Stereoselective Synthesis of the I–L Fragment of the Pacific Ciguatoxins


School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK
*
Author to whom correspondence should be addressed.
Toxins 2020, 12(12), 740; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120740
Submission received: 30 October 2020
/
Revised: 20 November 2020
/
Accepted: 20 November 2020
/
Published: 24 November 2020
(This article belongs to the Special Issue Ciguatoxins)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The I–L ring system found in all the Pacific ciguatoxins has been prepared from a tricyclic precursor in a highly stereoselective manner. Subtle differences in the reactivity of the enones present in the seven- and eight-membered rings of the tricyclic ether starting material have been exploited to allow selective protection of the enone in the eight-membered ring. Subsequent distereoselective allylation of the seven-membered ring has been accomplished by a palladium-mediated Tsuji-Trost reaction. The K-ring methyl and hydroxyl groups have been installed in a highly stereoselective manner by sequential conjugate reduction and enolate oxidation reactions. Ring L has been constructed by a use of a novel relay ring-closing metathesis reaction to complete the tetracyclic framework, which possesses the functionality necessary for elaboration of rings I and L and the introduction of ring M.
Keywords:
pacific ciguatoxins; natural product; polycyclic ether; ring-closing metathesis; Tsuji-Trost allylationKey Contribution: A tetracyclic lactone that corresponds to the I–L fragment that is common to all of the Pacific ciguatoxins has been prepared form a readily available tricyclic bis-enone by a 10-step sequence that involves a novel strategy involving relay ring-closing metathesis.
1. Introduction
1.1. Structures and Bioactivities of the Ciguatoxins
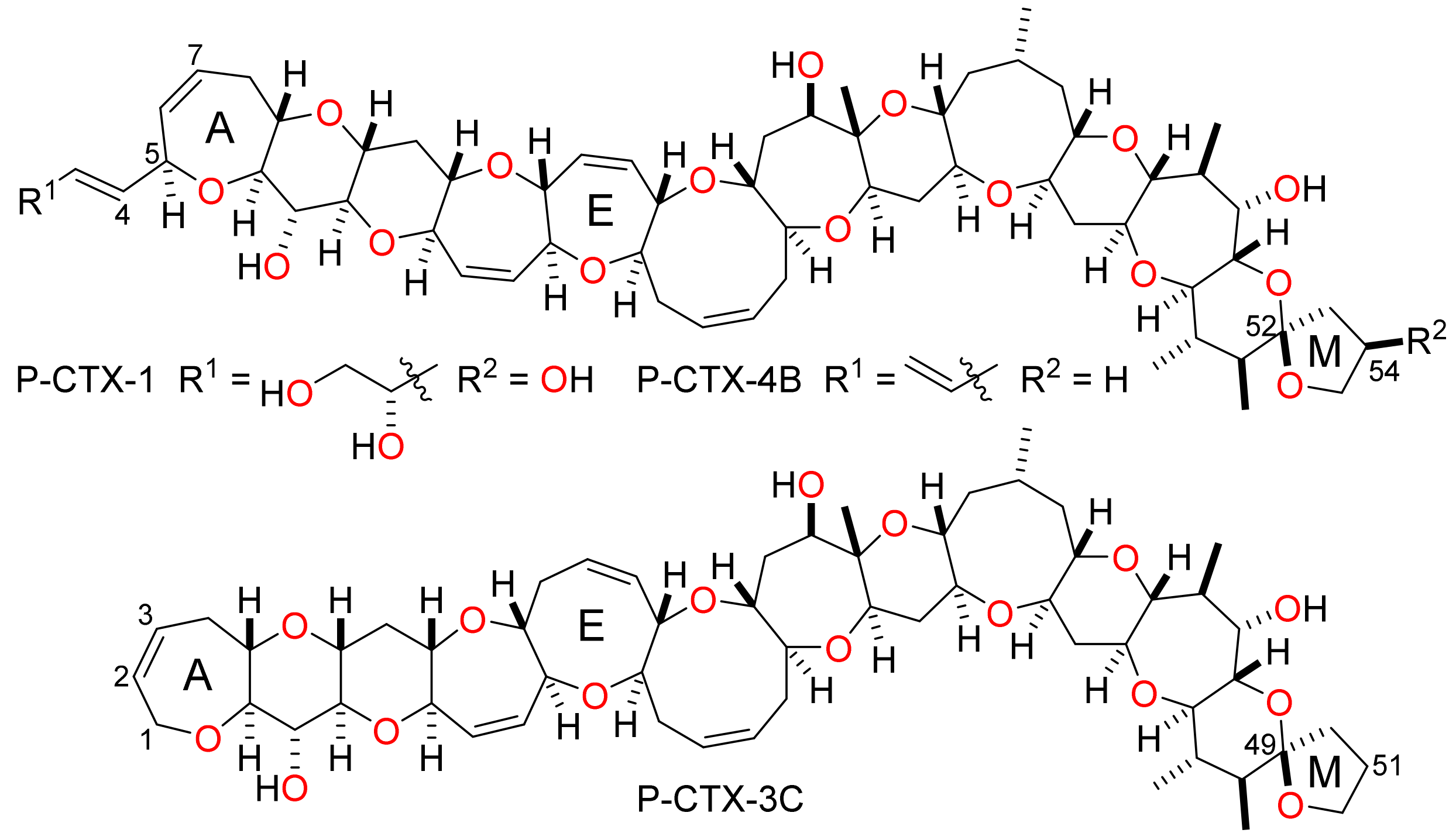
The ciguatoxins are a family of large and structurally complex fused polycyclic ether natural products of marine origin. Although the ciguatoxins had been implicated as the causative agents of ciguatera poisoning in humans since the 1960s, it was not until the late 1980s that the first structures of members of the ciguatoxin family—P-CTX-1 and P-CTX-4B—were fully elucidated by Yasumoto et al. (Figure 1) [1,2]. Subsequently, more than 30 other ciguatoxins have been isolated from marine dinoflagellates or from fish and marine organisms that accumulate the toxins by predation. The ciguatoxins can be grouped as Pacific, Caribbean or Indian according the ocean or sea where samples containing each toxin were first collected [3,4,5,6]. Pacific ciguatoxins are the most numerous and well-characterised of the ciguatoxins and this group accounts for more than three quarters of the ciguatoxins that are known [4,5,6]. Four Caribbean ciguatoxins have been characterized—C-CTX-1 and C-CTX-2 are equilibrating lactols and C-CTX-3 and C-CTX-4 are diastereomers that arise by reduction of C-CTX-1/C-CTX-2 [7,8]. A further eight Caribbean ciguatoxins have been detected by mass spectrometry but have yet to be characterised and the structures of the six ciguatoxins—I-CTX-1–6—isolated from samples collected in the Indian Ocean have yet to be elucidated [6,9,10].
The Pacific ciguatoxins can be divided into two distinct structural sub-groups. The first is exemplified by P-CTX-1 and P-CTX-4B, and the second by P-CTX-3C (Figure 1) [3,5]. Natural products in the first group have a four-carbon chain attached to the A-ring (C-5) and a seven-membered E-ring. This group comprises more than a dozen congeners that have a variety of structural modifications such as a hydroxyl substituent at C-54, inverted configuration at C-52, a seco M-ring or oxidation at the C-4 and/or C-7 positions. The second and larger group is based on a P-CTX-3C skeleton. Compounds in this group have an expanded (eight-membered) E-ring and lack the A-ring side chain found in P-CTX-1 and related ciguatoxins. Variations on the P-CTX-3C structure include A-ring and M-ring seco compounds, a C-49-epi compound (P-CTX-3B) and oxidised analogues in which there is a hydroxyl group at the C-51 position, and/or the A-ring alkene is replaced by a C-2 carbonyl group, a C-2 hydroxyl group or a 1,2-diol.
The ciguatoxins are extremely potent neurotoxins and some of them have been shown to have LD50 values of 4 μg/kg or lower in mice. Ingestion of fish or seafood that is contaminated by ciguatoxins causes a type of food poisoning known as ciguatera that can result in the victim experiencing a myriad of unpleasant and sometimes severe neurological (both peripheral and central nervous system), cardiovascular and gastrointestinal symptoms [11]. It has been estimated that tens or even hundreds of thousands of people suffer from ciguatera poisoning each year. Symptoms can persist for many months or even years and, in extreme cases, ciguatera food poisoning can be fatal. The pharmacological effects of the ciguatoxins result primarily from their ability to cause persistent activation of voltage-gated sodium channels in nerve membranes and thereby prevent the nerve repolarisation that is essential for normal nerve function [12,13]. There is also evidence that the ciguatoxins can block voltage-gated potassium channels [14].
1.2. Total Syntheses of Pacific Ciguatoxins
The ciguatoxins are daunting targets for total synthesis because of their size, their stereochemical complexity, the large number of rings they possess and the high proportion of synthetically challenging medium-sized cyclic ethers in their trans-fused polycyclic structures. The synthetic challenges presented by the ciguatoxins combined with their potent neurotoxic activities makes them highly alluring synthetic targets and many research groups have performed methodological studies in which substantial fragments or smaller sub-units of various ciguatoxins have been constructed. However, despite a continuous stream of highly innovative synthetic work spanning more than two decades, the ciguatoxins have proved to be very difficult to synthesise. The only successes in this area have been achieved by Hirama et al., who reported the first total synthesis of a ciguatoxin—P-CTX-3C—in 2001 followed by the total syntheses of 51-hydroxy-P-CTX-3C and P-CTX-1 in 2006 [15,16,17], and by Isobe et al., who completed a total synthesis of P-CTX-1 in 2009 [18]. These elegant and impressive syntheses are the only complete total syntheses of members of the ciguatoxin family of natural products to have been reported.
1.3. Proposed Synthetic Strategy and Previous Synthetic Work Concerning the Total Synthesis of P-CTX-3C and Related Ciguatoxins
We have been engaged in the development of new reactions, tactics and strategies for the rapid and efficient synthesis of fused polycyclic ether arrays for many years [19]. As part of this research program, we have invented and explored a variety of iterative and bidirectional strategies to assemble large fragments of fused polycyclic ether natural products of marine origin such as the brevetoxins, gambieric acids and ciguatoxins [20,21,22,23]. In our previous studies concerning the synthesis of P-CTX-3C, the A–E fragment of the natural product was assembled by a route in which ring-closing metathesis (RCM) reactions were used to construct the rings in an iterative manner [18]. We have also reported a novel bidirectional RCM approach to the synthesis of the tricyclic I–K fragment of P-CTX3-C from a simple monocyclic precursor and now present the results of studies concerning its elaboration to give an I–L fragment that can serve as an advanced intermediate for the preparation of the terminal fragment of any of the Pacific ciguatoxins [24].
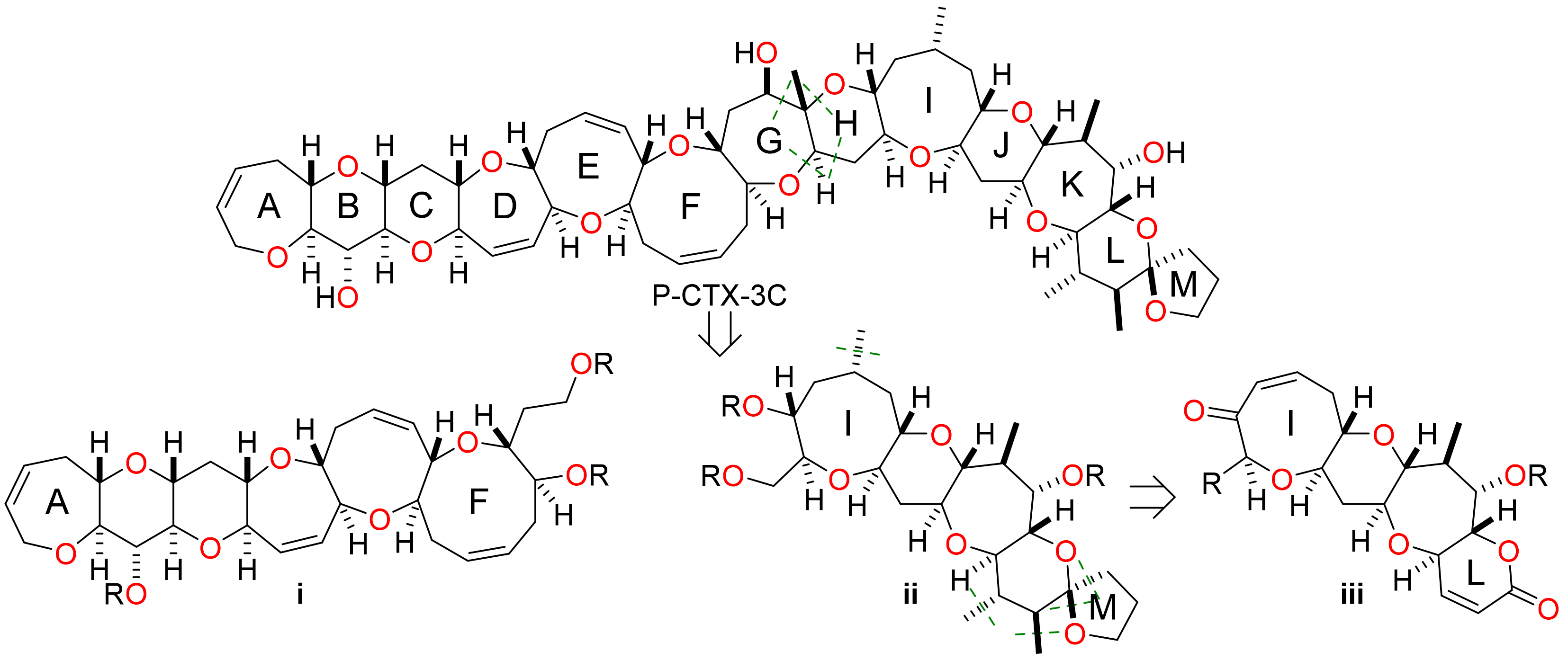
Retrosynthesis of P-CTX-3C commences with disconnection through rings G and H to give two fragments—the hexacyclic A–F fragment i and the pentacyclic I–M fragment ii—that are of similar size and structural complexity (Figure 2). The focus of the synthetic work described herein was the I–M fragment ii. Disconnection of this pentacyclic system by removal of the methyl substituents in rings I and scission of the M-ring spiroacetal leads to the tetracyclic fragment iii. This intermediate contains an α,β-unsaturated ketone in ring I and an α,β-unsaturated lactone in ring L to enable introduction of the methyl substituents by conjugate addition in the case of ring I and sequential conjugate addition and enolate alkylation in the case of ring L.
