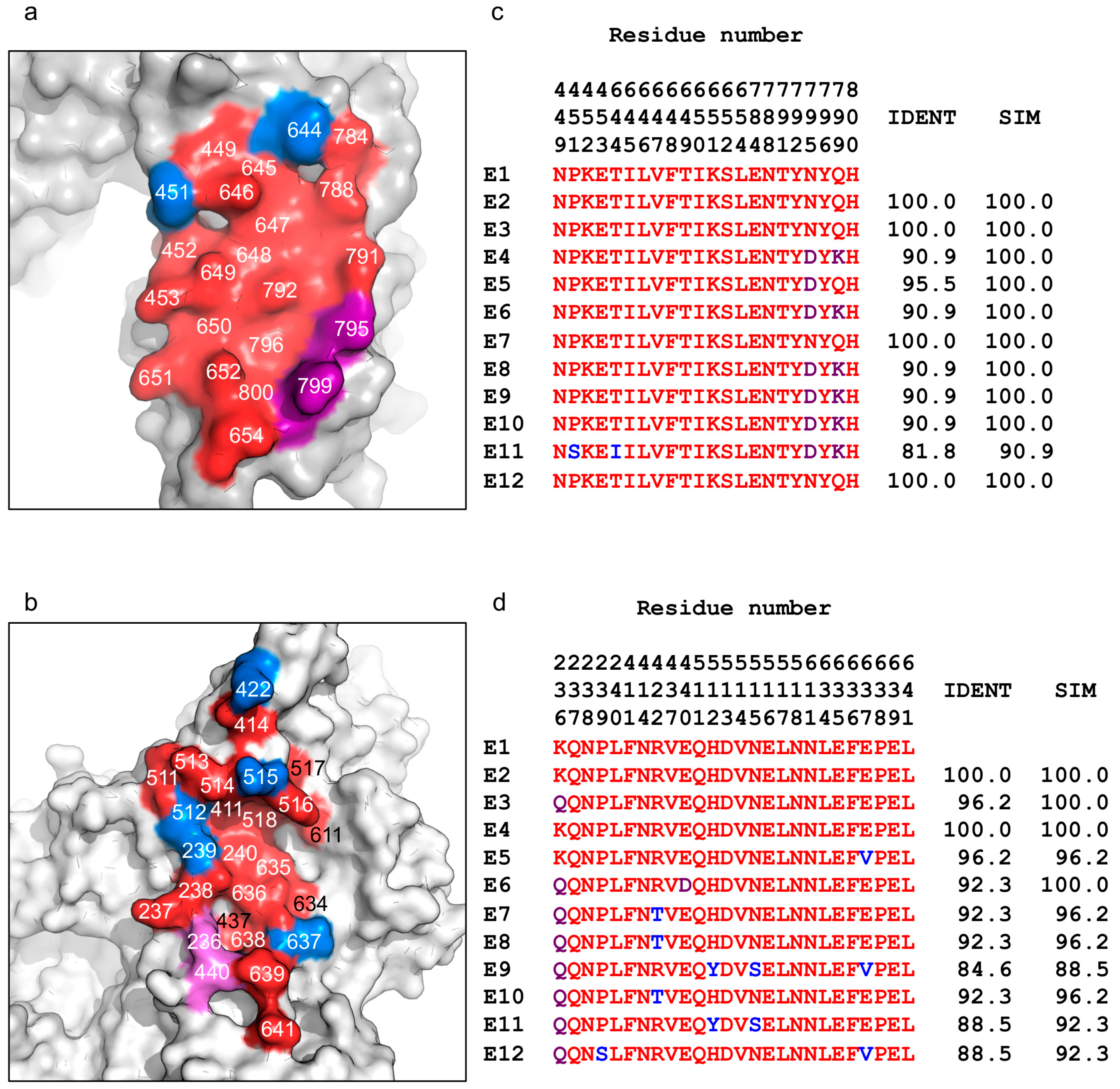
Twelve naturally occurring BoNT/E subtype variants have been reported to date, which pose significant challenges to developing broad-spectrum antitoxins against this toxin serotype. Fortunately, as shown in Figure 5, sequence analyses show that both JLE-E5 and JLE-E9-binding epitopes are quite highly conserved across the different BoNT/E subtypes, with sequence similarities of >90% and >88%, respectively. Notably, the JLE-E5-binding epitopes is identical in four other subtypes (E2, E3, E7, and E12), while a conserved mutation of N795D in subtype E5 and an additional charge replacement of Q799K in E4, E6, and E8–E10 may mildly affect JLE-E5 interactions (Figure 5a,c). The only exception is BoNT/E11, which has two unique amino acids at S451 and I644, which may weaken JLE-E5 binding. Hence, JLE-E5 is likely to bind well to most or all of the BoNT/E subtypes. The JLE-E9 binding epitope is less conserved compared to JLE-E5. This epitope is identical within subtypes E2 and E4 while there are 1–4 amino acid substitutions in the other BoNT/E subtypes that may weaken their recognition and neutralization by JLE-E9 (Figure 5b,d). Notably, our structures show that the epitopes recognized by these two VHHs are closely apposed on BoNT/E, thus making it feasible to create a simultaneous binding bivalent JLE-E5/JLE-E9 VNA by connecting them via a short peptide linker. Recent studies showed that bivalent VNAs with component VHHs that can bind simultaneously to the same toxin molecule have superior affinities and potency to neutralize toxin [20]. As shown in Tremblay et al. [51], a designer VNA optimized for simultaneous binding of JLE-E5 and JLE-E9 to BoNT/E1 displayed significantly improved antitoxin potency compared to a VNA that has the same VHH components but was not optimized for simultaneous binding. Beyond its superior antitoxin potency, this VNA will likely have strong binding to all BoNT/E subtypes due to the synergy between the two VHHs that should counteract a few subtle amino acid variations at individual epitopes, and therefore promises to be a broad-spectrum antitoxin against most or all BoNT/E subtypes.
