Immunotoxin Screening System: A Rapid and Direct Approach to Obtain Functional Antibodies with Internalization Capacities


Abstract
:1. Introduction
2. Current FDA-Approved Toxin-Mediated Therapeutics
3. Mode-of-Actions of Therapeutic Antibodies
4. Internalization Assays to Obtain Monoclonal Antibodies with Internalization Capacities
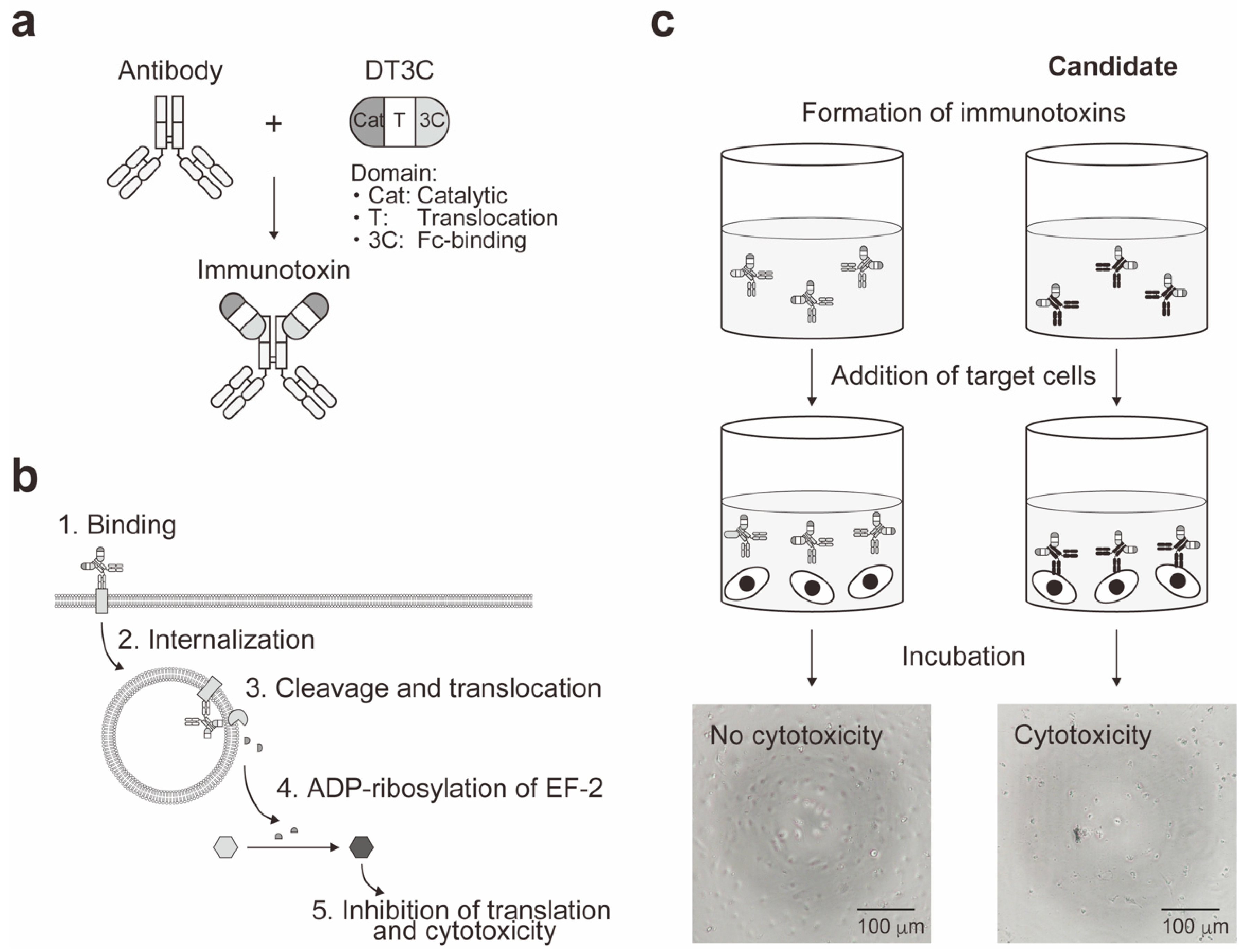
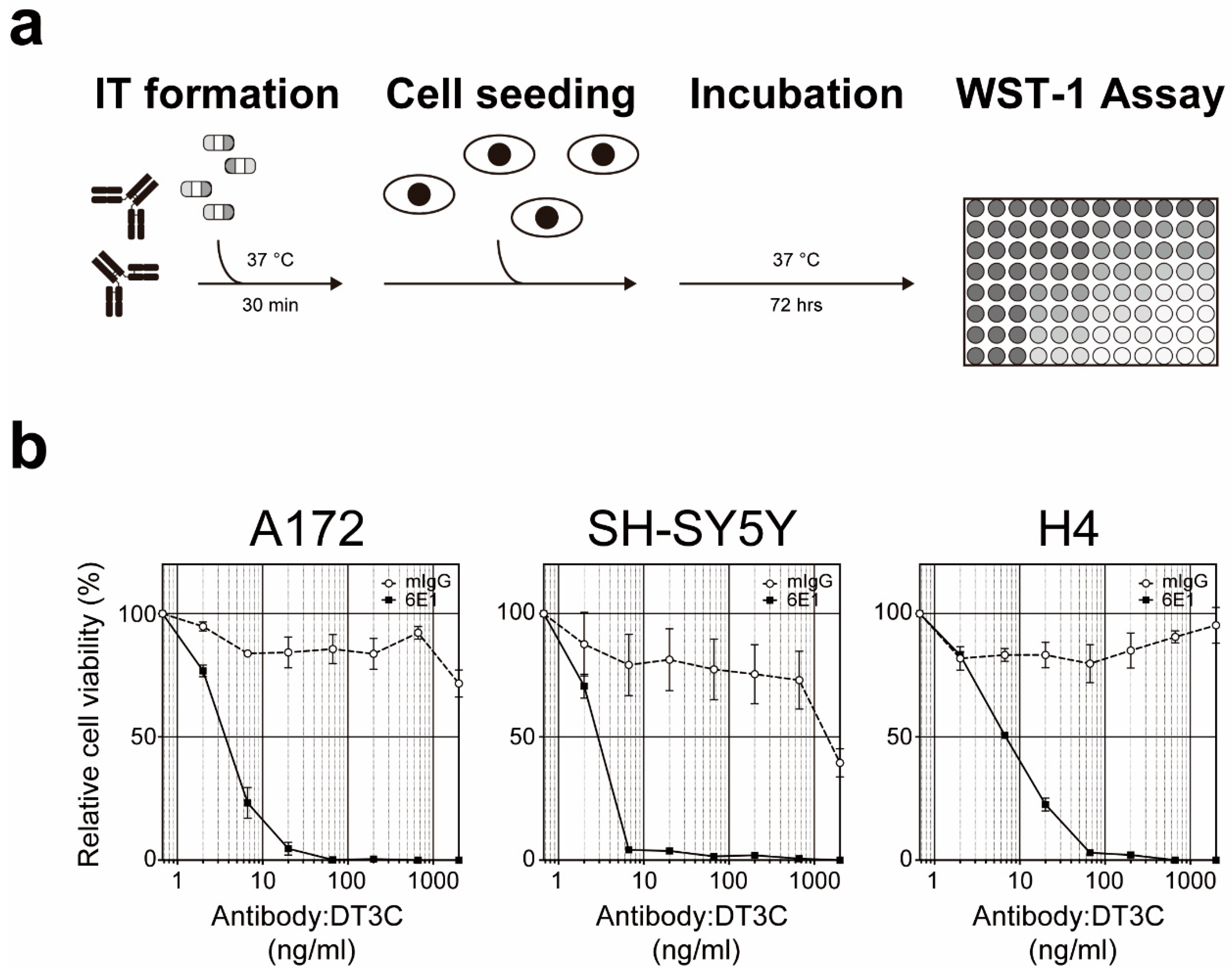
5. Cell-Based Immunotoxin Screening System
6. Application of Functional Monoclonal Antibodies as Antibody Drug Conjugates
7. Challenges for Next Generation of Antibody Drug Conjugates
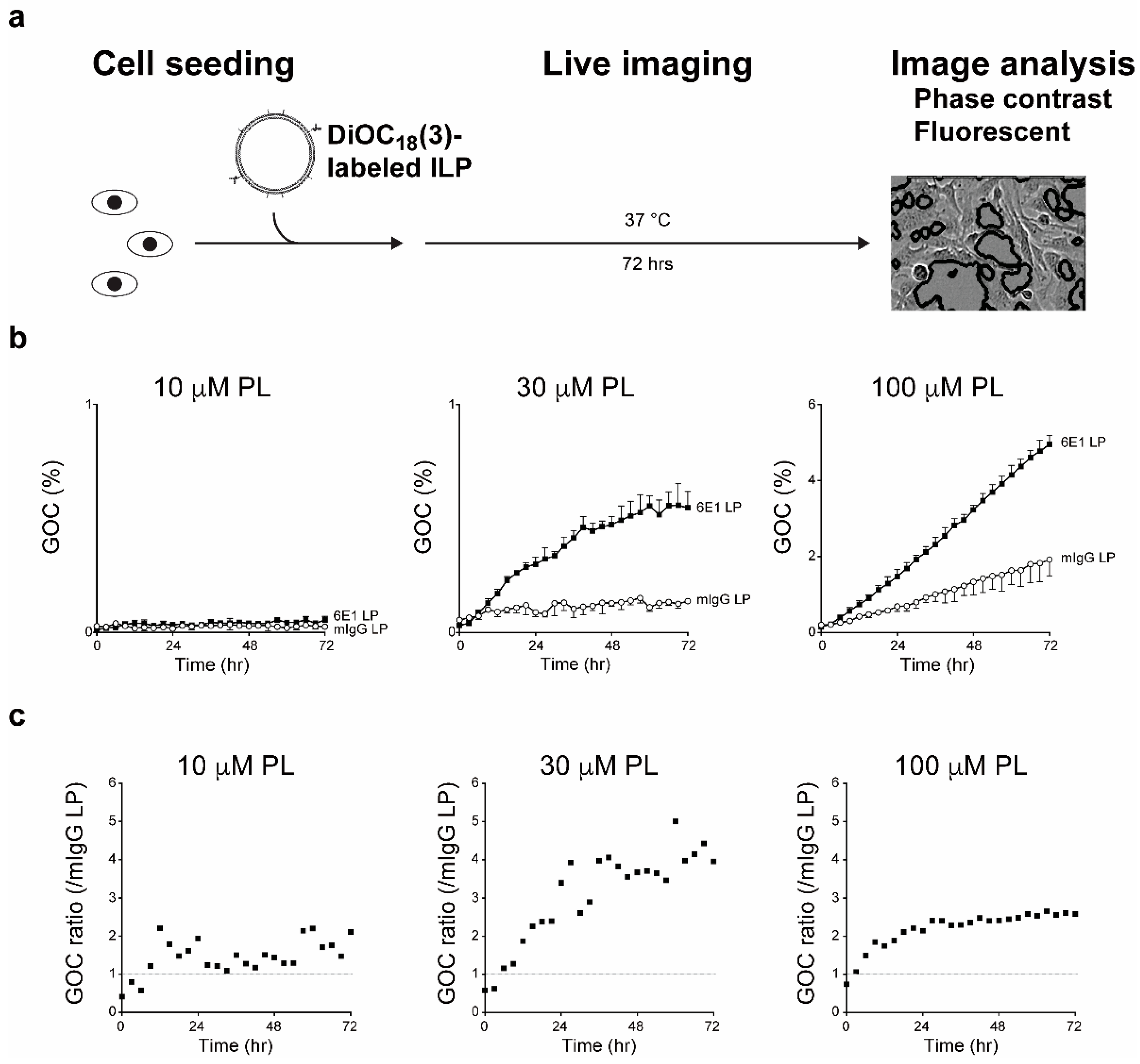
8. Application of Functional Monoclonal Antibodies as Immunoliposomes
9. Future Directions
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, K.; Jeppesen, M.G.; Malmgaard-Clausen, M.; Krzywkowski, K.; Kledal, T.N.; Rosenkilde, M.M. Novel chemokine-based immunotoxins for potent and selective targeting of cytomegalovirus infected cells. J. Immunol. Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidle, U.H.; Tiefenthaler, G.; Schiller, C.; Weiss, E.H.; Georges, G.; Brinkmann, U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genom. Proteom. 2014, 11, 25–38. [Google Scholar]
- Akbari, B.; Farajnia, S.; Khosroshahi, S.A.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef]
- Bortolotti, M.; Bolognesi, A.; Polito, L. Bouganin, an attractive weapon for immunotoxins. Toxins 2018, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Calafato, G.; Bolognesi, A. Ricin: An ancient story for a timeless plant toxin. Toxins 2019, 11, 324. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Q.; Zhu, Z.N.; Zheng, Y.T.; Shaw, P.C. Engineering of ribosome-inactivating proteins for improving pharmacological properties. Toxins 2020, 12, 167. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Shan, L.; Liu, Y.; Wang, P. Recombinant immunotoxin therapy of solid tumors: Challenges and strategies. J. Basic Clin. Med. 2013, 2, 1–6. [Google Scholar]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, P.; Fazio, V.M. Mouse monoclonal antibodies in biological research: Strategies for high-throughput production. Biotechnol. Lett. 2008, 30, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Layton, D.; Laverty, C.; Nice, E.C. Design and operation of an automated high-throughput monoclonal antibody facility. Biophys. Rev. 2013, 5, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Lansigan, F.; Stearns, D.M.; Foss, F. Role of denileukin diftitox in the treatment of persistent or recurrent cutaneous T-cell lymphoma. Cancer Manag. Res. 2010, 2, 53–59. [Google Scholar] [CrossRef]