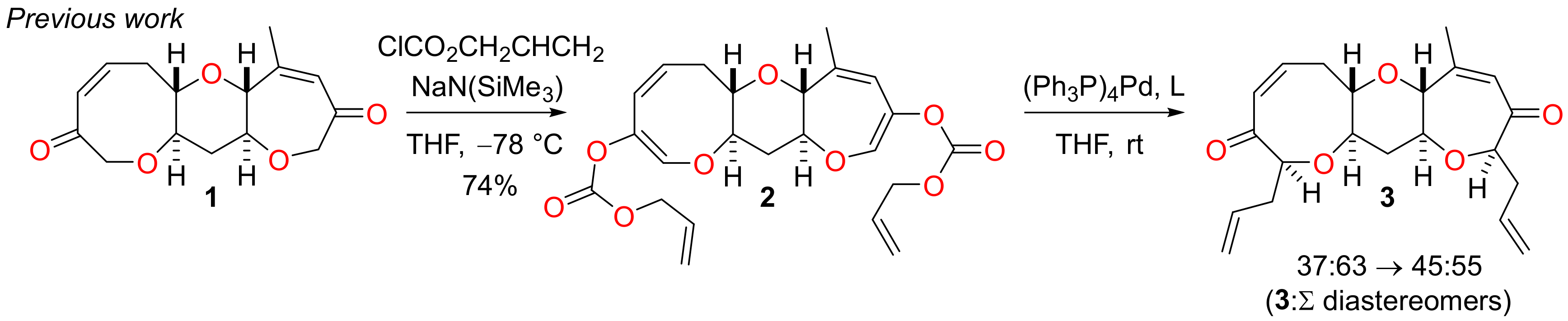
In previous work concerning the synthesis of the I–M fragment of P-CTX-3C and related ciguatoxins, we have demonstrated that the tricyclic sub-unit 1 can be prepared from a simple tetrahydropyranyl precursor by double ring-closing metathesis (RCM) and that subsequent bidirectional functionalisation of the diketone 1 is possible by sequential double allyl enol carbonate formation and double palladium-mediated Tsuji-Trost allylation (Scheme 1) [24,25]. Although it was possible to direct the double Tsuji-Trost reaction of the substrate 2 to give just two of the four possible diastereomeric products by the choice of a suitable chiral ligand, we were unable to identify a catalyst that would deliver the required diketone 3 as the sole or even predominant isomer. Thus, it was necessary to revise the synthetic strategy in order to allow rings I and K to be differentiated and functionalised independently thereafter. The following discussion provides an account of construction of the I–L sub-unit by such an approach.
2. Results and Discussion
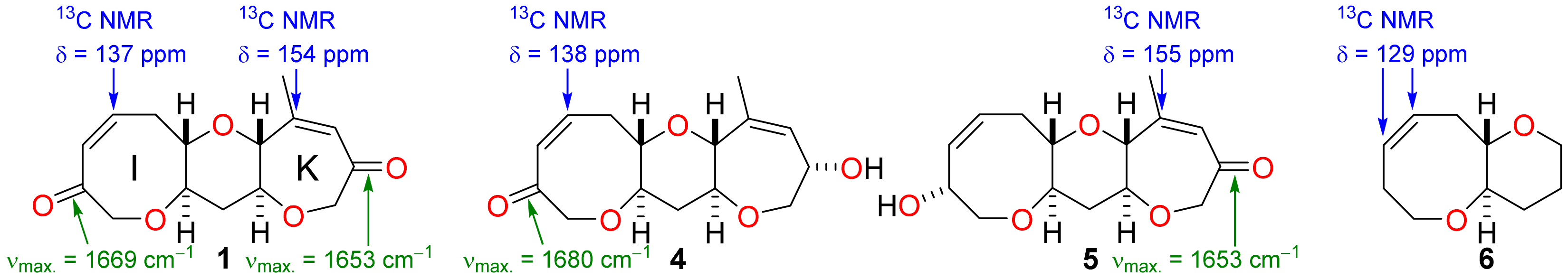
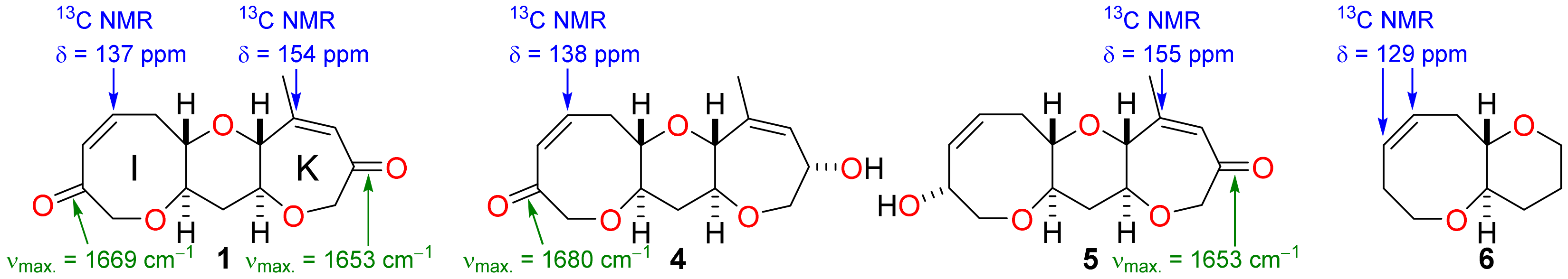
It was not possible to perform simultaneous and diastereoselective bidirectional functionalisation of the bis-enone 1 by double Tsuji-Trost allylation [24,25] and so it was necessary to elaborate rings I and K independently in order to extend the fused polyether framework (Scheme 2). The tricyclic bis-enone 1 is quasi-symmetric and so independent functionalisation of rings I and K by discrimination between reactive functional groups appears to be a challenging task at first glance. However, spectroscopic analysis of the bis-enone 1 reveals subtle structural differences between the enone functionality in rings I and K. In the IR spectrum of 1, the carbonyl group in ring I has a stretching frequency of 1669 cm−1 whereas the carbonyl group in ring K has a stretching frequency of 1653 cm−1—the identities of the carbonyl peaks in the IR spectrum of 1 were established unambiguously by comparison to data for the hydroxy ketones 4 and 5, produced by partial reduction of the diketone 1 (Figure 3). The IR data suggest that the degree of enone conjugation in the eight-membered I-ring is significantly lower than that in the seven-membered K-ring. This finding was further supported by 13C NMR spectroscopy where the β-carbon of the enone in ring I has a chemical shift (137 ppm) that is more typical of an unconjugated alkene at this position in an eight-membered cyclic ether, as can be seen by comparing the chemical shifts for the alkene carbons in the simple fused bicyclic ether 6 [26], whereas the corresponding carbon in ring K has a chemical shift (154 ppm) for the enone β-carbon that is consistent with a fully conjugated enone. The significant differences in the degree of conjugation in the enone groups suggested that it might be possible to functionalise them selectively by exploiting differences in their chemical reactivity. Indeed, there are pertinent examples of highly selective acid-mediated acetal formation in polycyclic substrates that contain both an enone and unconjugated ketone and so this approach was used to protect the I-ring carbonyl group selectively [27].
Completely selective protection of the I-ring carbonyl group as a dioxolane was accomplished by reaction of the bis-enone 1 with bis(trimethylsilyl)ethylene glycol in the presence of trimethylsilyl trifluoromethanesulfonate (Scheme 2) [28]. The enone 7 was then converted into the allyl enol carbonate 8 by deprotonation with sodium bis(trimethylsilyl)amide and enolate trapping with allyl chloroformate. Tsuji-Trost allylation was then performed thereafter by treatment of the allyl enol carbonate 8 with the complex generated from tetrakis(triphenylphosphine) palladium(0) and the (R)-t-butyl-PHOX ligand (9) in situ [24,25,29]. The reaction was highly stereoselective (>95:5 dr; minor isomer not detectable by 1H NMR analysis) and delivered the enone 10 in 95% yield (Scheme 2). The stereochemical outcome of this and subsequent diastereoselective reactions was confirmed by 1H NMR NOE analysis (for details, see Supplementary Materials).
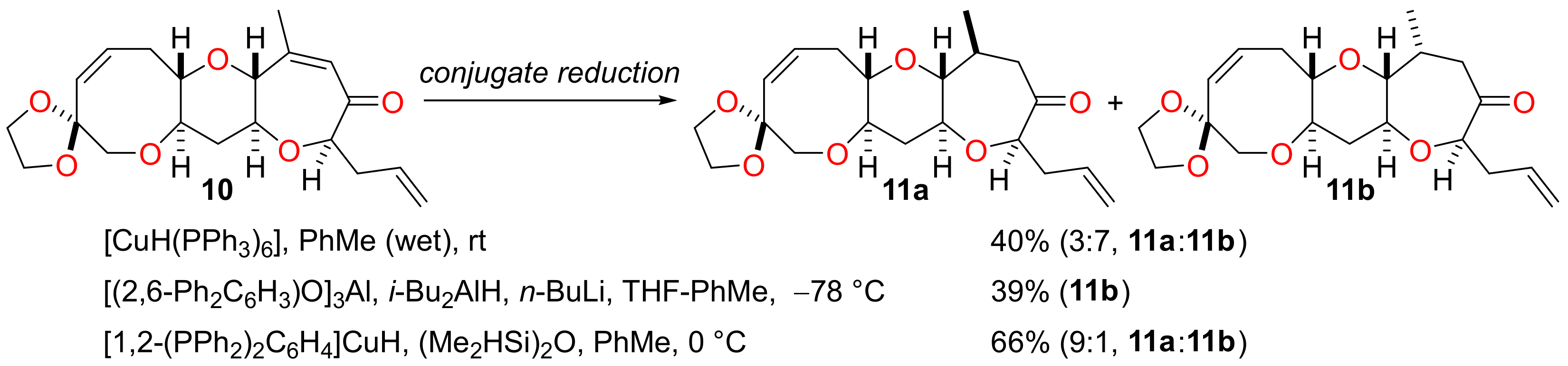
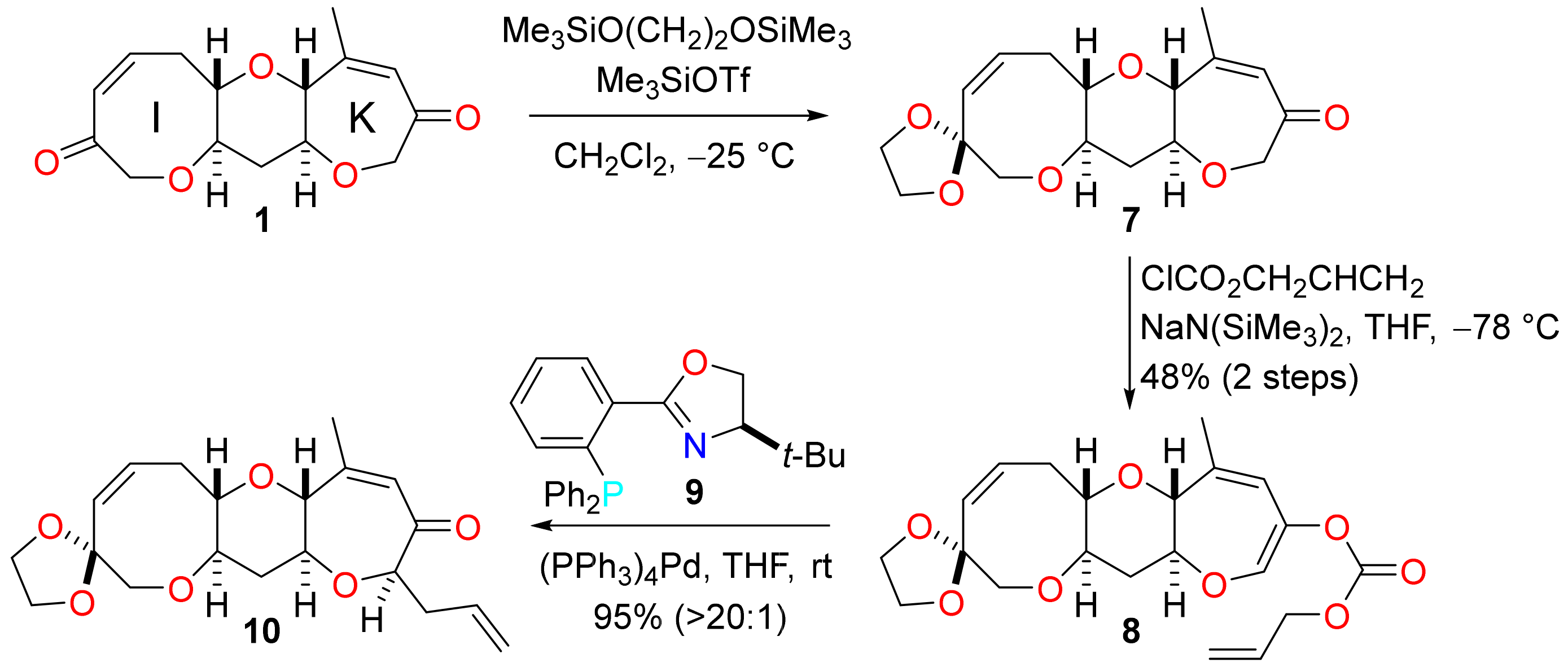
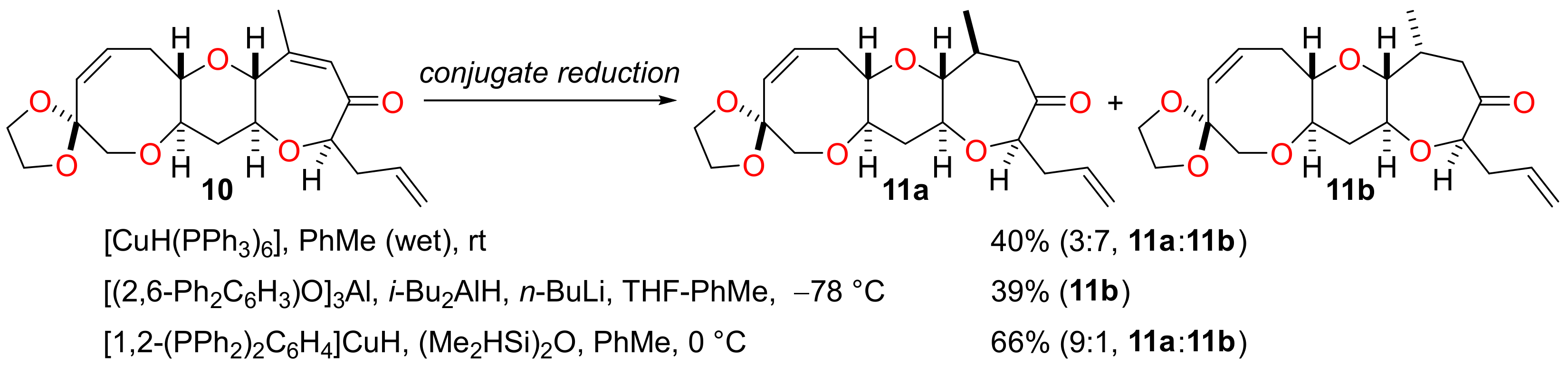
Ring K was elaborated further by conjugate reduction of the enone (Scheme 3). Reduction of the enone 10 with Stryker’s reagent [30] resulted in a mixture of the diastereomeric ketones 11a and 11b (3:7 ratio) in which the required diastereomer 11a was the minor component. This reaction also suffered from the problem that it did not proceed to completion, even when an excess of the reducing agent was used. A conjugate reduction protocol developed by Yamamoto et al. was deployed with the expectation that it would solve these reactivity and selectivity problems [31]. Coordination of the ketone to aluminium tris(2,6-diphenylphenoxide) followed by treatment with the reductant generated by the reaction of diisobutylaluminium hydride with n-butyllithium afforded the ketone 11b in 39% yield and the diketone (13% yield) resulting from cleavage of the acetal protecting group of 11b; none of the required diastereomer 11a was obtained from the reaction. Stereocontrolled conjugate reduction of the enone 10 to give the required ketone was finally performed in good yield by reaction with the ‘hot’ Stryker’s reagent generated from 1,2-bis(diphenylphosphino)benzene, copper(II) acetate hydrate and 1,1,3,3-tetramethyldisiloxane as described by Lipshutz and co-workers [32]. Reduction of the enone 10 with this reagent at 0 °C in toluene afforded a diastereomeric mixture of the ketones 11a and 11b (combined yield of 66%; 9:1 ratio) in which the required isomer was the major product. The diastereomeric ketones 11a and 11b could not be separated by standard silica gel chromatography.
