Ligand-Based Virtual Screening, Molecular Docking, Molecular Dynamics, and MM-PBSA Calculations towards the Identification of Potential Novel Ricin Inhibitors

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Protein Preparation and Redocking Procedure
2.2. LBVS, Ligand Preparation, and Target Prediction
2.3. Molecular Docking
2.4. Drug-Likeness Studies
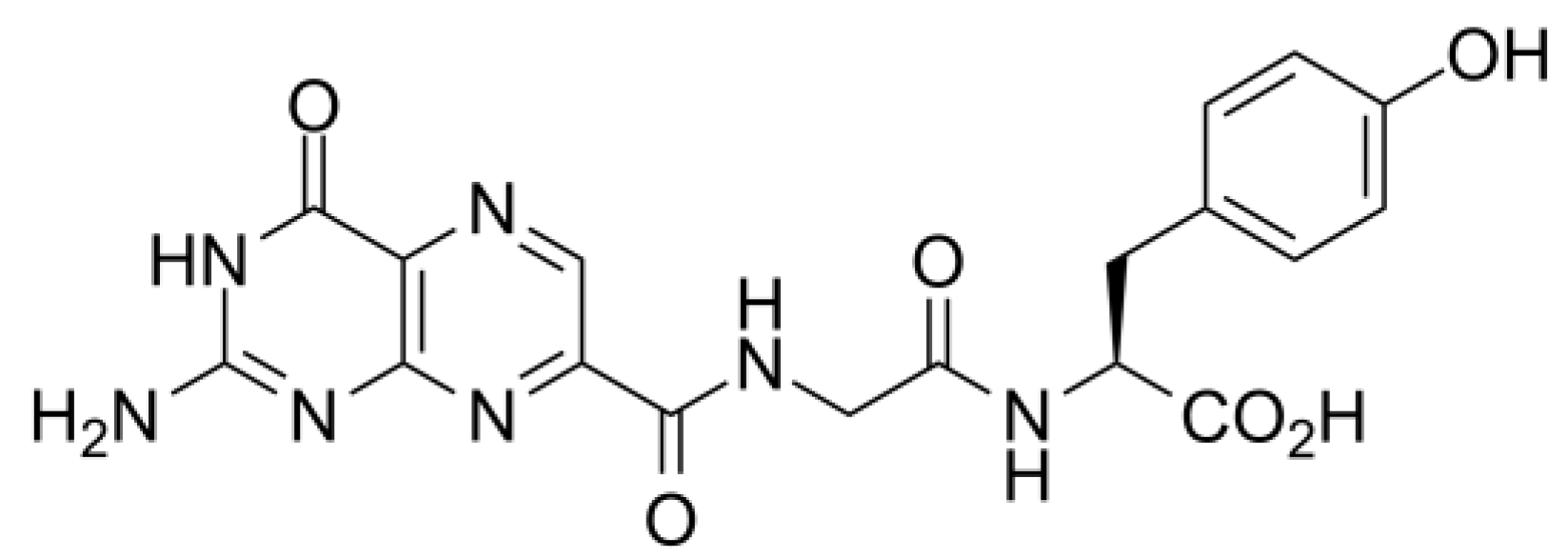
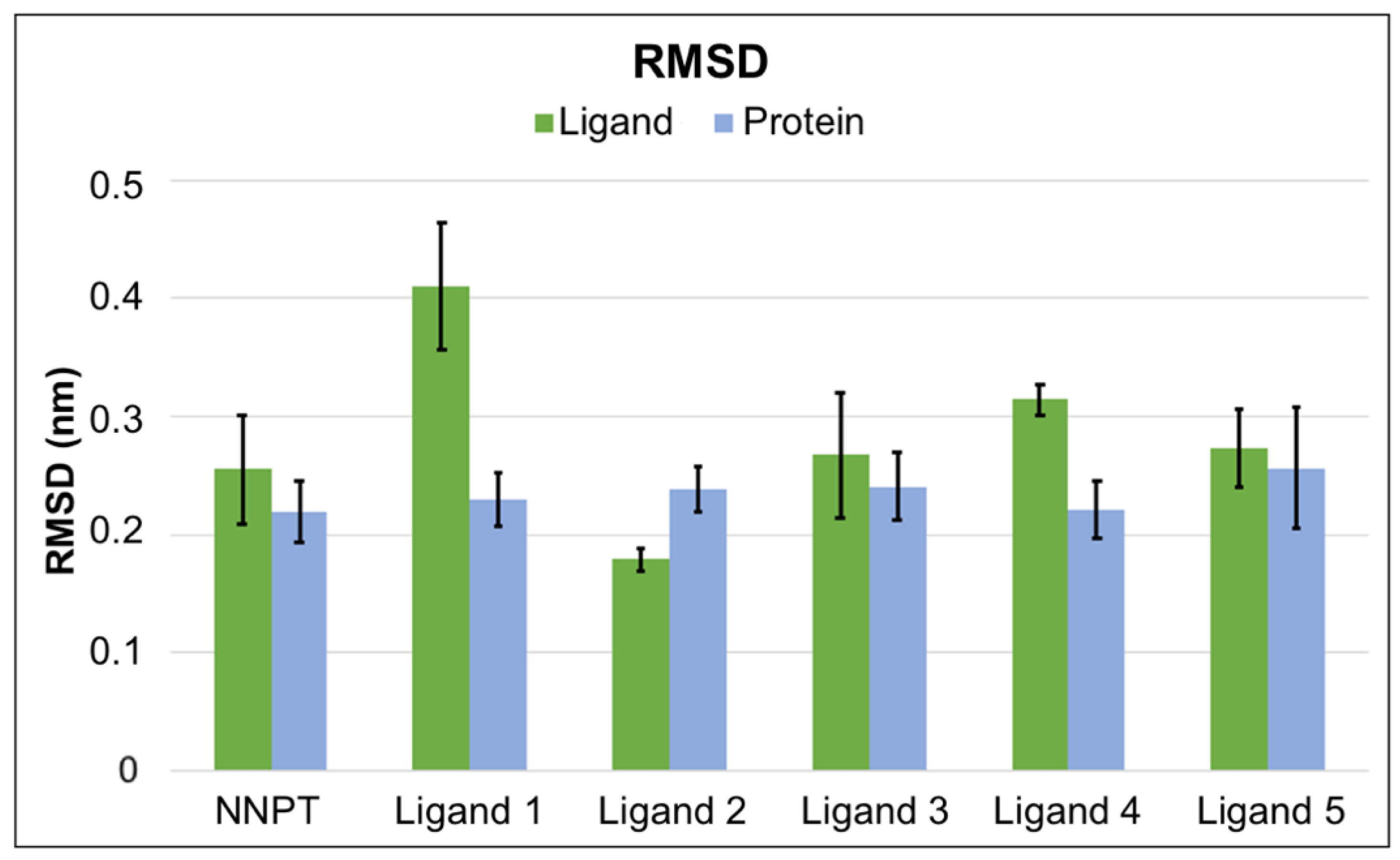
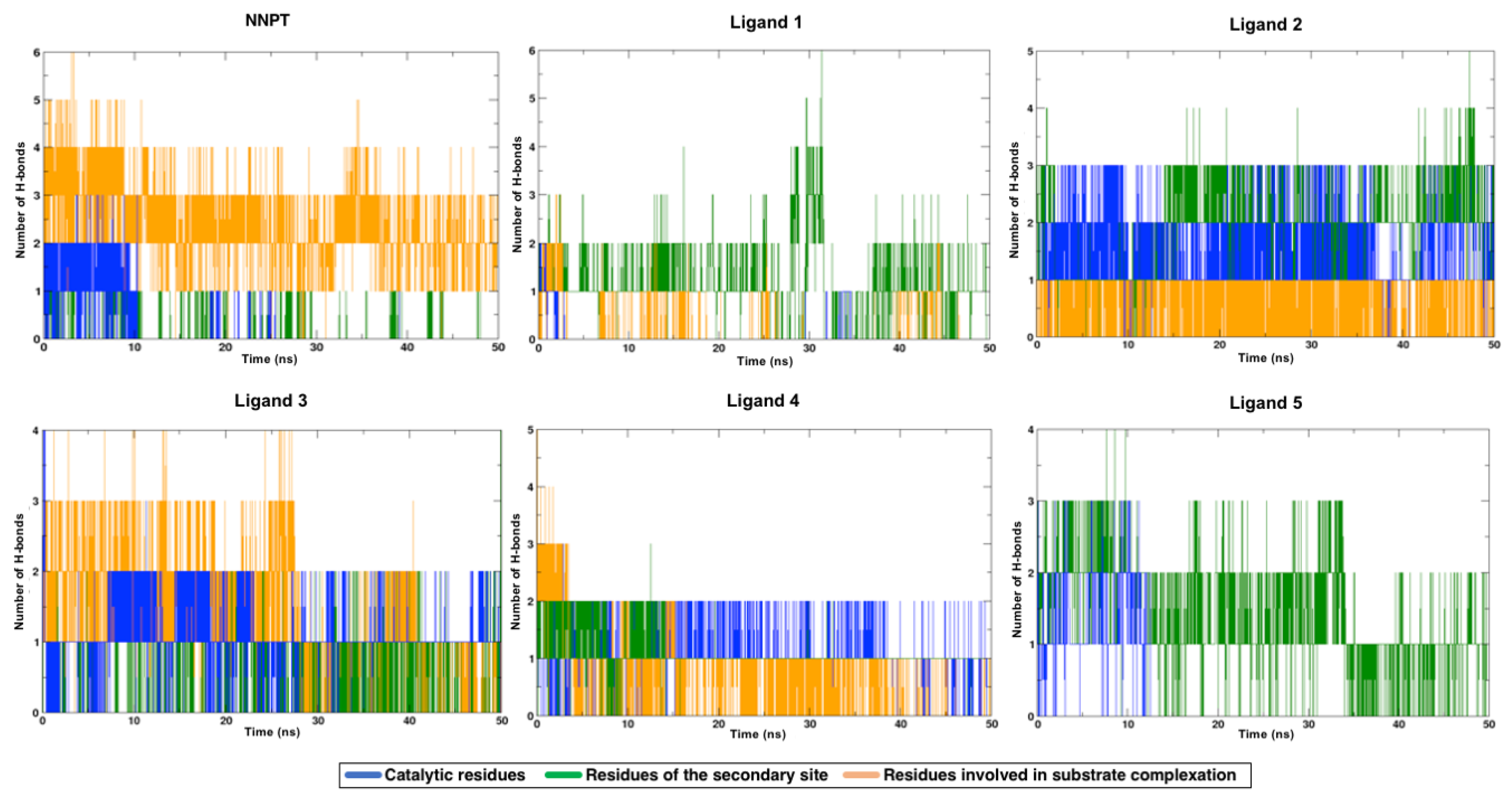
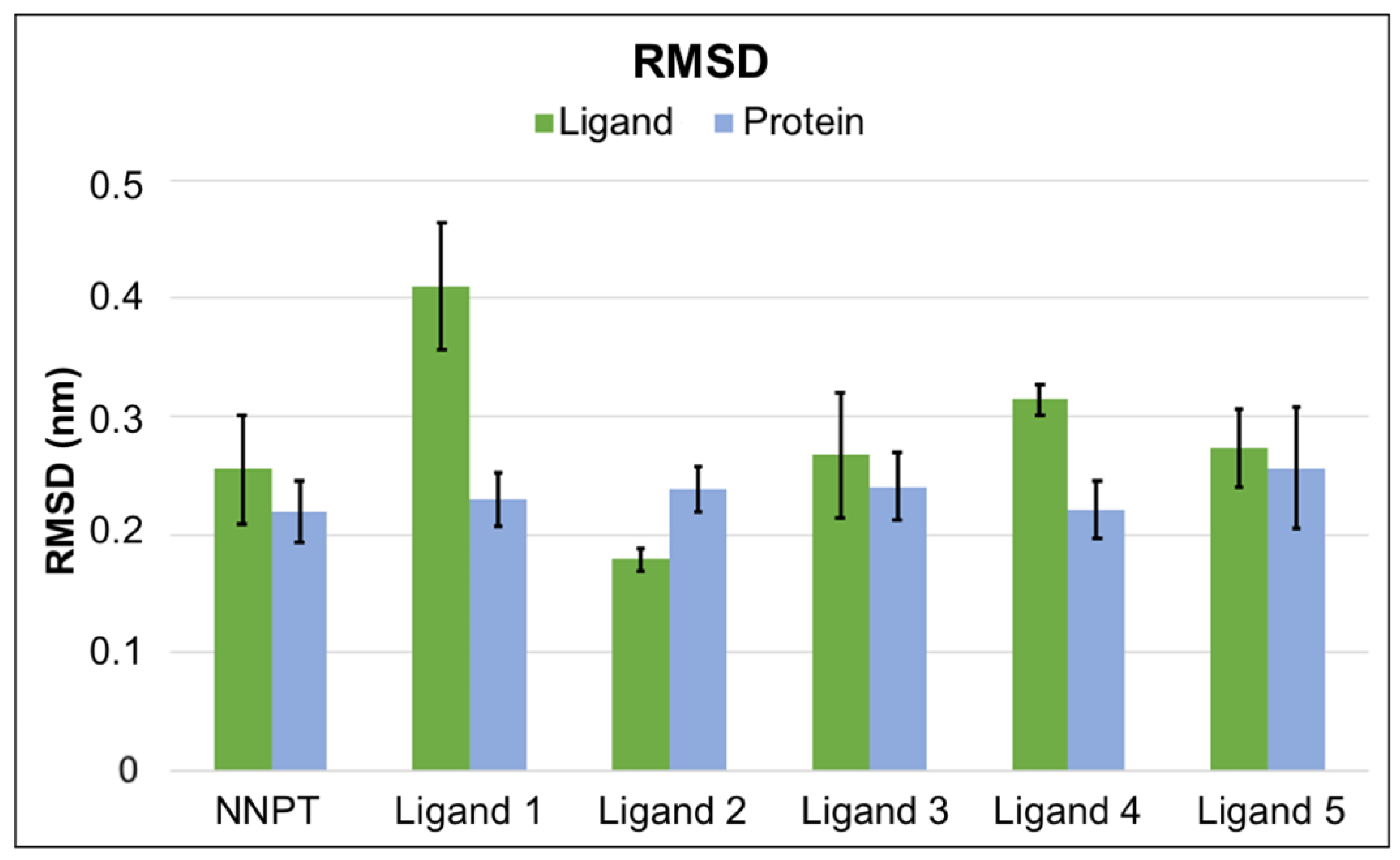
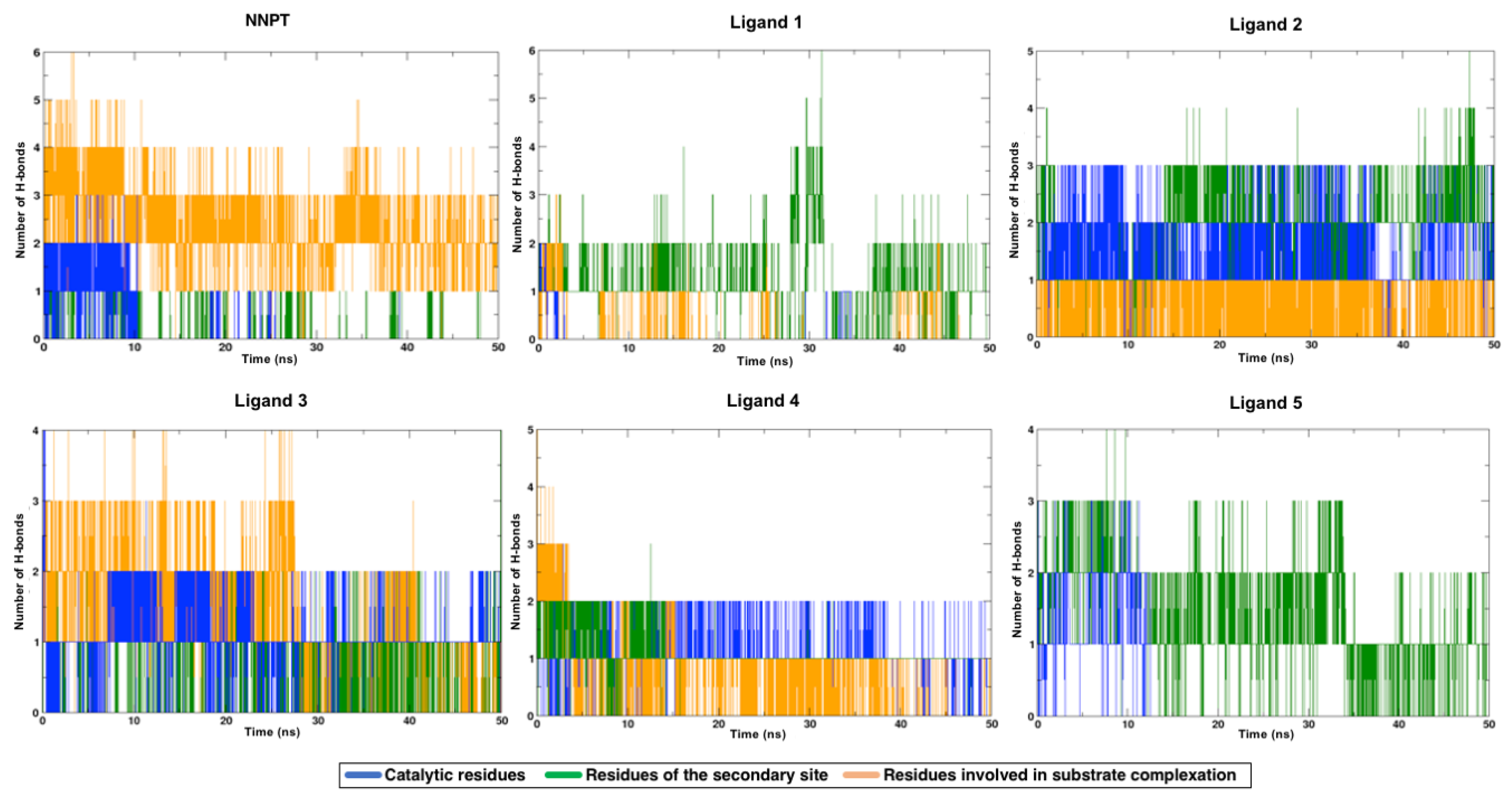
2.5. Molecular Dynamics Simulations
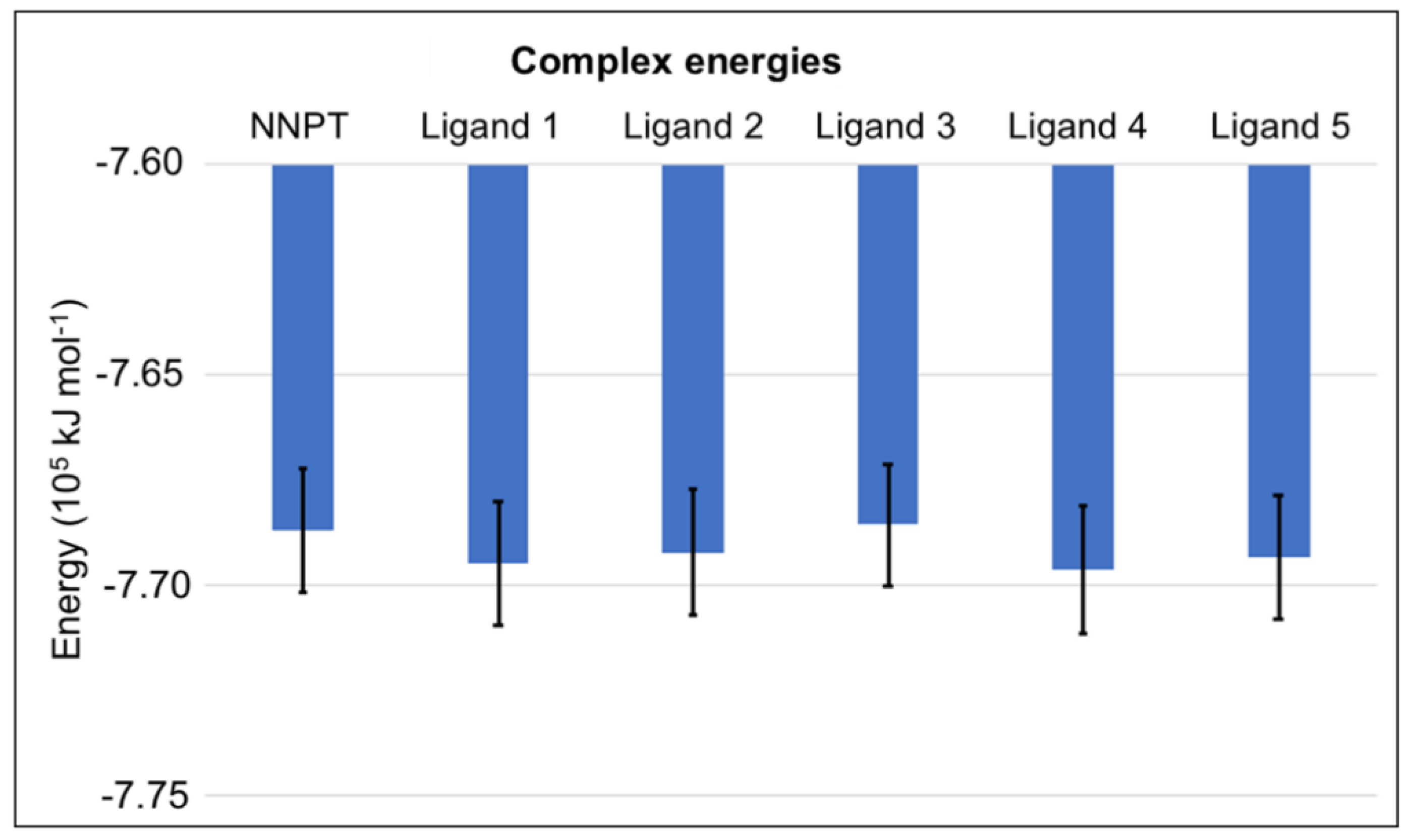
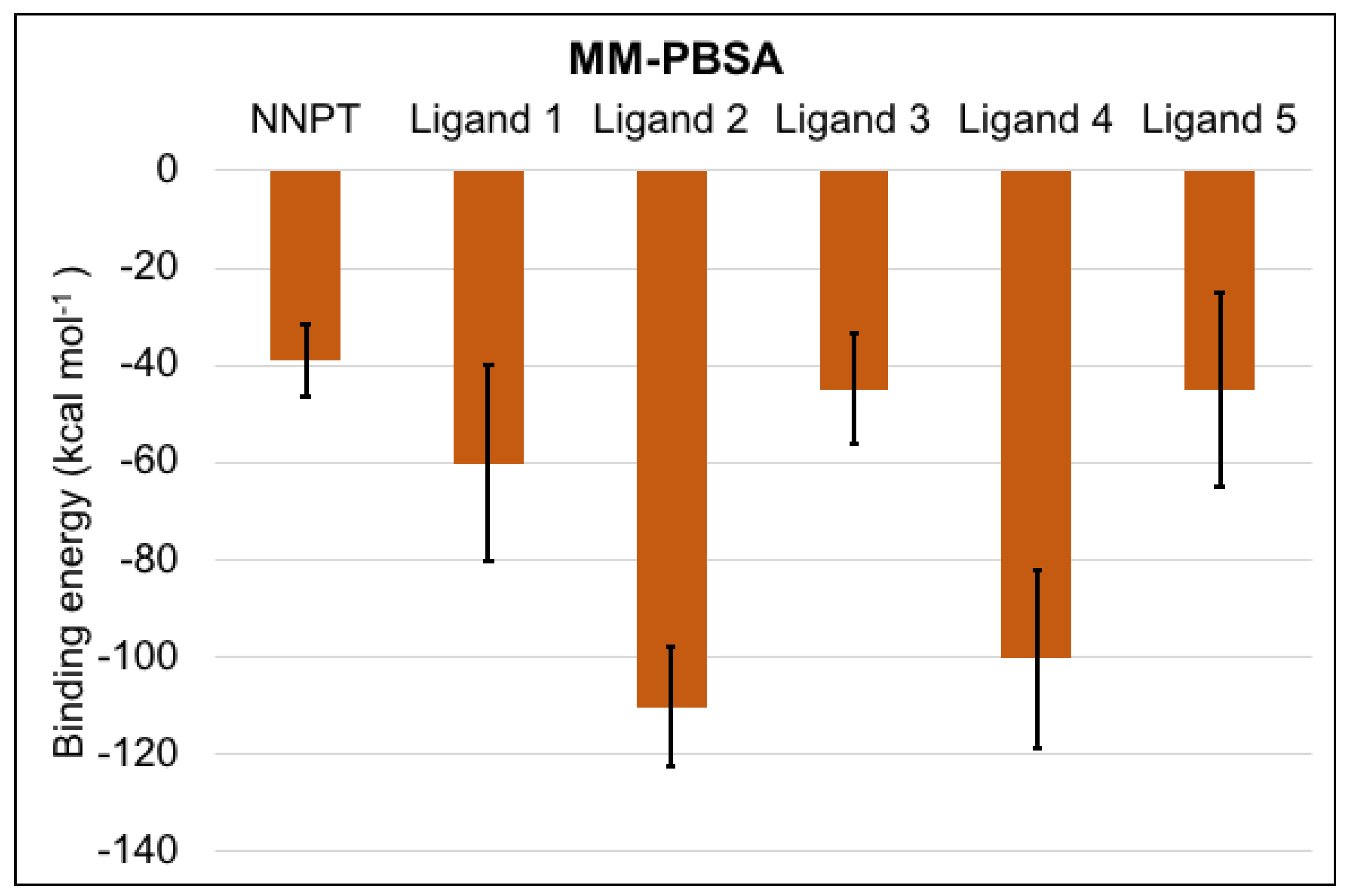
2.6. MM-PBSA Calculations
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Protein Preparation and Redocking Procedure
5.2. Selection of the Reference Ligand, LBVS and Ligand Preparation, and Target Prediction
5.3. Molecular Docking
5.4. Drug-Likeness Studies
5.5. MD Simulations