3. Conclusions
In summary, we report the crystal structures of two BoNT/E-neutralizing VHHs, JLE-E5 and JLE-E9, in complex with LCHN/E1 at 2.5 and 3.6 Å resolutions, respectively. Our structural data show that these VHHs recognize novel epitopes at the interface of the LC–HN domains via direct interactions with the membrane-interacting segment of the HN. Therefore, they are able to inhibit the acidic pH-triggered conformational change of BoNT/E1 and preclude its interaction with membrane. This hypothesis is supported by our biochemical studies showing that both VHHs stabilize BoNT/E1 at low pH and inhibit the binding of the toxin to lipid vesicles. Sequence analysis of the JLE-E5- and JLE-E9-binding epitopes reveal that these epitopes are quite conserved among BoNT/E subtypes, suggesting these VHHs are valuable building blocks for developing multivalent VNAs with broad protection capability. This study furthers our understanding of the membrane translocation of BoNTs and provides novel insights into antibody neutralization mechanisms targeting BoNT/E and potentially other BoNT subtypes.
4. Materials and Methods
4.1. Cloning, Expression, and Purification of Recombinant Proteins
Catalytically inactive BoNT/E1 (ciBoNT/E1) that carries three mutations (H212A/E213A/H216A), LCHN/E1 (M1–K845), VHH JLE-E5, and JLE-E9 were cloned into pGEX-6p-1 vector following the N-terminal GST and a PreScission cleavage site. The proteins were expressed in Escherichia coli strain BL21-Star (DE3) (Invitrogen). Transformed bacteria were grown at 37 °C in LB medium in the presence of ampicillin. Expression was induced with 1 mM isopropyl-b-D-thiogalactopyranoside (IPTG) when OD600 reached ≈0.6–0.8. Temperature was then decreased to 18 °C and expression was continued for 16 h. Cells were harvested by centrifugation and stored at −20 °C until use.
For protein purification, bacteria were resuspended in a buffer containing 50 mM Tris (pH 8.0), 400 mM NaCl, and 0.4 mM PMSF and lysed by sonication. The GST-tagged proteins were purified using glutathione Sepharose 4B resins (GE Healthcare, Marlborough, MA, USA) and eluted from the resins after on-column cleavage in PBS using PreScission protease. The proteins were further purified by Superdex-200 Increase or Superdex-75 size exclusion chromatography (SEC; GE Healthcare) in 150 mM NaCl and 10 mM HEPES (pH 7.4). The LCHN/E1–JLE-E5 and LCHN/E1–JLE-E9 complexes were prepared by mixing the purified LCHN/E1 and VHH at a molar ratio of 1:1.5 for 1 h on ice, followed by purification using Superdex-200 Increase SEC (10 mM HEPES, pH 7.4, and 150 mM NaCl). Each protein was concentrated to ≈5 mg/mL using Amicon Ultra centrifugal filters (Millipore, Burlington, VT, USA) and stored at −80 °C until further characterization or crystallization.
4.2. Membrane Depolarization Assay
Large unilamellar vesicles (LUV) were prepared as previously described [47]. Briefly, lipids (1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)) (Avanti Polar Lipid, Alabaster, USA) were dissolved in chloroform while GT1b trisodium salt (Santa Cruz Biotechnology, Dallas, TX, USA) was dissolved in methanol. The lipids at the indicated molar ratios were mixed and then dried under nitrogen gas and placed under vacuum overnight. The dried lipids were rehydrated and subjected to 5–10 rounds of freezing and thawing cycles. Liposomes were prepared by extrusion through a 200 nm pore membrane using an Avanti Mini Extruder according to the manufacturer’s instructions.