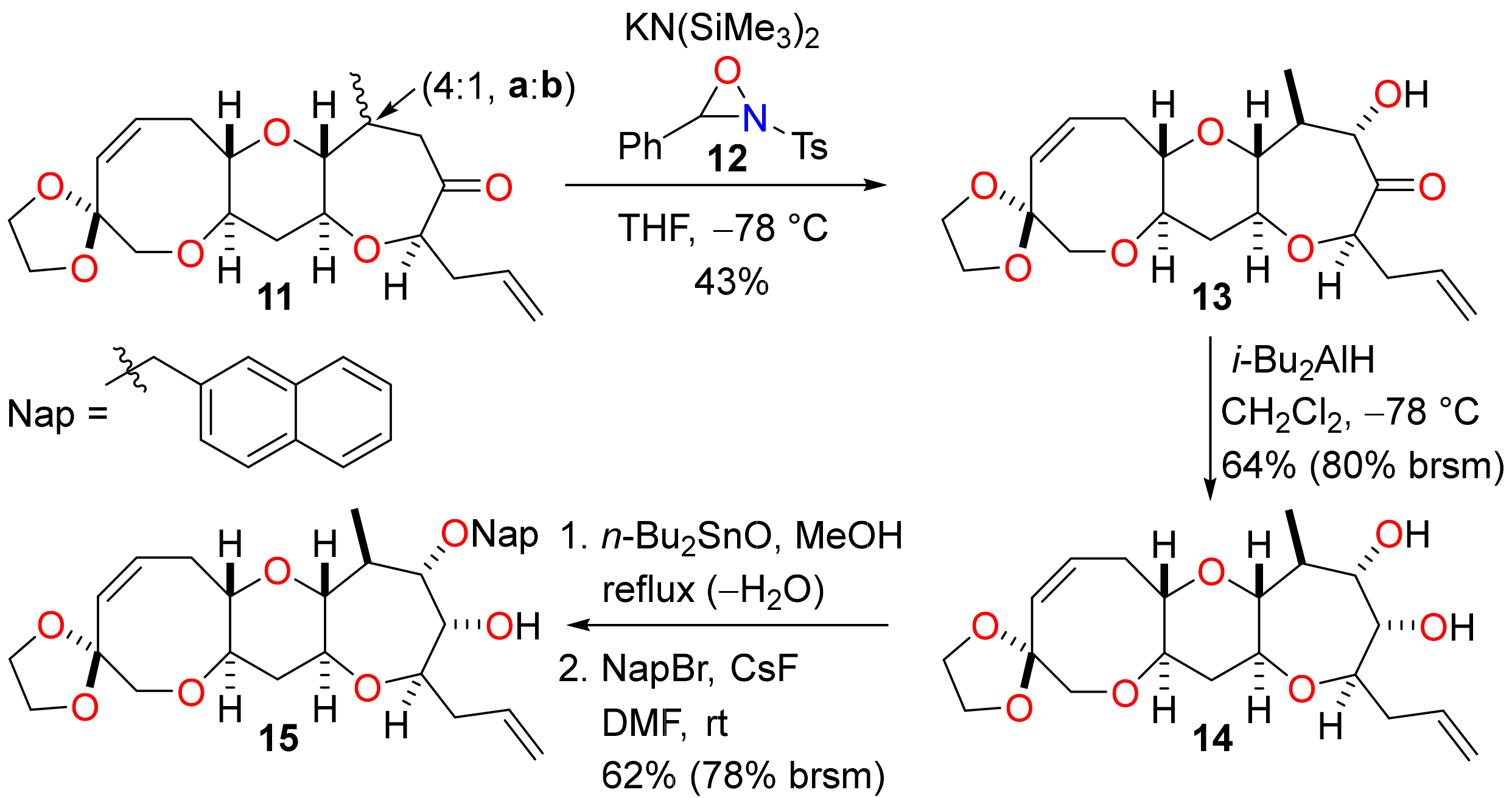
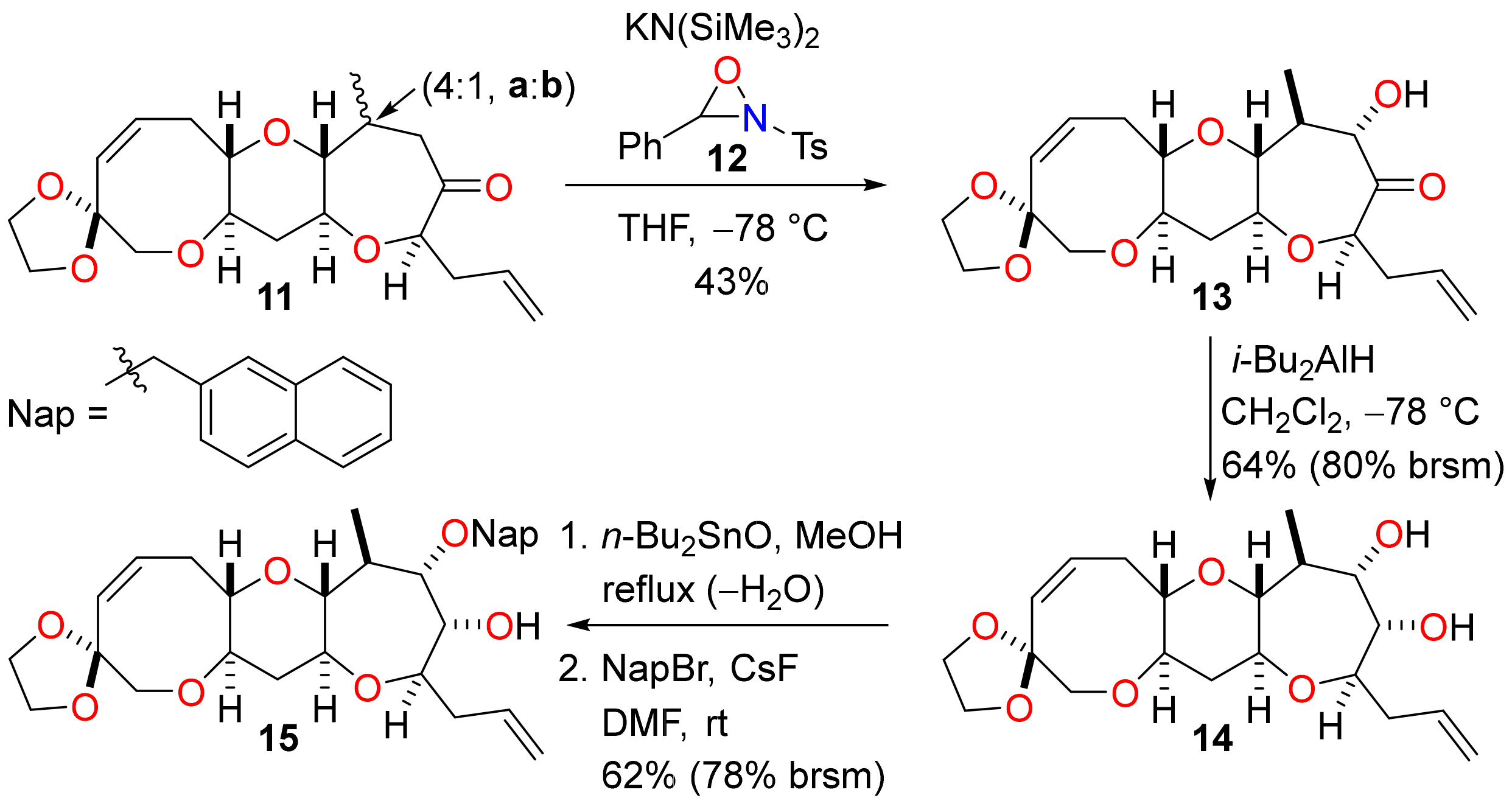
Stereoselective conjugate reduction of the enone 10 enabled the requisite K-ring hydroxyl group to be installed by enolate oxidation (Scheme 4). Regioselective deprotonation of a mixture of ketones 11a and 11b (4:1 ratio) with potassium bis(trimethylsilyl)amide and reaction of the resulting enolate with the N-sulfonyl oxaziridine 12 [33,34] produced the α-hydroxy ketone 13 in 43% yield (54% when adjusted to account for the isomer ratio of starting material 11) and with a very high level of diastereocontrol. The same transformation was performed by generation of the triethylsilyl enol ether and subsequent Rubottom oxidation with 3-chloroperoxybenzoic acid [35], but poor yields were obtained and so this sequence was not a viable alternative to direct enolate oxidation. The α-hydroxy ketone 13 was then subjected to stereoselective reduction with diisobutylaluminium hydride to give the diol 14 [36]. The stereochemical outcome of the reaction can be explained by a reaction mechanism in which complexation of the aluminium reagent to the hydroxyl and carbonyl groups produces a cyclic chelate and addition of diisobutylaluminium hydride occurs from the top face of the molecule (as drawn). At this stage, it was necessary to differentiate the hydroxyl groups of the syn diol 14 to enable construction of ring L. Selective protection of the hydroxyl group adjacent to the K-ring methyl substituent was accomplished by reaction of the diol 14 with dibutyltin oxide to give a cyclic dialkylstannylene acetal followed by regioselective fluoride-mediated alkylation with 2-bromomethylnaphthalene [37,38]. The Nap-protected alcohol 15 was obtained in reasonable yield and a small amount of unreacted diol 14 was recovered from the reaction.
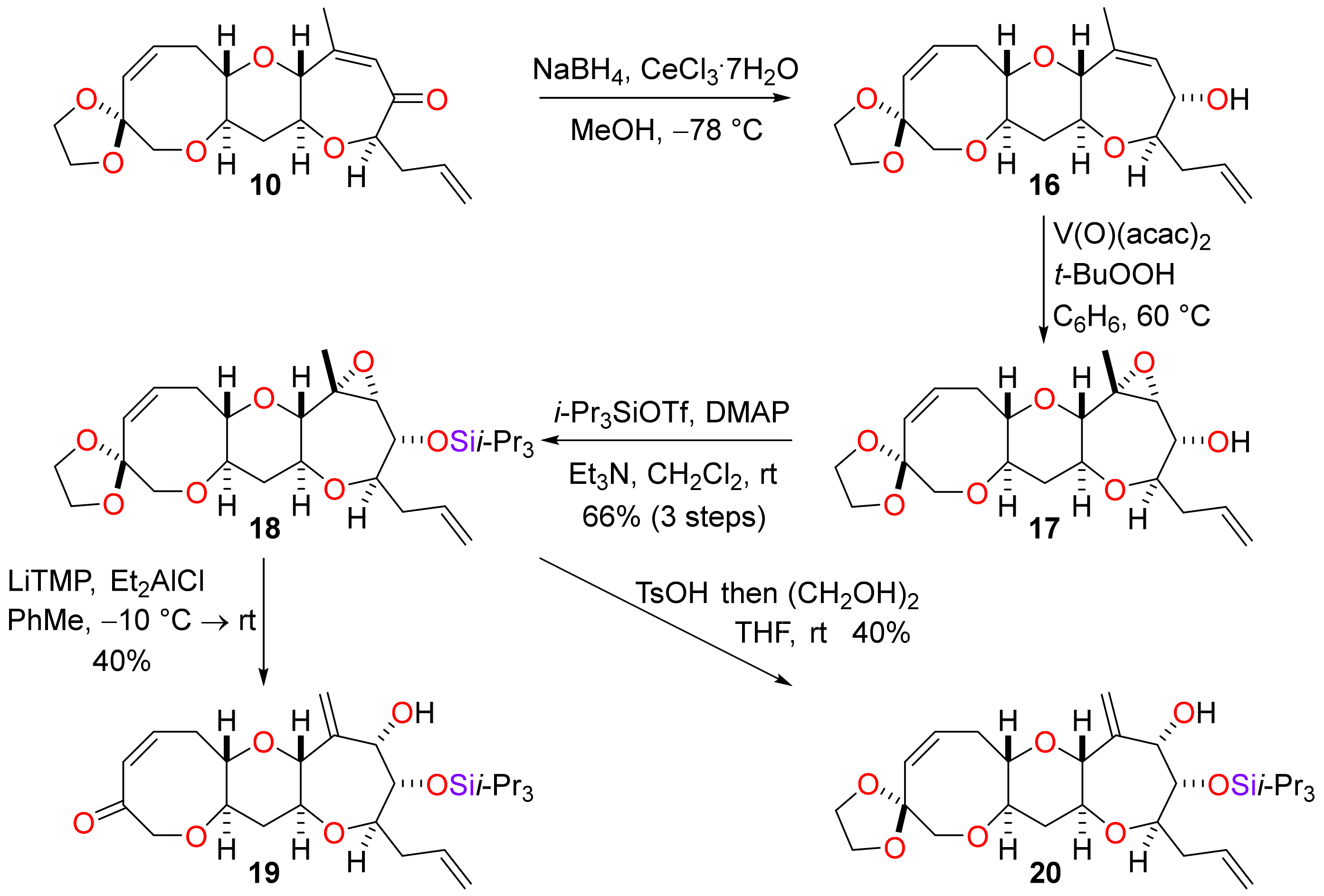
An alternative approach to functionalisation of ring K that did not involve sequential conjugate reduction and enolate oxidation was explored (Scheme 5). The tricyclic enone 10 was first subjected to Luche reduction which proceeded to give the allylic alcohol 16 in excellent yield and with a high level of diastereocontrol. Directed epoxidation of the allylic alcohol with t-butyl hydroperoxide mediated by vanadyl acetylacetonate produced the epoxide 17 in a highly stereoselective manner. Protection of the hydroxyl group as a triisopropylsilyl ether afforded the epoxide 18 in a yield of 66% from the enone 10. It was now possible to rearrange the epoxide to give an allylic alcohol by reaction of the epoxide 18 with lithium 2,2,6,6-tetramethylpiperidide in the presence of the Lewis acid diethylaluminium chloride [39]. The allylic alcohol 19, in which the acetal on ring I had been cleaved under the reaction conditions to reveal the enone, was obtained. Rearrangement of the epoxide 18 by treatment with p-toluenesulfonic acid was also successful; in this case ethylene glycol was added to the reaction mixture to ensure that the acetal protecting group was retained in product 20. In both cases, the yield of the rearranged product was modest. Unfortunately, subsequent chemoselective and stereoselective hydrogenation of the exocyclic alkene to produce the K-ring methyl substituent proved to be difficult to accomplish. It was expected that selective directed hydrogenation of the allylic alcohol 20 would be possible by use of Crabtree’s catalyst [40]. However, the hydrogenation reaction did not take place when a catalyst loading of 1 mol % was used and decomposition of the substrate occurred when higher catalysts loadings (≥10 mol %) were employed. When Wilkinson’s catalyst was used to hydrogenate the alkoxide generated by deprotonation of the allylic alcohol 20 with sodium hydride in the presence of 15-crown-5 [41,42], the terminal alkene in the side chain was reduced instead of the exocyclic 1,1-disubstituted alkene in ring K.