- Bacha, P.; Williams, D.P.; Waters, C.; Williams, J.M.; Murphy, J.R.; Strom, T.B. Interleukin 2 receptor-targeted cytotoxicity. Interleukin 2 receptor-mediated action of a diphtheria toxin-related interleukin 2 fusion protein. J. Exp. Med. 1988, 167, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Jen, E.Y.; Gao, X.; Li, L.; Zhuang, L.; Simpson, N.E.; Aryal, B.; Wang, R.; Przepiorka, D.; Shen, Y.L.; Leong, R.; et al. FDA approval summary: Tagraxofusp-erzs for treatment of blastic plasmacytoid dendritic cell neoplasm. Clin. Cancer Res. 2020, 26, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.Y.; Dinner, S.N. Moxetumomab pasudotox for hairy cell leukemia: Preclinical development to FDA approval. Blood Adv. 2019, 3, 2905–2910. [Google Scholar] [CrossRef]
- Kim, J.S.; Jun, S.Y.; Kim, Y.S. Critical issues in the development of immunotoxins for anticancer therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Richardson, N.C.; Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA approval summary: Brentuximab vedotin in first-line treatment of peripheral T-cell lymphoma. Oncologist 2019, 24, e180–e187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyraz, B.; Sendur, M.A.N.; Aksoy, S.; Babacan, T.; Roach, E.C.; Kizilarslanoglu, M.C.; Petekkaya, I.; Altundag, K. Trastuzumab emtansine (T-DM1) for HER2-positive breast cancer. Curr. Med. Res. Opin. 2013, 29, 405–414. [Google Scholar] [CrossRef]
- Wedam, S.; Fashoyin-Aje, L.; Gao, X.; Bloomquist, E.; Tang, S.; Sridhara, R.; Goldberg, K.B.; King-Kallimanis, B.L.; Theoret, M.R.; Ibrahim, A.; et al. FDA approval summary: Ado-trastuzumab emtansine for the adjuvant treatment of HER2-positive early breast cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Yurkiewicz, I.R.; Muffly, L.; Liedtke, M. Inotuzumab ozogamicin: A CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des. Devel. Ther. 2018, 12, 2293–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. 2019 FDA drug approvals. Nat. Rev. Drug Discov. 2019, 18, 85–89. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs. gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, M.; Guindon, J.; Brazas, L.; Moore, C.; Entwistle, J.; Cizeau, J.; Jewett, M.A.S.; MacDonald, G.C. A phase II study of oportuzumab monatox: An immunotoxin therapy for patients with noninvasive urothelial carcinoma in situ previously treated with Bacillus Calmette-Guérin. J. Urol. 2012, 188, 1712–1718. [Google Scholar] [CrossRef]