Liposomes composed of 70/20/10 mol% of DOPC/DOPS/GT1b were prepared in 100 mM KCl, 1 mM NaCl, and 10 mM HEPES (pH 7.0). To create a trans-negative membrane potential (−117 mV), liposomes were diluted in 100 mM NaCl, 1 mM KCl, and 10 mM sodium acetate (pH 4.6). Membrane potential was monitored using 1 µM 3,3′-diethyloxacarbocyanine iodide (DiOC2(3)) (Sigma-Aldrich, St. Louis, MO, USA) [61]. Valinomycin was added at time 0-s to give a final concentration of 5 µM. At 60-s, 100 nM of ciBoNTE, preincubated with 0–600 nM of JLE-E5 or JLE-E9, was added and the fluorescence intensity at 535 nm was monitored for 4 min with excitation at 488 nm. The reaction was stopped by adding 2 µM of gramicidin from Bacillus anerinolyticus (Sigma-Aldrich, St. Louis, USA). The fluorescence change relative to the maximal change in the presence of gramicidin was calculated as (F − Finitial)/(Ffinal − Finitial). The experiments were repeated three times independently.
4.3. 8-Anilinonaphthalene-1-Sulfonic Acid Binding Assay
ciBoNT/E, ciBoNT/E–VHH, or VHH was incubated at ≈0.35 µM with 70 µM ANS for 30 min at 37 °C in either 50 mM sodium acetate (pH 4.5–5.0) or 50 mM HEPES (pH 7). All buffers contained 100 mM NaCl. Fluorescence intensity was recorded at 25 °C using a SpectraMax M2e spectrophotometer (Molecular Devices, San Jose, CA, USA) with excitation at 370 nm and emission at 470 nm. The fluorescence intensity was corrected by subtraction of background fluorescence from ANS in a buffer without protein. Error bars indicate SD of three replicate measurements.
4.4. Thermal Denaturation Assay
The thermal stability of ciBoNT/E or ciBoNT/E–VHH was measured using a fluorescence-based thermal shift assay on a StepOne real-time PCR machine (Life Technologies, Carlsbad, CA, USA). ciBoNT/E with or without VHH was incubated for 30 min in a buffer containing 0.1 M NaCl combined with 50 mM sodium acetate (pH 4.5–5.0) or 50 mM sodium citrate (pH 6) or 50 mM HEPES (pH 7.0). Immediately before the experiment, the protein (0.7 µM) was mixed with the fluorescent dye SYPRO Orange (Sigma-Aldrich). The samples were heated from 20 to 90 °C in a standard ramp rate of 1.5 °C/min. The midpoint of the protein-melting curve, the TM, was determined using the analysis software provided by the instrument manufacturer. The experiment was performed in triplicate.
4.5. Crystallization
Initial crystallization screens were performed using a Gryphon crystallization robot (Art Robbins Instruments, Mountain View, CA, USA) and high-throughput crystallization screen kits (Hampton Research and Qiagen). Extensive manual optimizations were performed at 18 °C. The best single crystals of LCHN/E1–JLE-E5 were grown by the sitting-drop vapor diffusion method at a protein concentration of 5 mg/mL with a reservoir solution containing 0.15 M Ammonium sulfate, 14% PEG 4000, 0.1 M Hepes (pH 7.0) when proteins were mixed with reservoir solution at 1:1 (v/v) ratio. Crystals of LCHN/E1–JLE-E9 were obtained by the sitting-drop vapor diffusion method at a protein concentration of 6 mg/mL with a reservoir solution containing 12% PEG 20K, 0.1 M sodium citrate (pH 5.8) when proteins were mixed with reservoir solution at 2:1 (v/v) ratio.
4.6. Data Collection and Structure Determination
All crystals were cryoprotected in their original mother liquor supplemented with 15–20% (v/v) ethylene glycol. The X-ray diffraction data were collected at 100 K at the NE-CAT beamline 24-ID-E, Advanced Photon Source (APS). The data were processed with XDS as implemented in RAPD (https://github.com/RAPD/RAPD) [62]. Data collection statistics are summarized in Table S1. Structures of the LCHN/E1–JLE-E5 and LCHN/E1–JLE-E9 complexes were determined by molecular replacement using the Phaser software [63] with LCHN/E (PDB code 3FFZ) [28] and the homology models of JLE-E5 and JLE-E9 that were built based on a VHH in PDB 5L21 [41] as the search models. Manual model building and refinement were performed in COOT [64], PHENIX [65], and CCP4 suite [66] in an iterative manner. The refinement progress was monitored with the free R value using a 5% randomly selected test set [67]. The structures were validated through MolProbity [68] and showed excellent stereochemistry. Structural refinement statistics are listed in Table S1. All structure figures were prepared with PyMol (http://www.pymol.org).