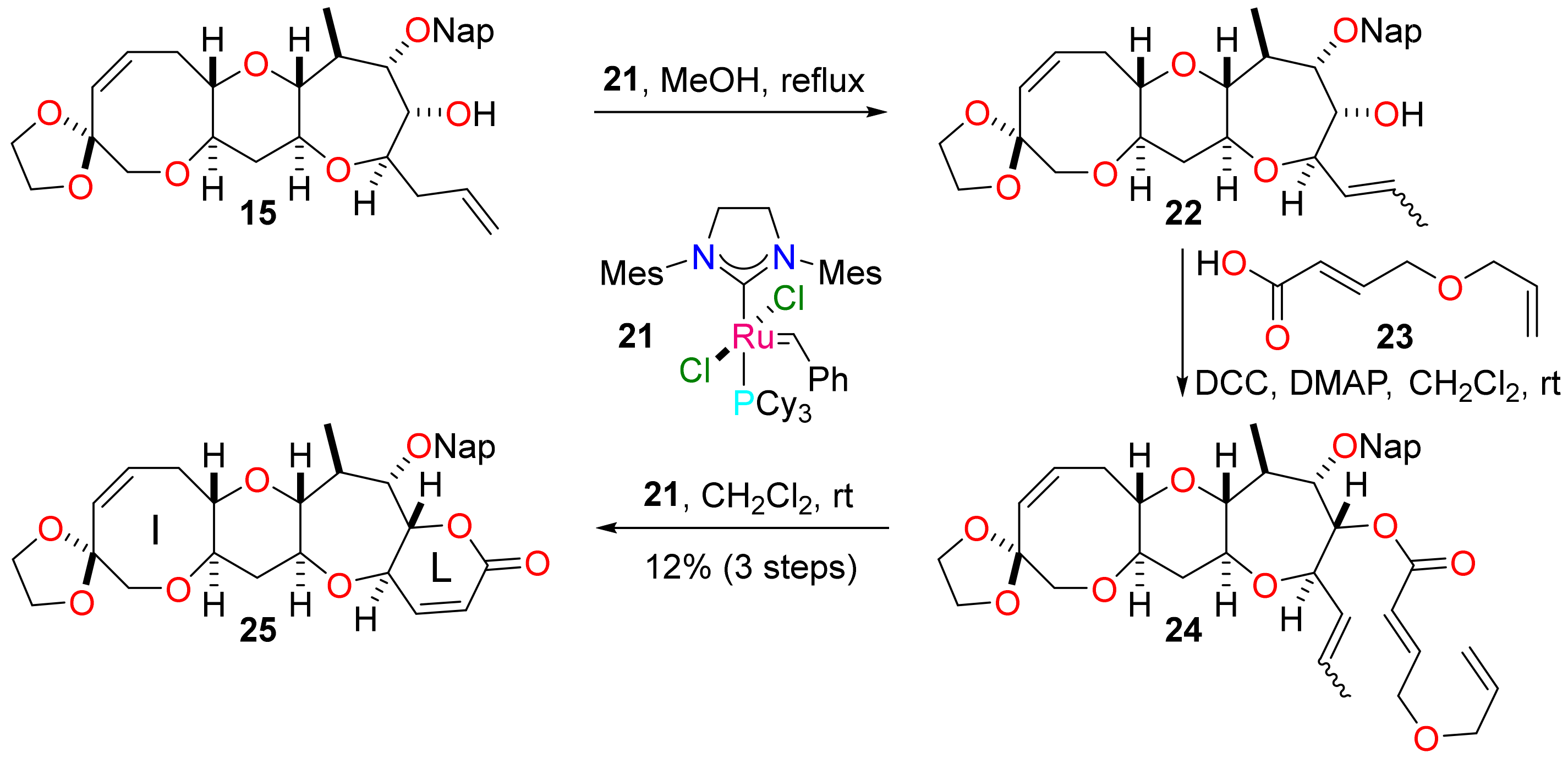
Construction of ring L to complete the tetracyclic ciguatoxin I–L core framework was accomplished as shown in Scheme 6. The allyl side chain in the alcohol 15 was subjected to isomerisation by treatment with the ruthenium complex 21 in methanol at reflux to give the alcohol 22 as a mixture of alkene isomers (E/Z) along with some side-products [43]. At this juncture, construction of ring L as a pentenolide by conversion of the alcohol 22 into a simple acrylate ester followed by direct RCM was considered. However, there were concerns regarding the rate of initiation of the metathesis reaction because the substrate would contain an acrylate and an internal alkene branched at the α position; consequently, a relay RCM reaction was employed as a precaution [44]. Subjection of the alcohol 22 to Steglich esterification with the known carboxylic acid 23 [45] delivered the relay metathesis precursor 24 and subsequent treatment with the ruthenium complex 21 resulted in relay RCM to give the lactone 25 corresponding to the tetracyclic framework found in the I–L ring system of the Pacific ciguatoxins. The modest yield for the three-step sequence implies an average of 50% per step; it can be accounted for by the scale on which the reactions were performed and by-product formation during isomerisation (15 → 22). The unsaturation in ring L would enable subsequent introduction of the requisite methyl substituents by conjugate addition with a suitable methyl nucleophile and trapping of the resulting enolate with a methyl electrophile [46].
3. Conclusions
The synthesis of the I–L ring system of the Pacific ciguatoxins has been accomplished in 10 steps starting from the tricyclic bis-enone 1. Elaboration of ring K by stereoselective Tsuji-Trost allylation was accomplished after selective protection of the I-ring enone by acetal formation. The configuration at the methyl-bearing stereogenic centre in ring K was controlled during conjugate reduction of the enone and the hydroxyl substituent was installed in a highly stereoselective manner by enolate oxidation according to the Davis procedure. Stereoselective ketone reduction followed by selective protection of the resulting 1,2-diol then allowed the metathesis trigger to be attached to the K-ring by esterification of the hydroxyl group. Relay RCM, mediated by the Grubbs second generation ruthenium complex, resulted in formation of ring L and completed the tetracyclic framework with the functionality necessary for subsequent elaboration of ring I, installation of the methyl substituents in ring L and construction of ring M.
4. Materials and Methods
Air- and moisture-sensitive reactions were performed under an atmosphere of argon in flame-dried apparatus. Tetrahydrofuran (THF), toluene, dichloromethane and diethyl ether were purified using a Pure-SolvTM 500 Solvent Purification System. Other dry solvents and starting materials were obtained from commercial sources and used as received unless stated otherwise. Petroleum ether (pet. ether) used for column chromatography was the 40–60 °C fraction. Triethylamine and 2,2,6,6-tetramethylpiperidine were distilled and stored under argon prior to use. n-Butyllithium solutions were titrated against diphenylacetic acid to obtain accurate molarity. 4 Å molecular sieves were oven dried prior to use.
Reactions were monitored by thin layer chromatography (TLC) using Merck silica gel 60 covered aluminium-backed plates F254. TLC plates were visualised under UV light and stained using potassium permanganate solution or acidic ethanolic anisaldehyde solution. Flash column chromatography was performed with silica gel (Geduran Si 60 35–70 µm) as solid support.
IR spectra were recorded using a Shimadzu FT IR-8400S ATR instrument (Shimadzu UK, Milton Keynes, UK). The IR spectrum of each compound (solid or liquid) was acquired directly on a thin layer at ambient temperature.
NMR spectra were recorded on Bruker Avance III 400 MHz and 500 MHz spectrometers (Bruker UK, Coventry, UK) at ambient temperature. 1H NMR chemical shifts are reported in ppm relative to CHCl3 (7.26) or CDCl2H (5.32) on the δ scale followed by integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br. = broad, app. = apparent or a combination of these) and coupling constant(s) J (Hz). 13C NMR spectra were recorded at 101 MHz and 126 MHz and chemical shifts are reported in ppm relative to CDCl3 (77.16) or CD2Cl2 (54.00) on the δ scale.
High resolution mass spectra (HRMS) were recorded by the University of Glasgow mass spectrometry service using a JEOL MStation JMS-700 instrument [positive electron impact ionisation (EI+)] (JEOL Ltd., Tokyo, Japan) or a Bruker micrOTOF-Q instrument [positive ion electrospray (ESI+)] (Bruker UK, Coventry, UK).
Optical rotations were recorded with an error of ≤ ±0.1 using an automatic polarimeter Autopol V (Rudolph Research Analytical, Hackettstown, USA) and melting points were recorded with an Electrothermal IA 9100 apparatus (Cole-Parmer, Stone, UK).
(1′S,3′R,9′S,11′R,15′Z)-4′-Methyl-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo-[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-one (7)
To a solution of the bis-enone 1 (870 mg, 3.13 mmol) in dichloromethane (63 mL) at −60 °C was added, 1,2-bis(trimethylsiloxy)ethane (0.81 mL, 3.3 mmol) and trimethylsilyl trifluoromethanesulfonate (0.06 mL, 0.3 mmol). The solution was allowed to warm to −25 °C and stirred for 16 h. The reaction mixture was diluted with diethyl ether (252 mL) and the mixture was washed with sat. aq. ammonium chloride (2 × 100 mL) and brine (100 mL). The organic phase was dried (MgSO4) and concentrated under reduced pressure to afford the acetal 7 (960 mg) as a brown oil. The unpurified acetal was used directly in the next reaction. For analytical purposes, a sample was purified by flash column chromatography (5–30% ethyl acetate in pet. ether) to afford 7 as a colourless solid. M.p. 145–146 °C; Rf = 0.43 (60% ethyl acetate in pet. ether); [α]D +22.6 (c = 0.50 in CHCl3, 29 °C); νmax 3028, 2953, 2932, 2884, 1655, 1443, 1348, 1115, 1086, 1074, 943, 893, 768 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.90 (1H, dq, J = 1.2, 1.2 Hz), 5.82 (1H, ddd, J = 11.6, 9.5, 7.5 Hz), 5.63 (1H, d, J = 11.6 Hz), 4.23 (1H, d, J = 18.3 Hz), 4.13 (1H, d, J = 18.3 Hz), 3.94–3.83 (4H, m), 3.67 (1H, ddq, J = 9.0, 1.2, 1.2 Hz), 3.61 (1H, d, J = 12.7 Hz), 3.50 (1H, d, J = 12.7 Hz), 3.47 (1H, ddd, J = 11.7, 9.3, 4.4 Hz), 3.33 (1H, ddd, J = 9.3, 5.8, 4.0 Hz), 3.30 (1H, ddd, J = 11.7, 9.0, 4.7 Hz), 2.89 (1H, ddd, J = 13.5, 9.5, 4.0 Hz), 2.56 (1H, ddd, J = 13.5, 7.5, 5.8 Hz), 2.36 (1H, ddd, J = 12.1, 4.7, 4.4 Hz), 1.97 (3H, dd, J = 1.2, 1.2 Hz), 1.61 (1H, ddd, J = 12.1, 11.7, 11.7 Hz); 13C NMR (101 MHz, CDCl3) δ 201.4, 155.1, 133.9, 130.3, 127.1, 108.0, 82.8, 82.5, 78.1, 77.6, 77.4, 74.2, 64.8, 64.6, 38.3, 30.1, 22.9; HRMS (ESI+) [C17H22O6Na]+ found 345.1299, [M + Na]+ calcld. 345.1309.