- Hawkins, R.E.; Gore, M.; Shparyk, Y.; Bondar, V.; Gladkov, O.; Ganev, T.; Harza, M.; Polenkov, S.; Bondarenko, I.; Karlov, P.; et al. A randomized phase II/III study of naptumomab estafenatox + IFNα versus IFNα in renal cell carcinoma: Final analysis with baseline biomarker subgroup and trend analysis. Clin. Cancer Res. 2016, 22, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Alewine, C.; Ahmad, M.; Peer, C.J.; Hu, Z.I.; Lee, M.J.; Yuno, A.; Kindrick, J.D.; Thomas, A.; Steinberg, S.M.; Trepel, J.B.; et al. Phase I/II study of the mesothelin-targeted immunotoxin LMB-100 with nab-paclitaxel for patients with advanced pancreatic adenocarcinoma. Clin. Cancer Res. 2020, 26, 828–836. [Google Scholar] [CrossRef]
- Thepen, T.; van Vuuren, A.J.; Kiekens, R.C.; Damen, C.A.; Vooijs, W.C.; van De Winkel, J.G. Resolution of cutaneous inflammation after local elimination of macrophages. Nat. Biotechnol. 2000, 18, 48–51. [Google Scholar] [CrossRef]
- Itoi, K.; Sugimoto, N.; Suzuki, S.; Sawada, K.; Das, G.; Uchida, K.; Fuse, T.; Ohara, S.; Kobayashi, K. Targeting of locus ceruleus noradrenergic neurons expressing human interleukin-2 receptor α-subunit in transgenic mice by a recombinant immunotoxin anti-Tac(Fv)-PE38: A study for exploring noradrenergic influence upon anxiety-like and depression-like behaviors. J. Neurosci. 2011, 31, 6132–6139. [Google Scholar] [PubMed] [Green Version]
- Sadraeian, M.; Guimarães, F.E.G.; Araújo, A.P.U.; Worthylake, D.K.; LeCour, L.J.; Pincus, S.H. Selective cytotoxicity of a novel immunotoxin based on pulchellin A chain for cells expressing HIV envelope. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Flavell, D.J. Countering immunotoxin immunogenicity. Br. J. Cancer 2016, 114, 1177–1179. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, Y.; Benhar, I. Addressing the immunogenicity of the cargo and of the targeting antibodies with a focus on deimmunized bacterial toxins and on antibody-targeted human effector proteins. Biomedicines 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; King, E.M.; Pastan, I. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am. J. Pathol. 2018, 188, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Ho, M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Li, J.; Zhu, X.; Tang, X.; Bao, Y.; Sun, X.; Huang, Y.; Tian, F.; Liu, X.; Yang, L. Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential. Int. J. Nanomed. 2017, 12, 1969–1983. [Google Scholar] [CrossRef] [Green Version]