4.7. Accession Code
Atomic coordinates and structure factors for the LCHN/E1–JLE-E5 and LCHN/E1–JLE-E9 complexes have been deposited in the Protein Data Bank under accession codes 7K84 and 7K7Y, respectively.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6651/12/10/616/s1, Figure S1: VHH JLE-E5 and JLE-E9 interact with LCHN/E1, Figure S2: LCHN/E1 interacts with symmetry molecule via LC/E–LC/E homo-dimerization, Table S1: Data collection and refinement statistics, Table S2: Buried surface area of JLE-E5 in complex with LCHN/E1, Table S3: Buried surface area of LCHN/E1 in complex with JLE-E5, Table S4: Buried surface area of JLE-E9 in complex with LCHN/E1, Table S5: Buried surface area of LCHN/E1 in complex with JLE-E9.
Author Contributions
Conceptualization, R.J., C.B.S., and K.-H.L.; methodology, K.-H.L.; software, K.-H.L. and K.P.; validation, K.-H.L., R.J., and C.B.S.; formal analysis, K.-H.L., R.J., and C.B.S.; investigation, K.-H.L.; resources, R.J. and C.B.S.; data curation, K.-H.L. and K.P.; writing—original draft preparation, K.-H.L.; writing—review and editing, R.J. and C.B.S.; visualization, K.-H.L.; supervision, R.J. and C.B.S.; project administration, R.J. and C.B.S.; funding acquisition, R.J. and C.B.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by National Institute of Allergy and Infectious Diseases (NIAID) grants R01AI139087, R21AI139690, and R21AI123920 to R.J.; and R01AI125704 to R.J. and C.B.S. NE-CAT at the Advanced Photon Source (APS) is supported by a grant from the National Institute of General Medical Sciences (P30 GM124165), and the Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Montecucco, C. Tables of Toxicity of Botulinum and Tetanus Neurotoxins. Toxins 2019, 11, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnon, S.S.; Schechter, R.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Hauer, J.; Layton, M.; et al. Botulinum Toxin as a Biological Weapon. JAMA 2001, 285, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Barash, J.R.; Conrad, F.; Lou, J.; Tam, C.C.; Cheng, L.; Arnon, S.S.; Marks, J.D. The Novel Clostridial Neurotoxin Produced by Strain IBCA10-7060 Is Immunologically Equivalent to BoNT/HA. Toxins 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.-I.; Martínez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Rasotto, M.B. On Botulinum Neurotoxin Variability. mBio 2015, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Peck, M.W.; Smith, T.J.; Anniballi, F.; Austin, J.W.; Bano, L.; Bradshaw, M.; Cuervo, P.; Cheng, L.W.; Derman, Y.; Dorner, B.G.; et al. Historical Perspectives and Guidelines for Botulinum Neurotoxin Subtype Nomenclature. Toxins 2017, 9, 38. [Google Scholar] [CrossRef]
- Dover, N.; Barash, J.R.; Hill, K.K.; Xie, G.; Arnon, S.S. Molecular Characterization of a Novel Botulinum Neurotoxin Type H Gene. J. Infect. Dis. 2013, 209, 192–202. [Google Scholar] [CrossRef]
- Mazuet, C.; Sautereau, J.; Legeay, C.; Bouchier, C.; Bouvet, P.; Popoff, M.R. An Atypical Outbreak of Food-Borne Botulism Due to Clostridium botulinum Types B and E from Ham. J. Clin. Microbiol. 2014, 53, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Meng, J.; Lawrence, G.W.; Zurawski, T.H.; Sasse, A.; Bodeker, M.O.; Gilmore, M.A.; Fernández-Salas, E.; Francis, J.; Steward, L.E.; et al. Novel Chimeras of Botulinum Neurotoxins A and E Unveil Contributions from the Binding, Translocation, and Protease Domains to Their Functional Characteristics. J. Boil. Chem. 2008, 283, 16993–17002. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Maditz, R.; Kuo, C.-L.; Fishman, P.S.; Shoemaker, C.B.; Oyler, G.A.; Weissman, A.M. Targeting botulinum neurotoxin persistence by the ubiquitin-proteasome system. Proc. Natl. Acad. Sci. USA 2010, 107, 16554–16559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Botulinum Neurotoxins B and E Translocate at Different Rates and Exhibit Divergent Responses to GT1b and Low pH. Biochemistry. 2012, 51, 5655–5662. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, B.Z. Type E botulism. Clin. Toxicol. 2010, 48, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Aureli, P.; Fenicia, L.; Pasolini, B.; Gianfranceschi, M.; McCroskey, L.M.; Hatheway, C.L. Two Cases of Type E Infant Botulism Caused by Neurotoxigenic Clostridium butyricum in Italy. J. Infect. Dis. 1986, 154, 207–211. [Google Scholar] [CrossRef]
- Artin, I.; Björkman, P.; Cronqvist, J.; Rådström, P.; Holst, E. First Case of Type E Wound Botulism Diagnosed Using Real-Time PCR. J. Clin. Microbiol. 2007, 45, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Maignel, J.; Lezmi, S.; Martin, V.; Splevins, A.; Shubber, S.; Kalinichev, M.; Foster, K.A.; Picaut, P.; Krupp, J. The Expanding Therapeutic Utility of Botulinum Neurotoxins. Toxins 2018, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin E. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 299. [Google Scholar]
- BAT Botulism Antitoxin Heptavalent (A, B, C, D, E, F, G)-(Equine). Available online: https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/bat-botulism-antitoxin-heptavalent-b-c-d-e-f-g-equine (accessed on 27 September 2020).