(1′S,3′R,9′S,11′R,15′Z)-4′-Methyl-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo-[9.6.0.03,9]heptadecane)-4′,6′,15′-trien-6′-yl prop-2-en-1-yl carbonate (8)
To a solution of the acetal 7 in THF (57 mL) was added, allyl chloroformate (1.58 mL, 14.9 mmol). The resultant solution was cooled to −78 °C and sodium bis(trimethylsilyl)amide (3.0 mL of a 2.0 M solution in THF, 6.0 mmol) was added dropwise. The reaction mixture was stirred for 4 h, diluted with 5% aq. potassium dihydrogen phosphate (240 mL) and the mixture was extracted with diethyl ether (3 × 60 mL). The combined organic extracts were washed with brine (3 × 60 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was dissolved in toluene (100 mL) and then concentrated under reduced pressure and the procedure was repeated. Residual material was purified by flash column chromatography (5–40% diethyl ether in pet. ether) to give the allylic carbonate 8 (610 mg, 48% over 2 steps) as a colourless oil. Rf = 0.57 (60% ethyl acetate in pet. ether); [α]D +96.1 (c = 1.00 in C6H6, 29 °C); νmax 2951, 2891, 1759, 1663, 1451, 1364, 1246, 1225, 1198, 1126, 1090, 1055, 995, 947, 770 cm−1; 1H NMR (500 MHz, CD2Cl2) δ 6.62 (1H, d, J = 1.6 Hz), 5.96 (1H, ddt, J = 17.3, 10.5, 5.7 Hz), 5.86 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.67 (1H, d, J = 11.5 Hz), 5.54 (1H, dqd, J = 1.6, 1.6, 1.3 Hz), 5.37 (1H, ddt, J = 17.3, 1.4, 1.4 Hz), 5.29 (1H, dddd, J = 10.5, 1.4, 1.4, 1.4 Hz), 4.64 (2H, ddd, J = 5.7, 1.4, 1.4 Hz), 3.95–3.87 (4H, m), 3.74 (1H, ddq, J = 7.9, 1.3, 1.3 Hz), 3.65 (1H, ddd, J = 11.4, 7.9, 4.9 Hz), 3.63 (1H, d, J = 12.8 Hz), 3.54 (1H, d, J = 12.8 Hz), 3.52 (1H, ddd, J = 11.8, 9.4, 4.4 Hz), 3.32 (1H, ddd, J = 9.4, 5.7, 4.1 Hz), 2.92 (1H, ddd, J = 13.4, 9.6, 4.1 Hz), 2.56 (1H, ddd, J = 13.4, 7.5, 5.7 Hz), 2.49 (1H, ddd, J = 12.1, 4.9, 4.4 Hz), 1.89 (3H, dd, J = 1.6, 1.3 Hz), 1.67 (1H, ddd, J = 12.1, 11.8, 11.4 Hz); 13C NMR (126 MHz, CD2Cl2) δ 155.0, 141.2, 138.5, 135.0, 134.2, 132.0, 130.7, 119.3, 118.8, 108.4, 81.8, 80.1, 78.0, 77.9, 74.6, 69.5, 65.1, 65.0, 38.7, 30.4, 21.9; HRMS (ESI+) [C21H26O8Na]+ found 429.1507, [M + Na]+ calcld. 529.1520.
(1′S,3′R,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-one (10)
A mixture of tetrakis(triphenylphosphine)palladium(0) (67 mg, 0.058 mmol, 5 mol%) and (4R)-t-butyl-2-[2-(diphenylphosphino)phenyl]-4,5-dihydroxazole (9) (56 mg, 0.14 mmol, 13 mol%) was dissolved in THF (20 mL). The solution was stirred for 45 min and a solution of the allylic carbonate 8 (471 mg, 1.16 mmol) in THF (38 mL) was added. The mixture was stirred at rt for a further 3.5 h and then adsorbed onto Celite. The crude material was dry loaded on to a column of silica gel and purified by flash column chromatography (5–20% ethyl acetate in pet. ether) to afford the enone 10 (398 mg, 95%, >20:1 dr) as a pale yellow oil. Rf = 0.57 (60% ethyl acetate in pet. ether); [α]D +15.0 (c = 0.50 in CHCl3, 30 °C); νmax 2949, 2888, 2855, 1657, 1443, 1279, 1111, 1088, 999, 949, 918 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.89 (1H, dq, J = 1.1, 1.1 Hz), 5.87 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.78 (1H, dddd, J = 17.0, 10.2, 6.8, 6.8 Hz), 5.70 (1H, d, J = 11.5 Hz), 5.12–5.00 (2H, m), 4.17 (1H, dd, J = 7.7, 4.4 Hz), 3.98–3.89 (4H, m), 3.68 (1H, ddq, J = 8.9, 1.1, 1.1 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.55 (1H, d, J = 12.7 Hz), 3.50 (1H, ddd, J = 11.7, 9.3, 4.4 Hz), 3.40 (1H, ddd, J = 9.3, 5.5, 4.1 Hz), 3.33 (1H, ddd, J = 11.3, 8.9, 4.7 Hz), 2.97 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.59 (1H, ddd, J = 13.6, 7.5, 5.5 Hz), 2.49 (1H, ddd, J = 14.3, 6.8, 4.4 Hz), 2.41 (1H, ddd, J = 12.2, 4.7, 4.4 Hz), 2.36 (1H, ddd, J = 14.3, 7.7, 6.8 Hz), 2.01 (3H, dd, J = 1.1, 1.1 Hz), 1.70 (1H, ddd, J = 12.2, 11.7, 11.3 Hz); 13C NMR (126 MHz, CDCl3) δ 203.6, 153.7, 134.0, 133.5, 130.4, 126.7, 117.7, 107.9, 86.5, 82.5, 82.5, 77.6, 77.0, 74.2, 64.8, 64.6, 38.2, 38.1, 30.1, 22.7; HRMS (ESI+) [C20H26O6Na]+ found 385.1606, [M + Na]+ calcld. 385.1622.
(1′S,3′R,4′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,⁹]heptadecan)-15′-en-6′-one (11a) and (1′S,3′R,4′R,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-en-6′-one (11b)
- (a)
- Conjugate reduction of10with of ‘hot’ Stryker’s reagent
To 1,2-bis(diphenylphosphino)benzene (491 mg, 1.10 mmol) and copper(II) acetate hydrate (220 mg, 1.10 mmol) was added degassed toluene (19 mL). The resultant mixture was stirred vigorously and sparged continuously with argon for 15 min. The mixture was then sonicated for a further 30 min, at which point a pale blue suspension had formed. Tetramethyldisiloxane (0.80 mL, 4.4 mmol) was added to the mixture and it was stirred vigorously for a further 30 min, at which point the suspension developed a green colour. After refrigeration for 16 h, a red stock solution (0.055 M) had formed.
To the enone 10 (18 mg, 50 µmol) was added ‘hot’ Stryker’s reagent (2.0 mL of a 0.055 M solution in toluene, 0.11 mmol). The solution was stirred at 0 °C for 48 h and then concentrated under reduced pressure. The residue was dissolved in THF (5 mL) and sat. aq. potassium fluoride (2 mL) was added. The biphasic mixture was stirred vigorously for 16 h and then diluted with water (10 mL). The mixture was extracted with diethyl ether (3 × 5 mL) and the combined organic extracts were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–20% ethyl acetate in pet. ether) to afford a diastereomeric mixture of the ketones 11a and 11b (12 mg, 66%, 9:1 a:b) as a colourless film.
- (b)
- Conjugate reduction of10with lithium n-butyl(diisobutyl)aluminium hydride
To a solution of 2,6-diphenylphenol (380 mg, 1.50 mmol) in toluene (2 mL) at 0 °C was added dropwise trimethylaluminium (0.25 mL of a 2.0 M solution in toluene, 0.50 mmol) and the mixture was stirred for 10 min. After gas evolution had subsided, a solution of the enone 10 (145 mg, 0.400 mmol) in toluene (1 mL) was added and the resultant solution cooled to −78 °C and stirred for 10 min.
To a solution of diisobutylaluminium hydride (0.60 mL of a 1.0 M solution in heptane, 0.60 mmol) in THF (2 mL) at 0 °C was added dropwise n-butyllithium (0.26 mL of a 2.33 M solution in hexane, 0.60 mmol) and solution was stirred for 10 min. The solution was added to the aforementioned toluene solution of the enone 10 and aluminium tris(2,6-diphenylphenoxide) at −78 °C and the mixture was stirred for 2 h at this temperature. The mixture was then diluted with diethyl ether (50 mL) and sat. aq. Rochelle salt (50 mL) was added and the mixture was stirred vigorously for 16 h. The phases were separated and the organic phase was washed with water (25 mL) and brine (25 mL), then dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (2.5–20% ethyl acetate in pentane) to afford the ketone 11b (57 mg, 39%) as a colourless solid and the diketone (17 mg, 12%), resulting from deacetalation of the ketone 11b, as a colourless film. 11a Rf = 0.20 (20% ethyl acetate in pet. ether); [α]D +117.5 (c = 1.00 in CHCl3, 27 °C); νmax 2080, 3028, 2959, 2934, 2916, 2876, 1713, 1643, 1260, 1088, 1059, 949, 912, 891, 802, 769 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.88 (1H, ddd, J = 11.6, 9.5, 7.5 Hz), 5.79 (1H, dddd, J = 17.1, 10.3, 6.9, 6.9 Hz), 5.67 (1H, d, J = 11.6 Hz), 5.11–5.04 (2H, m), 3.98–3.89 (4H, m), 3.80 (1H, dd, J = 7.9, 5.0 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.6, 9.3, 4.4 Hz), 3.28 (1H, ddd, J = 9.3, 5.8, 4.1 Hz), 2.96 (1H, ddd, J = 11.1, 9.3, 4.6 Hz), 2.89 (1H, ddd, J = 13.4, 9.5, 4.1 Hz), 2.88–2.78 (2H, m), 2.57 (1H, ddd, J = 13.4, 7.5, 5.8 Hz), 2.41–2.25 (3H, m), 2.12 (1H, dd, J = 11.5, 1.9 Hz), 1.69–1.57 (1H, m), 1.63 (1H, ddd, J = 12.4, 11.6, 11.1 Hz), 1.13 (3H, d, J = 6.6 Hz); 13C NMR (126 MHz, CDCl3) δ 215.2, 133.6, 133.1, 130.7, 118.0, 108.0, 86.5, 85.7, 81.6, 79.8, 77.9, 74.2, 64.8, 64.5, 45.1, 38.8, 37.2, 35.7, 30.1, 20.3; HRMS (ESI+) [C20H28O7Na]+ found 387.1766, [M + Na]+ calcld. 387.1778. 11b M.p. 108–110 °C; Rf = 0.25 (20% ethyl acetate in pet. ether); [α]D +87.1 (c = 2.00 in CHCl3, 29 °C); νmax 2957, 2936, 2884, 2855, 1711, 1641, 1288, 1090, 1061, 1042, 995, 949, 920, 770 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.87 (1H, ddd, J = 11.5, 9.6, 7.4 Hz), 5.80 (1H, dddd, J = 17.1, 10.2, 6.9, 6.9 Hz), 5.70 (1H, d, J = 11.5 Hz), 5.11–5.02 (2H, m), 3.98–3.90 (4H, m), 3.71 (1H, dd, J = 8.3, 4.2 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.41–3.34 (3H, m), 3.20 (1H, ddd, J = 11.1, 9.6, 5.0 Hz), 3.03–2.94 (1H, m), 3.00 (1H, dd, J = 11.7, 2.2 Hz), 2.50 (1H, ddd, J = 13.5, 7.4, 4.4 Hz), 2.42 (1H, ddddd, J = 13.4, 6.9, 4.2, 1.3, 1.3 Hz), 2.37 (1H, ddd, J = 12.1, 5.0, 4.2 Hz), 2.34–2.25 (3H, m), 1.65 (1H, ddd, J = 12.1, 11.3, 11.1 Hz), 0.86 (3H, d, J = 7.0 Hz); 13C NMR (126 MHz, CDCl3) δ 215.5, 133.9, 133.6, 130.9, 117.8, 107.8, 87.1, 83.1, 81.7, 77.7, 74.9, 74.4, 64.8, 64.6, 44.3, 38.5, 37.5, 31.9, 30.1, 12.3; HRMS (ESI+) [C20H28O6Na]+ found 387.1765, [M + Na]+ calcld. 387.1778.
(1′S,3′R,4′S,5′S,7′R,9′S,11′R,15′Z)-5′-Hydroxy-4′-methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-en-6′-one (13)
To a solution of the ketones 11a and 11b (18 mg of a 4:1 mixture, 50 µmol) in THF (2.9 mL) at −78 °C, a solution of potassium bis(trimethylsilyl)amide (0.10 mL of a 0.50 M solution in toluene, 50 µmol) was added and the mixture was stirred for 1 h. A solution of the oxaziridine 12 (14 mg, 50 µmol) in THF (1 mL) was then added to the mixture dropwise and the resulting mixture was stirred for 1 h. The reaction mixture was diluted with sat. aq. sodium thiosulfate (20 mL) and the extracted with diethyl ether (3 × 10 mL). The combined organic extracts were washed with sat. aq. ammonium chloride (10 mL) and brine (2 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–30% diethyl ether in pet. ether) to give the α-hydroxyketone 13 (8 mg, 43%) and recovered starting material 11a (2 mg, 11%) as colourless oils. Rf = 0.73 (60% ethyl acetate in pet. ether); [α]D +75.8 (c = 0.60 in CHCl3, 26 °C); νmax 3474, 2926, 2874, 2855, 1713, 1643, 1283, 1096, 1047, 949, 922, 770 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.88 (1H, ddd, J = 11.5, 9.5, 7.5 Hz), 5.78 (1H, ddd, J = 16.9, 9.8, 7.0 Hz), 5.67 (1H, d, J = 11.5 Hz), 5.13–5.06 (2H, m), 4.30 (1H, dd, J = 11.2, 6.4 Hz), 4.08 (1H, dd, J = 7.7, 5.0 Hz), 3.97–3.91 (4H, m), 3.66 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.7, 9.3, 4.5 Hz), 3.33 (1H, d, J = 6.4 Hz), 3.28 (1H, ddd, J = 9.3, 5.8, 4.2 Hz), 3.01–2.94 (2H, m), 2.90 (1H, ddd, J = 13.5, 9.5, 4.2 Hz), 2.58 (1H, ddd, J = 13.5, 7.5, 5.8 Hz), 2.48–2.32 (3H, m), 1.63 (1H, app. q, J = 11.7 Hz), 1.54–1.44 (1H, m), 1.28 (3H, d, J = 6.3 Hz); 13C NMR (101 MHz, CDCl3) δ 215.5, 133.6, 132.4, 130.6, 118.7, 108.1, 85.5, 83.6, 81.7, 79.8, 77.7, 75.1, 74.2, 64.8, 64.5, 43.2, 38.6, 37.9, 30.1, 15.3; HRMS (ESI+) [C20H28O7Na]+ found 403.1715, [M + Na]+ calcld. 403.1727.