- Bachanova, V.; Frankel, A.E.; Cao, Q.; Lewis, D.; Grzywacz, B.; Verneris, M.R.; Ustun, C.; Lazaryan, A.; McClune, B.; Warlick, E.D.; et al. Phase I study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies. Clin. Cancer Res. 2015, 21, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, J.U.; Todhunter, D.; Taras, E.; Bachanova, V.; Vallera, D.A. Development of a deimmunized bispecific immunotoxin dDT2219 against B-Cell malignancies. Toxins 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilides, C. Review of 90Y-ibritumomab tiuxetan as first-line consolidation radio-immunotherapy for B-cell follicular non-Hodgkin’s lymphoma. Cancer Manag. Res. 2009, 1, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. 90Y-ibritumomab tiuxetan: A nearly forgotten opportunity. Oncotarget 2016, 7, 7597–7609. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Choyke, P.L. Near-infrared photoimmunotherapy of cancer. Acc. Chem. Res. 2019, 52, 2332–2339. [Google Scholar] [CrossRef] [Green Version]
- Casalini, P.; Caldera, M.; Canevari, S.; Ménard, S.; Mezzanzanica, D.; Tosi, E.; Gadina, M.; Colnaghi, M.I. A critical comparison of three internalization assays applied to the evaluation of a given mAb as a toxin-carrier candidate. Cancer Immunol. Immunother. 1993, 37, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Liao-Chan, S.; Daine-Matsuoka, B.; Heald, N.; Wong, T.; Lin, T.; Cai, A.G.; Lai, M.; D’Alessio, J.A.; Theunissen, J.W. Quantitative assessment of antibody internalization with novel monoclonal antibodies against Alexa fluorophores. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Li, Y.; Corbett Liu, P.; Shen, Y.; Snavely, M.D.; Hiraga, K. A cell-based internalization and degradation assay with an activatable fluorescence-quencher probe as a tool for functional antibody screening. J. Biomol. Screen. 2015, 20, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Rigo, A.; Vinante, F. Flow Cytometry analysis of receptor internalization/shedding. Cytom. B 2017, 92B, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Wargalla, U.C.; Reisfeld, R.A. Rate of internalization of an immunotoxin correlates with cytotoxic activity against human tumor cells. Proc. Natl. Acad. Sci. USA 1989, 86, 5146–5150. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, E.; Guryev, E.; Yudintsev, A.; Vodeneev, V.; Deyev, S.; Balalaeva, I. HER2-specific recombinant immunotoxin 4D5scFv-PE40 passes through retrograde trafficking route and forces cells to enter apoptosis. Oncotarget 2017, 8, 22048–22058. [Google Scholar] [CrossRef] [Green Version]