- Garcia-Rodriguez, C.; Razai, A.; Geren, I.N.; Lou, J.; Conrad, F.; Wen, W.-H.; Farr-Jones, S.; Smith, T.J.; Brown, J.L.; Skerry, J.C.; et al. A Three Monoclonal Antibody Combination Potently Neutralizes Multiple Botulinum Neurotoxin Serotype E Subtypes. Toxins 2018, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.-H.; Tremblay, J.M.; Vazquez-Cintron, E.; Perry, K.; Ondeck, C.; Webb, R.P.; McNutt, P.M.; Shoemaker, C.B.; Jin, R. Structural Insights into Rational Design of Single-Domain Antibody-Based Antitoxins against Botulinum Neurotoxins. Cell Rep. 2020, 30, 2526–2539. [Google Scholar] [CrossRef] [Green Version]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2019, 34, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juárez-Moreno, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Masuyer, G.; Stenmark, P. Botulinum and Tetanus Neurotoxins. Annu. Rev. Biochem. 2019, 88, 811–837. [Google Scholar] [CrossRef]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; Dasgupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Genet. 1998, 5, 898–902. [Google Scholar] [CrossRef]
- Swaminathan, S.; Eswaramoorthy, S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat. Genet. 2000, 7, 693–699. [Google Scholar]
- Kumaran, D.; Eswaramoorthy, S.; Furey, W.; Navaza, J.; Sax, M.; Swaminathan, S. Domain Organization in Clostridium botulinum Neurotoxin Type E Is Unique: Its Implication in Faster Translocation. J. Mol. Boil. 2009, 386, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.R.; Liu, S.M.; Acharya, K.R. Variations in the Botulinum Neurotoxin Binding Domain and the Potential for Novel Therapeutics. Toxins 2018, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berntsson, R.P.-A.; Peng, L.; Svensson, L.M.; Dong, M.; Stenmark, P. Crystal Structures of Botulinum Neurotoxin DC in Complex with Its Protein Receptors Synaptotagmin I and II. Structure 2013, 21, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berntsson, R.P.-A.; Peng, L.; Dong, M.; Stenmark, P. Structure of dual receptor binding to botulinum neurotoxin B. Nat. Commun. 2013, 4, 2058. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Zhang, S.; Mahrhold, S.; Lam, K.-H.; Silva, D.V.; Bagramyan, K.; Perry, K.; Kalkum, M.; Rummel, A.; Dong, M.; et al. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat. Struct. Mol. Boil. 2016, 23, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Dupuy, J.; Imamura, A.; Kiso, M.; Stevens, R.C. Crystal Structure of Botulinum Neurotoxin Type A in Complex with the Cell Surface Co-Receptor GT1b—Insight into the Toxin–Neuron Interaction. PLoS Pathog. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Rummel, A.; Binz, T.; Brünger, A.T. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 2006, 444, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.-H.; Yao, G.; Jin, R. Diverse binding modes, same goal: The receptor recognition mechanism of botulinum neurotoxin. Prog. Biophys. Mol. Boil. 2015, 117, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Genet. 2003, 10, 13–18. [Google Scholar] [CrossRef]
- Pirazzini, M.; Tehran, D.A.; Leka, O.; Zanetti, G.; Rossetto, O.; Montecucco, C. On the translocation of botulinum and tetanus neurotoxins across the membrane of acidic intracellular compartments. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 467–474. [Google Scholar] [CrossRef]
- Breidenbach, M.A.; Brünger, A.T. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 2004, 432, 925–929. [Google Scholar] [CrossRef]
- Agarwal, R.; Schmidt, J.J.; Stafford, R.G.; Swaminathan, S. Mode of VAMP substrate recognition and inhibition of Clostridium botulinum neurotoxin F. Nat. Struct. Mol. Boil. 2009, 16, 789–794. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Levy, R.; Arndt, J.W.; Forsyth, C.M.; Razai, A.; Lou, J.; Geren, I.; Stevens, R.C.; Marks, J.D. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 2006, 25, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Lam, K.-H.; Weisemann, J.; Peng, L.; Krez, N.; Perry, K.; Shoemaker, C.B.; Dong, M.; Rummel, A.; Jin, R. A camelid single-domain antibody neutralizes botulinum neurotoxin A by blocking host receptor binding. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Thompson, A.A.; Fan, Y.; Lou, J.; Conrad, F.; Ho, M.; Pires-Alves, M.; Wilson, B.A.; Stevens, R.C.; Marks, J.D. A Single-Domain Llama Antibody Potently Inhibits the Enzymatic Activity of Botulinum Neurotoxin by Binding to the Non-Catalytic α-Exosite Binding Region. J. Mol. Boil. 2010, 397, 1106–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Mushrush, D.J.; Lacy, D.B.; Montal, M. Botulinum Neurotoxin Devoid of Receptor Binding Domain Translocates Active Protease. PLoS Pathog. 2008, 4, e1000245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Sambashivan, S.; Brünger, A.T.; Montal, M. Beltless translocation domain of botulinum neurotoxin A embodies a minimum ion-conductive channel. J. Boil. Chem. 2011, 287, 1657–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeda, F.J.; Olson, M.A. Structural predictions of the channel-forming region of botulinum neurotoxin heavy chain. Toxicon 1995, 33, 559–567. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Boil. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Lam, K.-H.; Guo, Z.; Krez, N.; Matsui, T.; Perry, K.; Weisemann, J.; Rummel, A.; Bowen, M.E.; Jin, R. A viral-fusion-peptide-like molecular switch drives membrane insertion of botulinum neurotoxin A1. Nat. Commun. 2018, 9, 5367. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, M.J.; Wentz, T.G.; Zhang, S.; Lee, E.J.; Dong, M.; Sharma, S.; Doxey, A.C. Bioinformatic discovery of a toxin family in Chryseobacterium piperi with sequence similarity to botulinum neurotoxins. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Oblatt-Montal, M.; Yamazaki, M.; Nelson, R.; Montal, M. Formation of ion channels in lipid bilayers by a peptide with the predicted transmembrane sequence of botulinum neurotoxin A. Protein Sci. 1995, 4, 1490–1497. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Garcia-Rodriguez, C.; Geren, I.; Lou, J.; Marks, J.D.; Nakagawa, T.; Montal, M. Molecular Architecture of Botulinum Neurotoxin E Revealed by Single Particle Electron Microscopy. J. Boil. Chem. 2007, 283, 3997–4003. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, J.M.; Vazquez-Cintron, E.; Lam, K.-H.; Mukherjee, J.; Bedenice, D.; Ondeck, C.A.; Conroy, M.T.; Bodt, S.M.L.; Winner, B.M.; Webb, R.P.; et al. Camelid VHH Antibodies that Neutralize Botulinum Neurotoxin Serotype E Intoxication or Protease Function. Toxins (Basel) 2020, 12, 611. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Geren, I.N.; Lou, J.; Conrad, F.; Forsyth, C.; Wen, W.; Chakraborti, S.; Zao, H.; Manzanarez, G.; Smith, T.J.; et al. Neutralizing human monoclonal antibodies binding multiple serotypes of botulinum neurotoxin. Protein Eng. Des. Sel. 2010, 24, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brünger, A.T.; Breidenbach, M.A.; Jin, R.; Fischer, A.; Santos, J.S.; Montal, M. Botulinum Neurotoxin Heavy Chain Belt as an Intramolecular Chaperone for the Light Chain. PLoS Pathog. 2007, 3, e113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.; Eswaramoorthy, S.; Kumaran, D.; Binz, T.; Swaminathan, S. Structural Analysis of Botulinum Neurotoxin Type E Catalytic Domain and Its Mutant Glu212→Gln Reveals the Pivotal Role of the Glu212 Carboxylate in the Catalytic Pathway. Biochemistry 2004, 43, 6637–6644. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Mushrush, D.J.; Koteiche, H.A.; Sammons, M.A.; Link, A.J.; Mchaourab, H.S.; Lacy, D.B. Studies of the Mechanistic Details of the pH-dependent Association of Botulinum Neurotoxin with Membranes. J. Boil. Chem. 2011, 286, 27011–27018. [Google Scholar] [CrossRef] [Green Version]
- Pirazzini, M.; Henke, T.; Rossetto, O.; Mahrhold, S.; Krez, N.; Rummel, A.; Montecucco, C.; Binz, T. Neutralisation of specific surface carboxylates speeds up translocation of botulinum neurotoxin type B enzymatic domain. FEBS Lett. 2013, 587, 3831–3836. [Google Scholar] [CrossRef]
- Fischer, A.; Montal, M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 10447–10452. [Google Scholar] [CrossRef] [Green Version]