(1′S,3′R,4′S,5′S,6′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-ene-5′,6′-diol (14)
To a solution of α-hydroxyketone 13 (31 mg, 81 µmol) in dichloromethane (2.9 mL) at −78°C, diisobutylaluminium hydride (0.32 mL of a 1.0 M solution in dichloromethane, 0.32 mmol) was added and the resultant solution stirred for 2 h. Further diisobutylaluminium hydride (0.32 mL of a 1.0 M solution in dichloromethane, 0.32 mmol) was added and the mixture was stirred for a further 2 h. The reaction mixture was diluted with sat. aq. Rochelle salt (20 mL) and ethyl acetate (20 mL) and stirred vigorously for 1 h. The phases were separated and the organic phase was washed with brine (2 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (20–100% diethyl ether in pet. ether) to afford the diol 14 (20 mg, 64%) and starting material 13 (6 mg, 19%) as colourless films. Rf = 0.30 (60% ethyl acetate in pet. ether); [α]D +18.4 (c = 0.50 in CHCl3, 27 °C); νmax 3447, 2926, 2874, 1647, 1456, 1285, 1090, 1042, 1013, 916, 770 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.90 (1H, ddd, J = 11.4, 9.6, 7.6 Hz), 5.85 (1H, dddd, J = 17.1, 10.2, 6.9, 6.9 Hz), 5.67 (1H, d, J = 11.4 Hz), 5.17–5.04 (2H, m), 3.94 (4H, m), 3.86 (1H br s), 3.71 (1H, d, J = 9.4 Hz), 3.65 (1H, d, J = 12.7 Hz), 3.56 (1H, ddd, J = 7.6, 5.5, 5.5 Hz), 3.55 (1H, d, J = 12.7), 3.48 (1H, ddd, J = 11.6, 9.3, 4.4 Hz), 3.37 (1H, ddd, J = 11.4, 9.3, 4.6 Hz), 3.22 (1H, ddd, J = 9.3, 5.9, 4.1 Hz), 2.88 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.72 (1H, dd, J = 9.3, 9.0 Hz), 2.59 (1H, ddd, J = 13.6, 7.6, 5.9 Hz), 2.39–2.24 (3H, m), 2.21 (1H, br s), 2.12 (1H, ddq, 9.4, 9.0, 6.7 Hz), 1.94 (1H, br s), 1.48 (1H, ddd, J = 12.1, 11.6, 11.4 Hz), 1.15 (3H, d, J = 6.7 Hz); 13C NMR (101 MHz, CDCl3) δ 134.8, 133.4, 130.8, 117.5, 108.2, 84.8, 82.7, 81.7, 78.3, 78.2, 77.7, 74.1, 73.4, 64.8, 64.5, 39.4, 39.0, 38.0, 30.3, 15.4; HRMS (ESI+) [C20H30O7Na]+ found 405.1871, [M + Na]+ calcld. 405.1884.
(1′S,3′R,4′S,5′S,6′R,7′R,9′S,11′R,15′Z)-4′-Methyl-5′-[(naphthalen-2-yl)methoxy]-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-15′-en-6′-ol (15)
To a solution of diol 14 (20 mg, 52 µmol) in methanol (5.2 mL), dibutyltin oxide (14 mg, 58 µmol) was added. The resultant suspension was stirred at reflux for 2 h to form a colourless solution. The mixture was concentrated under reduced pressure and dried by azeotropic distillation with toluene. The mixture was redissolved in DMF (5.2 mL) and 2-(bromomethyl)naphthalene (14 mg, 62 µmol) and caesium fluoride (9 mg, 0.06 mmol) were added. The reaction mixture was stirred for 16 h and then quenched with water carefully. The mixture was extracted with diethyl ether (3 × 10 mL) and the combined organic extracts were washed with brine (3 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (20–100% diethyl ether in pet. ether) to afford the naphthyl ether 15 (17 mg, 62%) and recovered diol 14 (4 mg, 20%) as colourless films. Rf = 0.39 (80% diethyl ether in pet. ether); [α]D +14.8 (c = 1.00 in CHCl3, 27 °C); νmax 3466, 2928, 2874, 2853, 1643, 1456, 1090, 949, 916, 1051, 818, 752 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.87–7.80 (3H, m), 7.76 (1H, s), 7.52–7.44 (3H, m), 5.91 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.83 (1H, dddd, J = 17.2, 10.3, 6.6, 6.6 Hz), 5.69 (1H, d, J = 11.5 Hz), 5.13–5.02 (2H, m), 4.74 (1H, d, J = 11.3 Hz), 4.65 (1H, d, J = 11.3 Hz), 3.98–3.92 (4H, m), 3.90 (1H, dd, J = 5.0, 2.2 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.59 (1H, ddd, J = 7.9, 5.0, 5.0 Hz), 3.55 (1H, d, J = 12.7), 3.53 (1H, dd, J = 8.8, 2.2 Hz), 3.50 (1H, ddd, J = 11.6, 9.5, 4.3 Hz), 3.49 (1H, ddd, J = 11.6, 9.4, 4.5 Hz), 3.24 (1H, ddd, J = 9.4, 5.7, 4.1 Hz), 2.92 (1H, ddd, J = 13.7, 9.6, 4.1 Hz), 2.78 (1H, dd, J = 9.5, 7.7 Hz), 2.59 (1H, ddd, J = 13.7, 7.5, 5.7 Hz), 2.39–2.19 (4H, m), 1.46 (1H, ddd, J = 11.6, 11.6, 11.6 Hz), 1.18 (3H, d, J = 6.9 Hz); 13C NMR (101 MHz, CDCl3) δ 135.4, 134.9, 133.5, 133.3, 133.2, 130.9, 128.4, 128.0, 127.9, 127.1, 126.4, 126.3, 126.2, 117.2, 108.1, 85.4, 82.0, 81.8, 81.8, 78.2, 77.3, 74.3, 74.1, 72.6, 64.8, 64.5, 39.0, 39.0, 36.8, 30.3, 16.4; HRMS (ESI+) [C31H38O7Na]+ found 545.2497, [M + Na]+ calcld. 545.2510.
(1′S,3′R,6′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-ol (16)
To a solution of the enone 10 (292 mg, 0.806 mmol) in methanol (16 mL), cerium(III) chloride heptahydrate (361 mg, 0.969 mmol) was added. The solution was cooled to −78 °C and sodium borohydride (37 mg, 0.97 mmol) was added and the mixture was stirred for 2 h. The mixture was diluted with ethyl acetate (80 mL) and washed with sat. aq. ammonium chloride (3 × 40 mL). The organic phase was dried (MgSO4) and concentrated under reduced pressure to afford the crude allylic alcohol 16 (294 mg) as a colourless gum. The residue was used directly in the next reaction without purification. For analytical purposes, a sample of the product was purified by flash column chromatography (25–75% diethyl ether in pentane) to afford the allylic alcohol 16 as a colourless gum. Rf = 0.68 (60% ethyl acetate in pet. ether); [α]D +13.5 (c = 1.00 in CHCl3, 21 °C); νmax 2994, 2942, 2880, 1701, 1641, 1377, 1101, 1087, 984, 959, 854 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.91 (1H, dddd, J = 17.1, 10.3, 6.9, 6.9 Hz), 5.88 (1H, ddd, J = 11.4, 9.6, 7.6 Hz), 5.69 (1H, d, J = 11.4 Hz), 5.47 (1H, ddq, J = 2.2, 1.6, 1.6 Hz), 5.17–5.00 (2H, m), 4.05 (1H, dd, J = 8.8, 2.2 Hz), 3.99–3.89 (4H, m), 3.66 (1H, ddq, J = 9.0, 1.6, 1.6 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.55 (1H, d, J = 12.7 Hz), 3.45 (1H, ddd, J = 11.7, 9.2, 4.4 Hz), 3.35–3.23 (1H, m), 3.29 (1H, ddd, J = 11.2, 9.0, 4.7 Hz), 3.26 (1H, ddd, J = 9.2, 5.5, 4.1 Hz), 2.94 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.55 (1H, ddd, J = 13.6, 7.6, 5.5 Hz), 2.49 (1H, ddddd, J = 14.6, 6.9, 3.5, 1.3, 1.3 Hz), 2.30 (1H, ddd, J = 12.1, 4.7, 4.4 Hz), 2.20 (1H, ddddd, J = 14.6, 8.2, 6.9, 1.3, 1.3 Hz), 1.78 (3H, dd, J = 1.6, 1.6 Hz), 1.58 (1H, ddd, J = 12.1, 11.7, 11.2 Hz); 13C NMR (101 MHz, CDCl3) δ 137.7, 135.3, 133.6, 130.8, 129.0, 117.0, 108.0, 83.9, 82.2, 81.7, 78.2, 77.8, 74.2, 73.4, 64.7, 64.6, 38.9, 37.8, 30.1, 21.9; HRMS (ESI+) [C20H28O6Na]+ found 387.1771, [M + Na]+ calcld. 387.1778
(1′S,3′S,4′R,6′R,7′R,8′R,10′S,12′R,16′Z)-4′-Methyl-8′-(prop-2-en-1-yl)-2′,5′,9′,13′-tetraoxaspiro(1,3-dioxolane-2,15′-tetracyclo[10.6.0.03,10.04,6]octadecane)-16′-en-7′-ol (17)
To a stirred solution of allylic alcohol 16 (294 mg) in benzene (41 mL), vanadyl acetylacetonate (42 mg, 0.16 mmol, >20 mol%) was added. A teal solution formed after 10 min and then t-butyl hydroperoxide (0.32 mL of a 5.0 M in decane, 1.6 mmol) was added to form a red solution. The solution was stirred at 60 °C for 2 h during which time the solution turned yellow. The reaction mixture was diluted with diethyl ether (40 mL) and washed with sat. aq. sodium sulfite (2 × 40 mL), sat. aq. ammonium chloride / 10% aq. ammonium hydroxide (4:1, 2 × 40 mL) and brine (2 × 40 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure to afford the epoxide 17 (290 mg) as a yellow oil. The residue was used directly in the next reaction without purification. For analytical purposes, a sample of the product was purified by flash column chromatography (25–75% diethyl ether in pentane) to afford the epoxide 17 as a colourless oil. Rf = 0.39 (60% ethyl acetate in pet. ether); [α]D +11.8 (c = 1.00 in CHCl3, 22 °C); νmax 3457, 2924, 2872, 2855, 1641, 1443, 1281, 1111, 1086, 1055, 1018, 882, 770 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 5.93–5.81 (2H, m), 5.66 (1H, d, J = 11.5 Hz), 5.12–5.02 (2H, m), 3.95–3.86 (4H, m), 3.75 (1H, ddd, J = 8.9, 8.2, 1.0 Hz), 3.59 (1H, d, J = 12.7 Hz), 3.52 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.6, 9.1, 4.5 Hz), 3.29 (1H, d, J = 9.1 Hz), 3.28–3.19 (2H, m), 3.15 (1H, ddd, J = 11.5, 9.1, 4.7 Hz), 3.08 (1H, d, J = 1.0 Hz), 2.86 (1H, ddd, J = 13.7, 9.6, 4.0 Hz), 2.60 (1H, ddd, J = 13.7, 6.9, 6.9 Hz), 2.53 (1H, ddd, J = 14.5, 6.9, 6.9 Hz), 2.26 (1H, ddd, J = 12.0, 4.7, 4.5 Hz), 2.12 (1H, ddd, J = 14.5, 7.7, 7.7 Hz), 2.02 (1H, d, J = 8.2 Hz), 1.42 (1H, ddd, J = 12.0, 11.6, 11.5 Hz), 1.40 (3H, s); 13C NMR (101 MHz, CD2Cl2) δ 135.8, 134.1, 130.7, 117.1, 108.5, 82.7, 82.2, 80.5, 78.0, 75.7, 74.4, 72.9, 67.2, 65.1, 64.9, 61.5, 38.9, 37.8, 30.5, 21.3; HRMS (EI+) [C20H28O7]+ found 380.1831, [M]+ calcld. 380.1835.
[(1′S,3′S,4′R,6′R,7′R,8′R,10′S,12′R,16′Z)-4′-Methyl-8′-(prop-2-en-1-yl)-2′,5′,9′,13′-tetraoxaspiro(1,3-dioxolane-2,15′-tetracyclo[10.6.0.03,10.04,6]octadecane)-16′-en-7′-yloxy]tris(propan-2-yl)silane (18)
The reaction was performed two batches. To a solution of the epoxy alcohol 17 (145 mg) in dichloromethane (20 mL), 4-dimethylaminopyridine (244 mg, 2.00 mmol), triethylamine (1.12 mL, 8.0 mmol) and triisopropylsilyl trifluoromethanesufonate (1.61 mL, 6.0 mmol) were added. The mixture was stirred for 16 h and diluted with diethyl ether (80 mL). The mixture was washed with sat. aq. ammonium chloride (3 × 50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (5–15% diethyl ether in pet. ether). The batches were combined to give the silyl ether 18 (284 mg, 66% over 3 steps) as a colourless oil. Rf = 0.51 (20% ethyl acetate in pet. ether); [α]D +39.2 (c = 0.50 in CHCl3, 27 °C); νmax 2943, 2893, 2866, 1641, 1464, 1109, 1089, 883, 912, 718 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 5.89 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.86 (1H, dddd, J = 17.1, 10.3, 6.8, 6.8 Hz), 5.66 (1H, d, J = 11.5 Hz), 5.09–4.98 (2H, m), 3.97 (1H, dd, J = 9.2, 1.0 Hz), 3.94–3.86 (4H, m), 3.59 (1H, d, J = 12.7 Hz), 3.52 (1H, d, J = 12.7 Hz), 3.45 (1H, ddd, J = 11.6, 9.2, 4.4 Hz), 3.29–3.25 (1H, m), 3.27 (1H, d, J = 9.2 Hz), 3.21 (1H, ddd, J = 9.2, 6.0, 4.0 Hz), 3.14 (1H, ddd, J = 11.5, 9.2, 4.7 Hz), 3.01 (1H, d, J = 1.0 Hz), 2.87 (1H, ddd, J = 13.5, 9.6, 4.0 Hz), 2.65–2.53 (2H, m), 2.25 (1H, ddd, J = 11.9, 4.7, 4.4 Hz), 2.02 (1H, ddddd, J = 14.4, 10.1, 6.8, 1.3, 1.3 Hz), 1.40 (1H, ddd, J = 11.9, 11.6, 11.5 Hz), 1.38 (3H, s), 1.16–1.02 (21H, m); 13C NMR (101 MHz, CD2Cl2) δ 136.4, 134.1, 130.7, 116.7, 108.5, 82.8, 82.3, 81.2, 78.1, 75.6, 74.4, 74.4, 67.4, 65.1, 64.9, 61.2, 38.9, 37.8, 30.6, 21.5, 18.6, 18.5, 13.6; HRMS (ESI+) [C29H48O7SiNa]+ found 559.3047, [M + Na]+ calcld. 559.3062.