- Derbyshire, E.J.; de Leij, L.; Wawrzynczak, E.J. Refinement of an indirect immunotoxin assay of monoclonal antibodies recognising the human small cell lung cancer cluster 2 antigen. Br. J. Cancer 1993, 67, 1242–1247. [Google Scholar] [CrossRef] [Green Version]
- Weltman, J.K.; Pedroso, P.; Johnson, S.A.; Davignon, D.; Fast, L.D.; Leone, L.A. Rapid screening with indirect immunotoxin for monoclonal antibodies against human small cell lung cancer. Cancer Res. 1987, 47, 5552–5556. [Google Scholar]
- Fukuhara, T.; Kim, J.; Hokaiwado, S.; Nawa, M.; Okamoto, H.; Kogiso, T.; Watabe, T.; Hattori, N. A novel immunotoxin reveals a new role for CD321 in endothelial cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awwad, S.; Angkawinitwong, U. Overview of Antibody Drug Delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Nishii, Y.; Nakamura, K.; Aoki, H.; Hirai, S.; Uchida, H.; Sakuma, Y.; Hamada, H. Development of a sensitive screening method for selecting monoclonal antibodies to be internalized by cells. Biochem. Biophys. Res. Commun. 2014, 454, 600–603. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Hou, S.C.; Chen, H.S.; Lin, H.W.; Chao, W.T.; Chen, Y.S.; Fu, C.Y.; Yu, C.M.; Huang, K.F.; Wang, A.H.; Yang, A.S. High throughput cytotoxicity screening of anti-HER2 immunotoxins conjugated with antibody fragments from phage-displayed synthetic antibody libraries. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of antibody phage display technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef] [Green Version]
- Babcook, J.S.; Leslie, K.B.; Olsen, O.A.; Salmon, R.A.; Schrader, J.W. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc. Natl. Acad. Sci. USA 1996, 93, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Tiller, T. Single B cell antibody technologies. N. Biotechnol. 2011, 28, 453–457. [Google Scholar] [CrossRef]
- Drachman, J.G.; Senter, P.D. Antibody-drug conjugates: The chemistry behind empowering antibodies to fight cancer. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 306–310. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Pagano, L.; Fianchi, L.; Caira, M.; Rutella, S.; Leone, G. The role of Gemtuzumab Ozogamicin in the treatment of acute myeloid leukemia patients. Oncogene 2007, 26, 3679–3690. [Google Scholar] [CrossRef] [Green Version]