- Puhar, A.; Johnson, E.; Rossetto, O.; Montecucco, C. Comparison of the pH-induced conformational change of different clostridial neurotoxins. Biochem. Biophys. Res. Commun. 2004, 319, 66–71. [Google Scholar] [CrossRef]
- Desmyter, A.; Spinelli, S.; Roussel, A.; Cambillau, C. Camelid nanobodies: Killing two birds with one stone. Curr. Opin. Struct. Boil. 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Michalek, M.; Sönnichsen, F.; Wechselberger, R.; Dingley, A.J.; Hung, C.-W.; Kopp, A.; Wienk, H.; Simanski, M.; Herbst, R.; Lorenzen, I.; et al. Structure and function of a unique pore-forming protein from a pathogenic acanthamoeba. Nat. Methods 2012, 9, 37–42. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Boil. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Boil. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Boil. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.; Ballard, C.C.; Cowtan, K.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of theCCP4 suite and current developments. Acta Crystallogr. Sect. D Boil. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Boil. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Crystal structure of the LCHN/E1–JLE-E5 complex. (a) Structures of BoNT/E1 (PDB code 3FFZ) and BoNT/A1 (PDB code 3BTA) holotoxins. (b) Cartoon representation of the LCHN/E1–JLE-E5 complex. Two orientations of the complex are presented at a rotation of 180° along the y-axis. Coloring scheme: LC, white; “belt”, yellow; HN, wheat; channel-forming helix, orange; BoNT-switch, green; amphipathic peptide, brown; JLE-E5, magenta. (c,d) Molecular interactions between LCHN/E1 and JLE-E5. Residues mediating electrostatic interactions (c) or hydrophobic interactions (d) between LCHN/E1 and JLE-E5 are shown as sticks. The complementary-determining regions (CDRs) are drawn in ribbon.
Figure 1.
Crystal structure of the LCHN/E1–JLE-E5 complex. (a) Structures of BoNT/E1 (PDB code 3FFZ) and BoNT/A1 (PDB code 3BTA) holotoxins. (b) Cartoon representation of the LCHN/E1–JLE-E5 complex. Two orientations of the complex are presented at a rotation of 180° along the y-axis. Coloring scheme: LC, white; “belt”, yellow; HN, wheat; channel-forming helix, orange; BoNT-switch, green; amphipathic peptide, brown; JLE-E5, magenta. (c,d) Molecular interactions between LCHN/E1 and JLE-E5. Residues mediating electrostatic interactions (c) or hydrophobic interactions (d) between LCHN/E1 and JLE-E5 are shown as sticks. The complementary-determining regions (CDRs) are drawn in ribbon.

Figure 2.
Crystal structure of LCHN/E1–JLE-E9. (a) Cartoon representation of the LCHN/E1–JLE-E9 complex. LCHN/E1 is colored as in Figure 1. JLE-E9 is colored in cyan. (b,c) Molecular interactions between LCHN/E1 and JLE-E9. Residues mediating electrostatic interactions (b) or hydrophobic interactions (c) between LCHN/E1 and JLE-E9 are shown as sticks. The CDRs are drawn in ribbon.
Figure 2.
Crystal structure of LCHN/E1–JLE-E9. (a) Cartoon representation of the LCHN/E1–JLE-E9 complex. LCHN/E1 is colored as in Figure 1. JLE-E9 is colored in cyan. (b,c) Molecular interactions between LCHN/E1 and JLE-E9. Residues mediating electrostatic interactions (b) or hydrophobic interactions (c) between LCHN/E1 and JLE-E9 are shown as sticks. The CDRs are drawn in ribbon.

Figure 3.
JLE-E5 and JLE-E9 inhibit the conformational change of catalytically inactive BoNT/E1 (ciBoNT/E) at acidic pH. (a) ANS fluorescence experiment. ciBoNT/E1 at 0.35 µM was incubated with an equimolar ratio of the indicated VHH in a buffer containing either 50 mM sodium acetate (pH 4.5–5) or HEPES (pH 7.0). All buffers contained 100 mM NaCl and 70 µM ANS. The mean values of fluorescence intensity at 470 nm are shown. (b) Thermal stability of ciBoNT/E1, ciBoNT/E1–JLE-E5, and ciBoNT/E1–JLE-E9. The thermal stability of the protein was measured using a fluorescence-based thermal shift assay on a StepOne real-time PCR system (ThermoFisher). Protein melting was monitored using SYPRO Orange dye as the temperature was increased in a linear ramp from 20 to 90 °C. The midpoint of the protein-melting curve (TM) was determined using software provided by the instrument manufacturer.