(1S,3R,5S,6S,7R,9S,11R,15Z)-5-Hydroxy-4-methylidene-7-(prop-2-en-1-yl)-6-{[tris(propan-2-yl)silyl]oxy}-2,8,12-trioxatricyclo[9.6.0.03,9]heptadec-15-en-14-one (19)
To a solution of 2,2,6,6-tetramethylpiperidine (0.05 mL, 0.3 mmol) in toluene (0.45 mL) at −10 °C, a solution of n-BuLi (0.5 mL of a 0.6 M solution in toluene and hexane, 3:2, 0.3 mmol) was added dropwise. The mixture was stirred for 30 min to produce an orange solution. Diethylaluminium chloride (0.30 mL of a 1.0 M solution in hexane, 0.30 mmol) was added dropwise and the mixture was stirred for a further 30 min resulting in the formation of a colourless solution. To the resultant solution of the complex was added a solution of the epoxide 18 (27 mg, 50 µmol) in toluene (0.70 mL). The reaction mixture was allowed to reach rt over 16 h and then diluted with diethyl ether (8 mL). Sat. aq. Rochelle salt (10 mL) was added and the mixture was stirred vigorously for 2 h. The phases were separated and the organic phase was washed with sat. aq. ammonium chloride (10 mL) and brine (10 mL), then dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–40% diethyl ether in pentane) to afford the allylic alcohol 19 (10 mg, 40%) as a yellow oil. Rf = 0.60 (60% ethyl acetate in pet. ether); [α]D +5.8 (c = 0.50 in CHCl3, 27 °C); νmax 3476, 2891, 2866, 1676, 1643, 1464, 1098, 1016, 997, 918, 883 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.47 (1H, ddd, J = 12.4, 8.8, 7.6 Hz), 5.87 (1H, dddd, J = 17.1, 10.2, 6.9, 6.6 Hz), 5.86 (1H, dddd, J = 12.4, 1.6, 1.6, 1.1 Hz), 5.45 (1H, ddd, J = 1.3, 1.3, 1.3 Hz), 5.39 (1H, ddd, J = 1.3, 1.3, 1.3 Hz), 5.13–5.04 (2H, m), 4.51 (1H, dd, J = 17.8, 1.1 Hz), 4.47 (1H, ddd, J = 2.5, 1.3, 1.3 Hz), 4.20 (1H, d, J = 17.8 Hz), 3.80 (1H, dd, J = 6.7, 2.5 Hz), 3.68 (1H, ddd, J = 11.2, 9.6, 4.3 Hz), 3.64 (1H, ddd, J = 9.6, 1.3, 1.3 Hz), 3.55 (1H, ddd, J = 9.7, 6.7, 3.0 Hz), 3.41 (1H, ddd, J = 11.4, 9.1, 4.2 Hz), 3.32 (1H, ddd, J = 9.1, 9.1, 1.6 Hz), 2.70 (1H, dddd, J = 14.9, 7.6, 1.6, 1.6 Hz), 2.64 (1H, dddd, J = 14.9, 9.1, 8.8, 1.6 Hz), 2.46 (1H, ddddd, J = 14.4, 6.9, 3.0, 1.3, 1.3 Hz), 2.34 (1H, ddd, J = 11.6, 4.3, 4.2 Hz), 2.16 (1H, ddddd, J = 14.4, 9.7, 6.6, 1.3, 1.3 Hz), 1.65 (1H, ddd, J = 11.6, 11.4, 11.2 Hz), 1.15–1.03 (21H, m); 13C NMR (101 MHz, CDCl3) δ 203.7, 145.1, 138.1, 135.3, 129.0, 117.7, 117.0, 85.3, 83.1, 82.0, 78.7, 78.6, 77.4, 77.0, 76.3, 38.3, 38.2, 35.1, 18.3, 13.0; HRMS (ESI+) [C27H44O6SiNa]+ found 515.2775, [M + Na]+ calcld. 515.2799.
(1′S,3′R,5′S,6′S,7′R,9′S,11′R,15′Z)-4′-Methylidene-7′-(prop-2-en-1-yl)-6′-{[tris(propan-2-yl)silyl]oxy}-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-15′-en-5′-ol (20)
To a solution of epoxide 18 (43 mg, 80 µmol) in THF (4 mL), p-toluenesulfonic acid (69 mg, 0.40 mmol) and 4 Å molecular sieves (3 g) were added. The resultant mixture was stirred for 16 h and dried (4 Å molecular sieves) ethylene glycol (1 mL) was added. The reaction mixture was stirred for a further 24 h and diluted with sat. aq. sodium bicarbonate (25 mL). The organic phase was extracted with diethyl ether (3 × 10 mL) and the combined organic extracts were washed with brine (3 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–30% diethyl ether in pet. ether) to afford allylic alcohol 20 (17 mg, 40%) and starting epoxide 18 (11 mg, 26%) as colourless films. Rf = 0.63 (60% ethyl acetate in pet. ether); [α]D +32.4 (c = 1.00 in CHCl3, 27 °C); νmax 3476, 2941, 2866, 1643, 1464, 1090, 1018, 901, 883 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.92 (1H, ddd, J = 11.4, 9.5, 7.5 Hz), 5.87 (1H, dddd, J = 17.1, 10.3, 7.1, 6.5 Hz), 5.68 (1H, d, J = 11.4 Hz), 5.45 (1H, ddd, J = 1.4, 1.4, 1.4 Hz), 5.35 (1H, ddd, J = 1.4, 1.4, 1.4 Hz), 5.12–5.02 (2H, m), 4.44 (1H, dddd, J = 4.1, 2.7, 1.4, 1.4 Hz), 3.98–3.91 (4H, m), 3.79 (1H, dd, J = 6.9, 2.7 Hz), 3.66 (1H, d, J = 12.8 Hz), 3.61 (1H, ddd, J = 9.6, 1.4, 1.4 Hz), 3.56 (1H, d, J = 12.8 Hz), 3.54–3.46 (3H, m), 3.38 (1H, ddd, J = 9.3, 5.5, 4.1 Hz), 2.96 (1H, ddd, J = 13.4, 9.5, 4.1 Hz), 2.62 (1H, ddd, J = 13.4, 7.5, 5.5 Hz), 2.45 (1H, ddddd, J = 14.5, 7.1, 2.8, 1.3, 1.3 Hz), 2.31 (1H, d, J = 4.1 Hz), 2.30 (1H, ddd, J = 11.7, 4.4, 4.4 Hz), 2.15 (1H, ddddd, J = 14.5, 9.6, 6.5, 1.4, 1.4 Hz), 1.53 (1H, app. dt, J = 11.7, 11.4 Hz), 1.13–1.04 (21H, m); 13C NMR (101 MHz, CDCl3) δ 145.6, 135.4, 133.5, 130.9, 117.0, 116.9, 108.0, 83.2, 82.0, 82.0, 78.6, 78.2, 77.8, 76.1, 74.1, 64.8, 64.6, 38.7, 38.3, 30.3, 18.3, 13.1; HRMS (ESI+) [C29H48O7SiNa]+ found 559.3041, [M + Na]+ calcld. 559.3062.
(1′R,3′S,5′Z,10′R,12′S,14′R,19′S,20′S,21′S)-21′-Methyl-20′-[(naphthalen-2-yl)methoxy]-2′,9′,13′,18′-tetraoxaspiro[1,3-dioxolane-2,7′-tetracyclo(10.9.0.03,10.014,19]henicosane)-5′,15′-dien-17′-one (25)
To a solution of alcohol 15 (8 mg, 0.02 mmol) in methanol (0.64 mL), the ruthenium complex 21 (3 mg, 0.03 mmol, 20 mol%) was added. The solution was stirred at reflux for 1 h and concentrated under reduced pressure. The residue was purified by flash column chromatography to afford an E / Z mixture of alcohol 22 (4 mg) as a colourless film. The residue was used directly in the next reaction without purification.
To a solution of (2E)-4-(prop-2-en-1-yloxy)but-2-enoic acid 23 (2 mg, 0.02 mmol) in dichloromethane (0.8 mL), N,N′-dicyclohexylcarbodiimide (3 mg, 0.02 mmol) and 4-dimethylaminopyridine (0.2 mg, 0.02 mmol) were added. The resultant suspension was added to 22 (4 mg) and the mixture stirred for 16 h. The mixture was diluted with diethyl ether (4.2 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography (5–60% diethyl ether in pet. ether) to give an isomeric mixture of esters 24 (2 mg) as a colourless film. The residue was used directly in the next reaction without purification.
To a solution of ester 24 (2 mg) in dichloromethane (16 mL), the ruthenium complex 21 (1 mg, 1 µmol) was added. The solution was stirred for 6 h at reflux and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–25% diethyl ether in dichloromethane) to deliver the tetracyclic lactone 25 (1 mg, 12% over 3 steps) as a colourless film. Rf = 0.23 (80% diethyl ether in pet. ether); [α]D +48 (c = 0.08 in CHCl3, 36 °C); νmax 2922, 2851, 1734, 1636, 1559, 1119, 1042, 818 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.88–7.81 (3H, m), 7.80 (1H, s), 7.54–7.42 (3H, m), 6.82 (1H, dd, J = 9.9, 1.6 Hz), 5.93 (1H, dd, J = 9.9, 2.5 Hz), 5.91 (1H, ddd, J = 11.4, 9.7, 7.4 Hz), 5.73 (1H, d, J = 11.4 Hz), 4.87 (1H, d, J = 11.5 Hz), 4.86 (1H, ddd, J = 10.5, 2.5, 1.6 Hz), 4.81 (1H, d, J = 11.5 Hz), 4.26 (1H, dd, J = 10.5, 0.7 Hz), 3.96 (4H, m), 3.90 (1H, ddd, J = 11.0, 9.3, 5.5 Hz), 3.83 (1H, dd, J = 2.6, 0.7 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.43 (1H, ddd, J = 12.4, 9.1, 4.3 Hz), 3.23 (1H, ddd, J = 9.1, 7.2, 5.5 Hz), 2.97 (1H, ddd, J = 13.5, 9.7, 7.2 Hz), 2.93 (1H, dd, J = 9.3, 5.7 Hz), 2.54 (1H, ddd, J = 13.5, 7.4, 5.5 Hz), 2.33 (1H, ddd, J = 11.8, 5.5, 4.3 Hz), 2.27 (1H, qdd, J = 7.4, 5.7, 2.6 Hz), 1.47 (1H, ddd, J = 12.4, 11.8, 11.0 Hz), 1.18 (3H, d, J = 7.4 Hz); 13C NMR (126 MHz, CDCl3) δ 163.4, 149.9, 135.6, 133.8, 133.3, 133.1, 130.8, 128.3, 128.1, 127.9, 126.8, 126.3, 126.1, 126.1, 119.4, 107.9, 86.9, 82.9, 81.8, 81.0, 77.7, 75.4, 74.4, 73.0, 69.8, 64.8, 64.7, 39.9, 39.0, 32.1, 19.9; HRMS (ESI+) [C31H34O8Na]+ found 557.2124, [M + Na]+ calcld. 557.2146.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6651/12/12/740/s1, 1H and 13C NMR spectra for key compounds, plus additional 1H NMR NOE data for selected intermediate compounds.
Author Contributions
J.S.C. conceived the synthetic route, supervised the project and wrote the paper; J.S.C and M.P. designed the experiments; M.P. performed all synthetic work in the laboratory. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by EPSRC, grant numbers EP/K503058 and EP/L50497X.