- Kikkawa, Y.; Enomoto-Okawa, Y.; Fujiyama, A.; Fukuhara, T.; Harashima, N.; Sugawara, Y.; Negishi, Y.; Katagiri, F.; Hozumi, K.; Nomizu, M.; et al. Internalization of CD239 highly expressed in breast cancer cells: A potential antigen for antibody-drug conjugates. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Enomoto-Okawa, Y.; Maeda, Y.; Harashima, N.; Sugawara, Y.; Katagiri, F.; Hozumi, K.; Hui, K.M.; Nomizu, M.; Ito, Y.; Kikkawa, Y. An anti-human lutheran glycoprotein phage antibody inhibits cell migration on laminin-511: Epitope mapping of the antibody. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Muguruma, K.; Yakushiji, F.; Kawamata, R.; Akiyama, D.; Arima, R.; Shirasaka, T.; Kikkawa, Y.; Taguchi, A.; Takayama, K.; Fukuhara, T.; et al. Novel hybrid compound of a plinabulin prodrug with an IgG binding peptide for generating a tumor selective noncovalent-type antibody-drug conjugate. Bioconjugate Chem. 2016, 27, 1606–1613. [Google Scholar] [CrossRef]
- Nishii, Y.; Yamaguchi, M.; Kimura, Y.; Hasegawa, T.; Aburatani, H.; Uchida, H.; Hirata, K.; Sakuma, Y. A newly developed anti-Mucin 13 monoclonal antibody targets pancreatic ductal adenocarcinoma cells. Int. J. Oncol. 2015, 46, 1781–1787. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. mAbs 2011, 3, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2016, 24, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA approval summary: (daunorubicin and cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Trompetero, A.; Gordillo, A.; Del Pilar, M.C.; Cristina, V.M.; Bustos Cruz, R.H. Alzheimer’s Disease and Parkinson’s Disease: A Review of current treatment adopting a nanotechnology approach. Curr. Pharm. Des. 2018, 24, 22–45. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal. Transduct. Target. Ther. 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Madhankumar, A.B.; Miller, P.A.; Duck, K.A.; Hafenstein, S.; Rizk, E.; Slagle-Webb, B.; Sheehan, J.M.; Connor, J.R.; Yang, Q.X. MRI contrast agent for targeting glioma: Interleukin-13 labeled liposome encapsulating gadolinium-DTPA. Neuro Oncol. 