Figure 3.
JLE-E5 and JLE-E9 inhibit the conformational change of catalytically inactive BoNT/E1 (ciBoNT/E) at acidic pH. (a) ANS fluorescence experiment. ciBoNT/E1 at 0.35 µM was incubated with an equimolar ratio of the indicated VHH in a buffer containing either 50 mM sodium acetate (pH 4.5–5) or HEPES (pH 7.0). All buffers contained 100 mM NaCl and 70 µM ANS. The mean values of fluorescence intensity at 470 nm are shown. (b) Thermal stability of ciBoNT/E1, ciBoNT/E1–JLE-E5, and ciBoNT/E1–JLE-E9. The thermal stability of the protein was measured using a fluorescence-based thermal shift assay on a StepOne real-time PCR system (ThermoFisher). Protein melting was monitored using SYPRO Orange dye as the temperature was increased in a linear ramp from 20 to 90 °C. The midpoint of the protein-melting curve (TM) was determined using software provided by the instrument manufacturer.

Figure 4.
Membrane depolarization assay. Liposomes composed of 70/20/10 mol% of DOPC/DOPS/GT1b were polarized at a negative internal voltage by adding valinomycin in the presence of a transmembrane KCl gradient. Membrane potential was monitored using the fluorescence dye DiOC2(3). (a) ciBoNT/E1 was added at the indicated buffer pH. The experiment was performed in duplicate. (b,c) Effect of JLE-E5 (b) or JLE-E9 (c) on membrane depolarization of ciBoNT/E1. Indicated concentrations of the VHHs were preincubated with ciBoNT/E before the measurement. The data are presented as ± S.D; n = 3.
Figure 4.
Membrane depolarization assay. Liposomes composed of 70/20/10 mol% of DOPC/DOPS/GT1b were polarized at a negative internal voltage by adding valinomycin in the presence of a transmembrane KCl gradient. Membrane potential was monitored using the fluorescence dye DiOC2(3). (a) ciBoNT/E1 was added at the indicated buffer pH. The experiment was performed in duplicate. (b,c) Effect of JLE-E5 (b) or JLE-E9 (c) on membrane depolarization of ciBoNT/E1. Indicated concentrations of the VHHs were preincubated with ciBoNT/E before the measurement. The data are presented as ± S.D; n = 3.

Figure 5.
Sequence conservation of the VHH-binding epitopes across BoNT/E subtypes. (a,b) Sequence conservation of (a) JLE-E5- and (b) JLE-E9-binding epitopes are plotted on the LCHN/E1 structure. Identical, conserved, and variable residues at the binding interface are colored red, purple, and blue, respectively. (c,d) Amino acid sequence alignment among all BoNT/E subtypes. Only amino acids forming the JLE-E5 (c) or JLE-E9-binding (d) epitopes on BoNT/E are presented with residue numbers as indicated (displayed vertically, top down, for each residue position). The percentage of sequence identity (IDENT) and similarity (SIM) are listed. Sequence alignments were performed using Clustal Omega.
Figure 5.
Sequence conservation of the VHH-binding epitopes across BoNT/E subtypes. (a,b) Sequence conservation of (a) JLE-E5- and (b) JLE-E9-binding epitopes are plotted on the LCHN/E1 structure. Identical, conserved, and variable residues at the binding interface are colored red, purple, and blue, respectively. (c,d) Amino acid sequence alignment among all BoNT/E subtypes. Only amino acids forming the JLE-E5 (c) or JLE-E9-binding (d) epitopes on BoNT/E are presented with residue numbers as indicated (displayed vertically, top down, for each residue position). The percentage of sequence identity (IDENT) and similarity (SIM) are listed. Sequence alignments were performed using Clustal Omega.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lam, K.-H.; Perry, K.; Shoemaker, C.B.; Jin, R. Two VHH Antibodies Neutralize Botulinum Neurotoxin E1 by Blocking Its Membrane Translocation in Host Cells. Toxins 2020, 12, 616. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100616
AMA Style
Lam K-H, Perry K, Shoemaker CB, Jin R. Two VHH Antibodies Neutralize Botulinum Neurotoxin E1 by Blocking Its Membrane Translocation in Host Cells. Toxins. 2020; 12(10):616. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100616
Chicago/Turabian StyleLam, Kwok-Ho, Kay Perry, Charles B. Shoemaker, and Rongsheng Jin. 2020. "Two VHH Antibodies Neutralize Botulinum Neurotoxin E1 by Blocking Its Membrane Translocation in Host Cells" Toxins 12, no. 10: 616. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100616
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.