Acknowledgments
The authors gratefully acknowledge studentship funding from EPSRC to support M.P. The authors also thank Alistair Boyer (University of Glasgow) for providing the carboxylic acid 23.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References and Note
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Yasumoto, T. Structures of Ciguatoxin and Its Congener. J. Am. Chem. Soc. 1989, 111, 8929–8931. [Google Scholar] [CrossRef]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Fukui, M.; Yasumoto, T. Structures and Configurations of Ciguatoxin from the Moray Eel Gymnothorax javanicus and Its Likely Precursor from the Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1990, 112, 4380–4386. [Google Scholar] [CrossRef]
- Yasumoto, T.; Igarashi, T.; Legrand, A.-M.; Cruchet, P.; Chinain, M.; Fujita, T.; Naoki, H. Structural Elucidation of Ciguatoxin Congeners by Fast-Atom Bombardment Tandem Mass Spectroscopy. J. Am. Chem. Soc. 2000, 122, 4988–4989. [Google Scholar] [CrossRef]
- Nicholson, G.M.; Lewis, R.J. Ciguatoxins: Cyclic Polyether Modulators of Voltage-Gated Ion Channel Function. Mar. Drugs 2006, 4, 82–118. [Google Scholar] [CrossRef] [Green Version]
- Soliño, L.; Costa, P.R. Differential Toxin Profiles of Ciguatoxins in Marine Organisms: Chemistry, Fate and Global Distribution. Toxicon 2018, 150, 124–143. [Google Scholar] [CrossRef]
- Pasinszki, T.; Lako, J.; Dennis, T.E. Advances in Detecting Ciguatoxins in Fish. Toxins 2020, 12, 494. [Google Scholar] [CrossRef]
- Lewis, R.J.; Vernoux, J.-P.; Brereton, I.M. Structure of Caribbean Ciguatoxin Isolated from Caranx latus. J. Am. Chem. Soc. 1998, 120, 5914–5920. [Google Scholar] [CrossRef]
- Kryuchkov, F.; Robertson, A.; Miles, C.O.; Mudge, E.M.; Uhlig, S. LC-HRMS and Chemical Derivatization Strategies for the Structure Elucidation of Caribbean Ciguatoxins: Identification of C-CTX-3 and -4. Mar. Drugs 2020, 18, 182. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, B.; Hurbungs, M.; Jones, A.; Lewis, R.J. Multiple Ciguatoxins Present in Indian Ocean Reef Fish. Toxicon 2002, 1347–1353. [Google Scholar] [CrossRef]
- Diogène, J.; Reverté, L.; Rambla-Alegre, M.; del Río, V.; de la Iglesia, P.; Campàs, M.; Palacios, O.; Flores, C.; Caixach, J.; Ralijaona, C.; et al. Identification of Ciguatoxins in a Shark Involved in a Fatal Food Poisoning in the Indian Ocean. Sci. Rep. 2017, 7, 8240. [Google Scholar] [CrossRef]
- Friedman, M.A.; Fernandez, M.; Backer, L.C.; Dickey, R.W.; Bernstein, J.; Schrank, K.; Kibler, S.; Stephan, W.; Gribble, M.O.; Bienfang, P.; et al. An Updated Review of Ciguatera Fish Poisoning: Clinical, Epidemiological, Environmental, and Public Health Management. Mar. Drugs 2017, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Frelin, C.; Durand-Clément, M.; Bidard, J.-N.; Lazdunski, M. The Molecular Basis of Ciguatoxin Action. In Marine Toxins; Hall, S., Strichartz, G., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1990; Volume 418, pp. 192–199. [Google Scholar]
- Bidard, J.-N.; Vijverberg, H.M.P.; Frelin, C.; Chungue, E.; Legrand, A.-M.; Bagnis, R.; Lazdunski, M. Ciguatoxin Is a Novel Type of Na+ Channel Toxin. J. Biol. Chem. 1984, 259, 8353–8357. [Google Scholar] [PubMed]
- Hidalgo, J.; Liberona, J.J.; Molgó, J.; Jaimovich, E. Pacific Ciguatoxin-1b effect over Na+ and K+ Currents, Inositol 1,4,5-Triphosphate Content and Intracellular Ca2+ Signals in Cultured Rat Myotubes. Br. J. Pharmacol. 2002, 137, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirama, M.; Oishi, T.; Uehara, H.; Inoue, M.; Maruyama, M.; Oguri, H.; Satake, M. Total Synthesis of Ciguatoxin CTX3C. Science 2001, 294, 1904–1907. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Miyazaki, K.; Uehara, H.; Maruyama, M.; Hirama, M. First- and Second-Generation Total Synthesis of Ciguatoxin CTX3C. Proc. Natl. Acad. Sci. USA 2004, 101, 12013–12018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Miyazaki, K.; Ishihara, Y.; Tatami, A.; Ohnuma, Y.; Kawada, Y.; Komano, K.; Yamashita, S.; Lee, N.; Hirama, M. Total Synthesis of Ciguatoxin and 51-HydroxyCTX3C. J. Am. Chem. Soc. 2006, 128, 9352–9354. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, A.; Isobe, M. Total Synthesis of Ciguatoxin. Angew. Chem. Int. Ed. 2009, 49, 2941–2945. [Google Scholar] [CrossRef]
- Clark, J.S. Construction of Fused Polycyclic Ethers by Strategies Involving Ring-Closing Metathesis. Chem. Commun. 2006, 3571–3581. [Google Scholar] [CrossRef]
- Clark, J.S.; Grainger, D.M.; Ehkirch, A.A.-C.; Blake, A.J.; Wilson, C. Synthesis of the Fused Polycyclic Ether Core of Hemibrevetoxin B by Two-Directional Ring-Closing Metathesis. Org. Lett. 2007, 9, 1033–1036. [Google Scholar] [CrossRef]
- Clark, J.S.; Kimber, M.C.; Robertson, J.; McErlean, C.S.P.; Wilson, C. Rapid Two-Directional Synthesis of the F–J Fragment of the Gambieric Acids by Iterative Doube Ring-Closing Metathesis. Angew. Chem. Int. Ed. 2005, 44, 6157–6162. [Google Scholar] [CrossRef]
- Clark, J.S.; Romiti, F.; Sieng, B.; Paterson, L.C.; Stewart, A.; Chaudhury, S.; Thomas, L.H. Synthesis of the A–D Ring System of the Gambieric Acids. Org. Lett. 2015, 17, 4694–4697. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Conroy, J.; Blake, A.J. Rapid Synthesis of the A–E Fragment of CTX3C. Org. Lett. 2007, 9, 2091–2094. [Google Scholar] [CrossRef]
- Skardon-Duncan, J.; Sparenberg, M.; Bayle, A.; Alexander, S.; Clark, J.S. Steroselective Synthesis of Medium-Sized Cyclic Ethers by Sequential Ring-Closing Metathesis and Tsuji-Trost Allylation. Org. Lett. 2018, 20, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Popadynec, M.; Gibbard, H.; Clark, J.S. Bidirectional Synthesis of the IJK Fragment of Ciguatoxin CTX3C by Sequential Double Ring-Closing Metathesis and Tsuji-Trost Allylation. Org. Lett. 2020, 22, 3734–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.S.; Kettle, J.G. Enantioselective Synthesis of Medium-Ring Sub-Units of Brevetoxin A by Ring-Closing Metathesis. Tetrahedron Lett. 1997, 17, 127–130. [Google Scholar] [CrossRef]
- Ciceri, P.; Demnitz, F.W.J. An Efficient, Rapid and Highly Selective Preparation of the Wieland-Miescher Ketone-9-Ethylene Ketal. Tetrahedron Lett. 1997, 38, 389–390. [Google Scholar] [CrossRef]
- Tsunoda, T.; Suzuki, M.; Noyori, R. A Facile Procedure for Acetalization Under Aprotic Conditions. Tetrahedron Lett. 1980, 21, 1357–1358. [Google Scholar] [CrossRef]
- Behenna, D.C.; Stoltz, B.M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, W.S.; Brestensky, D.M.; Stryker, J.M. Selective Hydride-Mediated Conjugate Reduction of α,β-Unsaturated Carbonyl Compounds Using [(Ph3P)CuH]6. J. Am. Chem. Soc. 1988, 110, 291–293. [Google Scholar] [CrossRef]
- Saito, S.; Yamamoto, H. Efficient Conjugate Reduction of α,β-Unsaturated Carbonyl Compounds by Complexation with Aluminum Tris(2,6-diphenylphenoxide). J. Org. Chem. 1996, 61, 2928–2929. [Google Scholar] [CrossRef]
- Baker, B.A.; Bošković, Ž.V.; Lipshutz, B.H. (BDP)CuH: A “Hot” Stryker’s Reagent for Use in Achiral Conjugate Reductions. Org. Lett. 2008, 10, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; Vishwakarma, L.C.; Billmers, J.M.; Finn, J. Synthesis of α-Hydroxy Carbonyl Compounds (Acyloins): Direct Oxidation of Enolates Using 2-Sulfonyloxaziridines. J. Org. Chem. 1984, 49, 3241–3243. [Google Scholar] [CrossRef]
- Davis, F.A.; Sheppard, A.C. Applications of Oxaziridines in Organic Synthesis. Tetrahedron 1989, 45, 5703–5742. [Google Scholar] [CrossRef]
- Rubottom, G.M.; Vazquez, M.A.; Pelegrina, D.R. Peracid Oxidation of Trimethylsilyl Enol Ethers: A Facile α-Hydroxylation Procedure. Tetrahedron Lett. 1974, 15, 4319–4322. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohanna, J.; Zhou, J.; Iyer, K.; Rainier, J.D. Total Synthesis of Brevenal. J. Am. Chem. Soc. 2011, 133, 3208–3216. [Google Scholar] [CrossRef] [Green Version]
- Nashed, M.A.; Anderson, L. Organotin Derivatives and the Selective Acylation and Alkylation of the Equatorial Hydroxy Group in a Vicinal, Equatorial-Axial Pair. Tetrahedron Lett. 1976, 17, 3503–3506. [Google Scholar] [CrossRef]
- Zou, X.; Qin, C.; Pereira, C.L.; Tian, G.; Hu, J.; Seeberger, P.H.; Yin, J. Synergistic Glycosylation as Key to the Chemical Synthesis of an Outer Core Octasaccharide of Helicobacter pylori. Chem. Eur. J. 2018, 24, 2868–2872. [Google Scholar] [CrossRef]
- Yasuda, A.; Tanaka, S.; Oshima, K.; Yamamoto, H.; Nozaki, H. Organoaluminum Reagents of Type R1R2NAlEt2 Which Allow Regiospecific Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc. 1974, 96, 6513–6514. [Google Scholar] [CrossRef]
- Crabtree, R.H.; Davis, M.W. Directing Effects in Homogeneous Hydrogenation with [Ir(cod)(PCy3)(py)]PF6. J. Org. Chem. 1986, 51, 2655–2661. [Google Scholar] [CrossRef]
- Thompson, H.W.; McPherson, E. Control of Hydrogenation Stereochemistry by Intramolecular Anionic Coordination to Homogeneous Catalysts. J. Am. Chem. Soc. 1974, 96, 6232–6233. [Google Scholar] [CrossRef]
- Machado, A.S.; Olesker, A.; Castillon, S.; Lukacs, G. Hydroxy Group Directed Hydrogenation with Rhodium and Iridium Catalysts. Synthesis of a Protected Chiral Carbocyclic Analogue of Daunosamine. J. Chem. Soc. Chem. Commun. 1985, 6, 330–332. [Google Scholar] [CrossRef]
- Hanessian, S.; Giroux, S.; Larsson, A. Efficient Allyl to Propenyl Isomerization in Functionally Diverse Compounds with a Thermally Modified Grubbs Second-Generation Catalyst. Org. Lett. 2006, 8, 5481–5484. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Jeffrey, C.S.; Tennakoon, M.A.; Wang, J.; Zhao, H. Relay Ring-Closing Metathesis (RRCM): A Strategy for Directing Metal Movement Throughout Olefin Metathesis Sequences. J. Am. Chem. Soc. 2004, 126, 10210–12211. [Google Scholar] [CrossRef] [PubMed]
- Harding, K.E.; Clement, B.A.; Moreno, L.; Peter-Katalinic, J. Synthesis of Some Polyfunctionalized Bicyclo[3.3.1 ]nonane-2,9-diones and Bicyclo[4.3.1]decane-2,10-diones. J. Org. Chem. 1981, 46, 940–948. [Google Scholar] [CrossRef]
- For installation of the C-47 and C-48 methyl substituents and the M-ring spiroacetal in a related but simpler α,β-unsaturated lactone, see: Domon, D.; Fujiwara, K.; Ohtaniuchi, Y.; Takezawa, A.; Takeda, S.; Kawasaki, H.; Murai, A.; Kawai, H.; Suzuki, T. Synthesis of the C42–C52 Part of Ciguatoxin CTX3C. Tetrahedron Lett. 2005, 46, 8279–8283. [Google Scholar]
Figure 1.
Representative Pacific ciguatoxins.
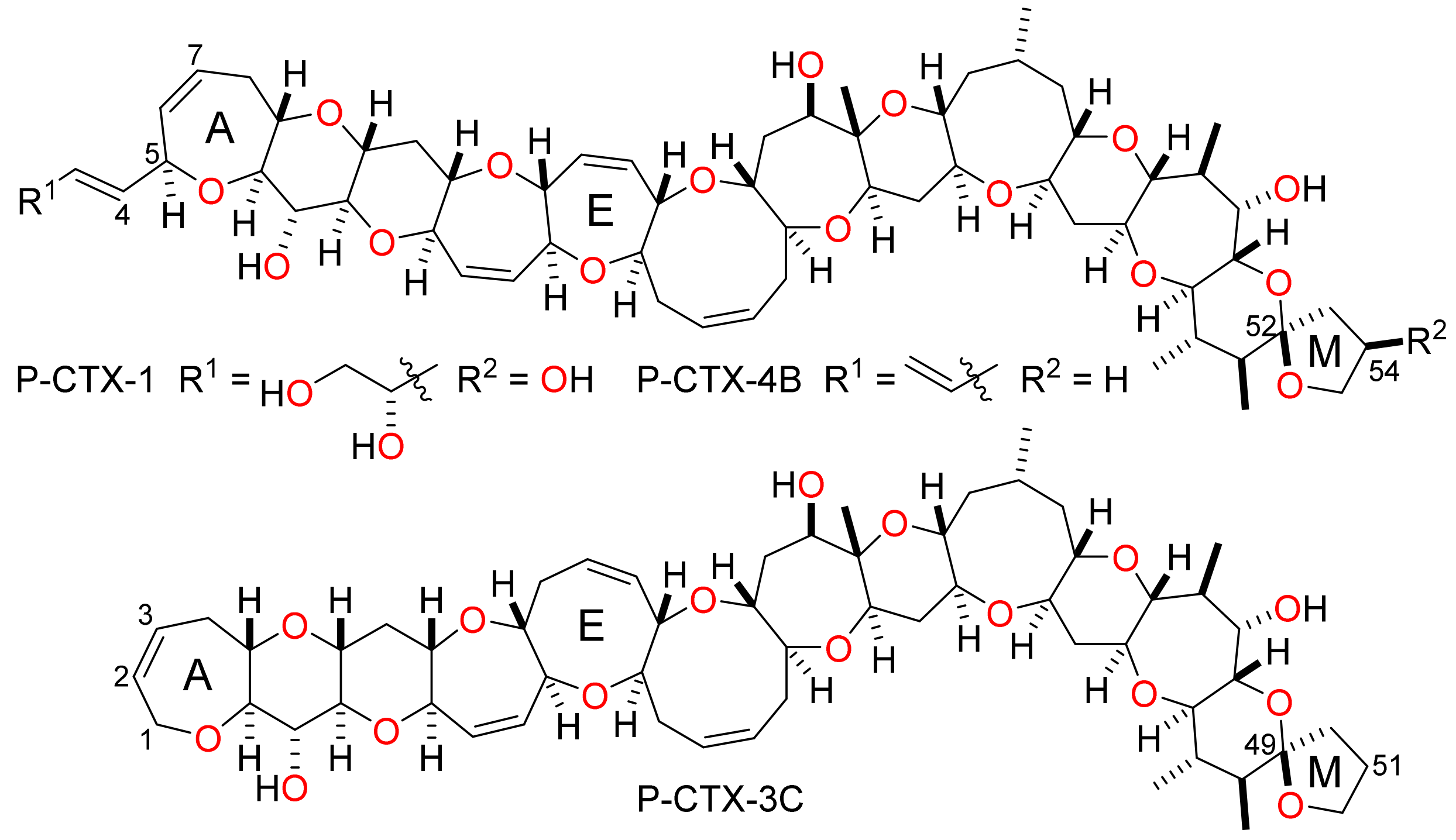
Figure 2.
Retrosynthetic analysis of P-CTX-3C (1) to reveal the key A–F and I–L fragments.

Scheme 1.
Functionalisation of the I–K fragment by bidirectional double Tsuji-Trost allylation.

Figure 3.
IR and 13C data for the bis-enone 1 and comparison to data for the related compounds 4–6.
Scheme 2.
Selective protection of the I-ring carbonyl group and subsequent Tsuji-Trost allylation of ring K.
Scheme 2.
Selective protection of the I-ring carbonyl group and subsequent Tsuji-Trost allylation of ring K.
Scheme 3.
Conjugate reduction of the enone 10.
Scheme 4.
Hydroxylation and further elaboration of ring K.
Scheme 5.
An alternative approach to the elaboration of ring K.

Scheme 6.
Use of relay RCM to construct ring L and form the I–L ring system of the Pacific ciguatoxins.
Scheme 6.
Use of relay RCM to construct ring L and form the I–L ring system of the Pacific ciguatoxins.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Clark, J.S.; Popadynec, M. Stereoselective Synthesis of the I–L Fragment of the Pacific Ciguatoxins. Toxins 2020, 12, 740. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120740
AMA Style
Clark JS, Popadynec M. Stereoselective Synthesis of the I–L Fragment of the Pacific Ciguatoxins. Toxins. 2020; 12(12):740. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120740
Chicago/Turabian StyleClark, J. Stephen, and Michael Popadynec. 2020. "Stereoselective Synthesis of the I–L Fragment of the Pacific Ciguatoxins" Toxins 12, no. 12: 740. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120740
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.