2016, 18, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, R.; Oda, Y.; Omata, D.; Nishiie, N.; Koshima, R.; Shiono, Y.; Sawaguchi, Y.; Unga, J.; Naoi, T.; Negishi, Y.; et al. Tumor growth suppression by the combination of nanobubbles and ultrasound. Cancer Sci. 2016, 107, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hamamichi, S.; Asano, M.; Hori, Y.; Matsui, J.; Iwata, M.; Funahashi, Y.; Umeda, I.O.; Fujii, H. Radiolabeled liposome imaging determines an indication for liposomal anticancer agent in ovarian cancer mouse xenograft models. Cancer Sci. 2016, 107, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.L.; Binderup, T.; Rasmussen, P.; Henriksen, J.R.; Elema, D.R.; Kjær, A.; Andresen, T.L. In vivo evaluation of PEGylated 64Cu-liposomes with theranostic and radiotherapeutic potential using micro PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Koshkaryev, A.; Sawant, R.; Deshpande, M.; Torchilin, V. Immunoconjugates and long circulating systems: Origins, current state of the art and future directions. Adv. Drug Deliv. Rev. 2013, 65, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Eloy, J.O.; Petrilli, R.; Trevizan, L.N.F.; Chorilli, M. Immunoliposomes: A review on functionalization strategies and targets for drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Tran, S.; DeGiovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Hong, K.; Kirpotin, D.B.; Colbern, G.; Shalaby, R.; Baselga, J.; Shao, Y.; Nielsen, U.B.; Marks, J.D.; Moore, D.; et al. Anti-HER2 immunoliposomes: Enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 2002, 8, 1172–1181. [Google Scholar]
- Eloy, J.O.; Petrilli, R.; Chesca, D.L.; Saggioro, F.P.; Lee, R.J.; Marchetti, J.M. Anti-HER2 immunoliposomes for co-delivery of paclitaxel and rapamycin for breast cancer therapy. Eur. J. Pharm. Biopharm. 2017, 115, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoeds, M.H.; Steerenberg, P.A.; Hendriks, J.J.G.W.; Dekker, L.C.; van Hoesel, Q.G.C.M.; Crommelin, D.J.A.; Storm, S. Immunoliposome-mediated targeting of doxorubicin to human ovarian carcinoma in vitro and in vivo. Br. J. Cancer 1996, 74, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood-brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X. Current strategies for brain drug delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Villaseñor, R.; Lampe, J.; Schwaninger, M.; Collin, L. Intracellular transport and regulation of transcytosis across the blood–brain barrier. Cell. Mol. Life Sci. 2019, 76, 1081–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulgar, V.M. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front. Neurosci. 2019, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Zhang, Y.; Schlachetzki, F.; Pardridge, W.M. Global non-viral gene transfer to the primate brain following intravenous administration. Mol. Ther. 2003, 7, 11–18. [Google Scholar] [CrossRef]
- Boado, R.J.; Pardridge, W.M. The trojan horse liposome technology for nonviral gene transfer across the blood-brain barrier. J. Drug Deliv. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | Targeting Moiety | Toxin Moiety | Tumor Type | Approval Year | References |
|---|---|---|---|---|---|
| Cytotoxins | |||||
| Denileukin diftitox (Ontak®) | IL2 | DT (DAB389) | CTCL | 1999 | [16] |
| Tagraxofusp-erzs (Elzonris®) | IL3 | DT (DAB389) | BPDCN | 2018 | [18] |
| Immunotoxin | |||||
| Moxetumomab pasudotox (Lumoxiti®) | Anti-CD22 dsFv | PE (PE38) | HCL | 2018 | [19] |
| Antibody Drug Conjugates | |||||
| Gemtuzumab ozogamicin (Mylotarg®) | Humanized anti-CD33 mAb | Ozogamicin | AML | 2000-approved 2010-withdrawn 2017-reapproved | [21] |
| Brentuximab vedotin (Acetris®) | Chimeric anti-CD30 mAb | MMAE | ALCL, HL, PTCL | 2011 | [22,23] |
| Trastuzumab emtansine (Kadcyla®) | Humanized anti-HER2 mAb | DM1 | HER2+ BC | 2013 | [24,25] |
| Inotuzumab ozogamicin (Besponsa®) | Humanized anti-CD22 mAb | Ozogamicin | ALL | 2017 | [26] |
| Polatuzumab vedotin (PolivyTM) | Humanized anti-CD79B mAb | MMAE | DLBCL | 2019 | [27] |
| Enfortumab vedotin (PadcevTM) | Human anti-nectin-4 mAb | MMAE | UC | 2019 | [27] |
| Trastuzumab deruxtecan (Enhertu®) | Humanized anti-HER2 mAb | Deruxtecan | HER2+ BC | 2019 | [27] |
| Sacituzumab govitecan (TrodelvyTM) | Humanized anti-Trop-2 mAb | SN-38 | Triple-negative BC | 2020 | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamamichi, S.; Fukuhara, T.; Hattori, N. Immunotoxin Screening System: A Rapid and Direct Approach to Obtain Functional Antibodies with Internalization Capacities. Toxins 2020, 12, 658. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100658
Hamamichi S, Fukuhara T, Hattori N. Immunotoxin Screening System: A Rapid and Direct Approach to Obtain Functional Antibodies with Internalization Capacities. Toxins. 2020; 12(10):658. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100658
Chicago/Turabian StyleHamamichi, Shusei, Takeshi Fukuhara, and Nobutaka Hattori. 2020. "Immunotoxin Screening System: A Rapid and Direct Approach to Obtain Functional Antibodies with Internalization Capacities" Toxins 12, no. 10: 658. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12100658