5.6. MM-PBSA Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The Mechanism of Action of Ricin and Related Toxic Lectins on Eukaryotic Ribosomes. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Audi, J.; Belson, M. Ricin Poisoning—A comprehensive review. JAMA 2005, 294. [Google Scholar] [CrossRef]
- Argent, R.H.; Roberts, L.M.; Wales, R.; Robertus, J.D.; Lord, J.M. Introduction of a disulfide bond into ricin A chain decreases the cytotoxicity of the ricin holotoxin. J. Biol. Chem. 1994, 269, 26705–26710. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.H.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome Inactivation by the Toxic Lectins Abrin and Ricin. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef]
- Franke, H.; Scholl, R.; Aigner, A. Ricin and Ricinus communis in pharmacology and toxicology-from ancient use and “Papyrus Ebers” to modern perspectives and “poisonous plant of the year 2018”. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1181–1208. [Google Scholar] [CrossRef] [Green Version]
- Chemical Weapons Convention | OPCW. Available online: https://www.opcw.org/chemical-weapons-convention (accessed on 30 October 2020).
- Nehring, C. Umbrella or pen? The murder of Georgi Markov. New facts and old questions. J. Intell. Hist. 2017, 16, 47–58. [Google Scholar] [CrossRef]
- Benner, K.; Draper, R. Arrest Is Made In Connection To Ricin Letter Sent to Trump. New York Times, 21 September 2020; p. 20. [Google Scholar]
- Patel, V.R.; Dumancas, G.G.; Viswanath, L.C.K.; Maples, R.; Subong, B.J.J. Castor Oil: Properties, Uses, and Optimization of Processing Parameters in Commercial Production. Lipid Insights 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Sturm, M.B.; Almo, S.C.; Schramm, V.L. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 20276–20281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiget, P.A.; Manzano, L.A.; Pruet, J.M.; Gao, G.; Saito, R.; Monzingo, A.F.; Jasheway, K.R.; Robertus, J.D.; Anslyn, E.V. Sulfur incorporation generally improves Ricin inhibition in pterin-appended glycine-phenylalanine dipeptide mimics. Bioorg. Med. Chem. Lett. 2013, 23, 6799–6804. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Pruet, J.M.; Manzano, L.A.; Jasheway, K.; Monzingo, A.F.; Wiget, P.A.; Kamat, I.; Anslyn, E.V.; Robertus, J.D. Peptide-conjugated pterins as inhibitors of ricin toxin A. J. Med. Chem. 2013, 56, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruet, J.M.; Saito, R.; Manzano, L.A.; Jasheway, K.R.; Wiget, P.A.; Kamat, I.; Anslyn, E.V.; Robertus, J.D. Optimized 5-membered heterocycle-linked pterins for the inhibition of Ricin Toxin A. ACS Med. Chem. Lett. 2012, 3, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.H.; Cheng, B.; Lin, Y.S.; Gu, Q.; He, X.H.; Xu, J. Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Choe, H.; Hong, S. Virtual screening and biochemical evaluation to identify new inhibitors of mammalian target of rapamycin (mTOR). Bioorg. Med. Chem. Lett. 2014, 24, 835–838. [Google Scholar] [CrossRef]
- Nagpal, I.; Raj, I.; Subbarao, N.; Gourinath, S. Virtual Screening, Identification and In Vitro Testing of Novel Inhibitors of O-Acetyl-L-Serine Sulfhydrylase of Entamoeba histolytica. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- De Paula, R.L.; De Almeida, J.S.F.D.; Cavalcante, S.F.A.; Gonçalves, A.S.; Simas, A.B.C.; Franca, T.C.C.; Valis, M.; Kuca, K.; Nepovimova, E.; Granjeir, J.M. Molecular Modeling and In Vitro Studies of a Neutral Oxime as a Potential Reactivator for Acetylcholinesterase Inhibited by Paraoxon. Molecules 2018, 23, 2954. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D. e-LEA3D: A computational-aided drug design web server. Nucleic Acids Res. 2010, 38, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Schrödinger LigPrep. Schrödinger Release 2019-4; LLC: New York, NY, USA, 2019. [Google Scholar]
- Abad-Zapatero, C. A Sorcerer’s apprentice and The Rule of Five: From rule-of-thumb to commandment and beyond. Drug Discov. Today 2007, 12, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Hollis, T.; Svinth, M.; Day, P.; Monzingo, A.F.; Milne, G.W.; Robertus, J.D. Structure-based identification of a ricin inhibitor. J. Mol. Biol. 1997, 266, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Watt, B.; Wahome, P.G.; Mantis, N.J.; Robertus, J.D. Identification of new classes of ricin toxin inhibitors by virtual screening. Toxicon 2010, 56, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Ferreira Neto, D.C.; Alencar Lima, J.; Sobreiro Francisco Diz de Almeida, J.; Costa França, T.C.; Jorge do Nascimento, C.; Figueroa Villar, J.D. New semicarbazones as gorge-spanning ligands of acetylcholinesterase and potential new drugs against Alzheimer’s disease: Synthesis, molecular modeling, NMR, and biological evaluation. J. Biomol. Struct. Dyn. 2018, 36, 4099–4113. [Google Scholar] [CrossRef] [PubMed]
- Chaves, E.J.F.; Padilha, I.Q.M.; Araújo, D.A.M.; Rocha, G.B. Determining the Relative Binding Affinity of Ricin Toxin A Inhibitors by Using Molecular Docking and Nonequilibrium Work. J. Chem. Inf. Model. 2018, 58, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Kooistra, A.J.; Ritschel, T.; Leurs, R.; de Esch, I.J.; de Graaf, C. Molecular interaction fingerprint approaches for GPCR drug discovery. Curr. Opin. Pharmacol. 2016, 30, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A. Approaches to Virtual Screening and Screening Library Selection. Curr. Pharm. Des. 2013, 19, 4787–4796. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Spoel, D.; van der Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.A.S.T.; Horta, B.A.C.; De Alencastro, R.B. MKTOP: A program for automatic construction of molecular topologies. J. Braz. Chem. Soc. 2008, 19, 1433–1435. [Google Scholar] [CrossRef]
- Abraham, M.J.; van der Spoel, D.; Lindahl, E.; Hess, B. GROMACS User Manual version 2018.8; Royal Institue of Technology and Uppsala University: Uppsala, Sweden, 2019. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Turner, P.J. XMGRACE; Version 5.1.25; Center for Coastal and Land-margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Gilson, M.K.; Honig, B. Calculation of the total electrostatic energy of a macromolecular system: Solvation energies, binding energies, and conformational analysis. Proteins Struct. Funct. Bioinform. 1988, 4, 7–18. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Aynechi, T.; Case, D.A.; Kuntz, I.D. Estimation of Absolute Free Energies of Hydration Using Continuum Methods: Accuracy of Partial Charge Models and Optimization of Nonpolar Contributions. J. Chem. Theory Comput. 2006, 2, 128–139. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa: A GROMACS Tool for High- Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Criterion |
|---|---|
| First criterion | Largest number of catalytic residues interacting with the pose (Glu177 and Arg180) |
| Second criterion | Largest number of residues located in the secondary pocket interacting with the pose (Asp75, Asn78, Asp96, and Asp100) |
| Third criterion | Largest number of residues involved in substrate complexation interacting with the pose (Tyr80, Val81, Gly121, Tyr123, Asn209, Trp211) |
| Fourth criterion | Lowest MolDock score |
| Group | Molecule 1 | MolDock Score (kcal mol−1) | Residues Forming H-bonds with the Pose 3 |
|---|---|---|---|
| --- | NNPT 2 | −138.40 | Arg180Asn78Tyr80Val81 |
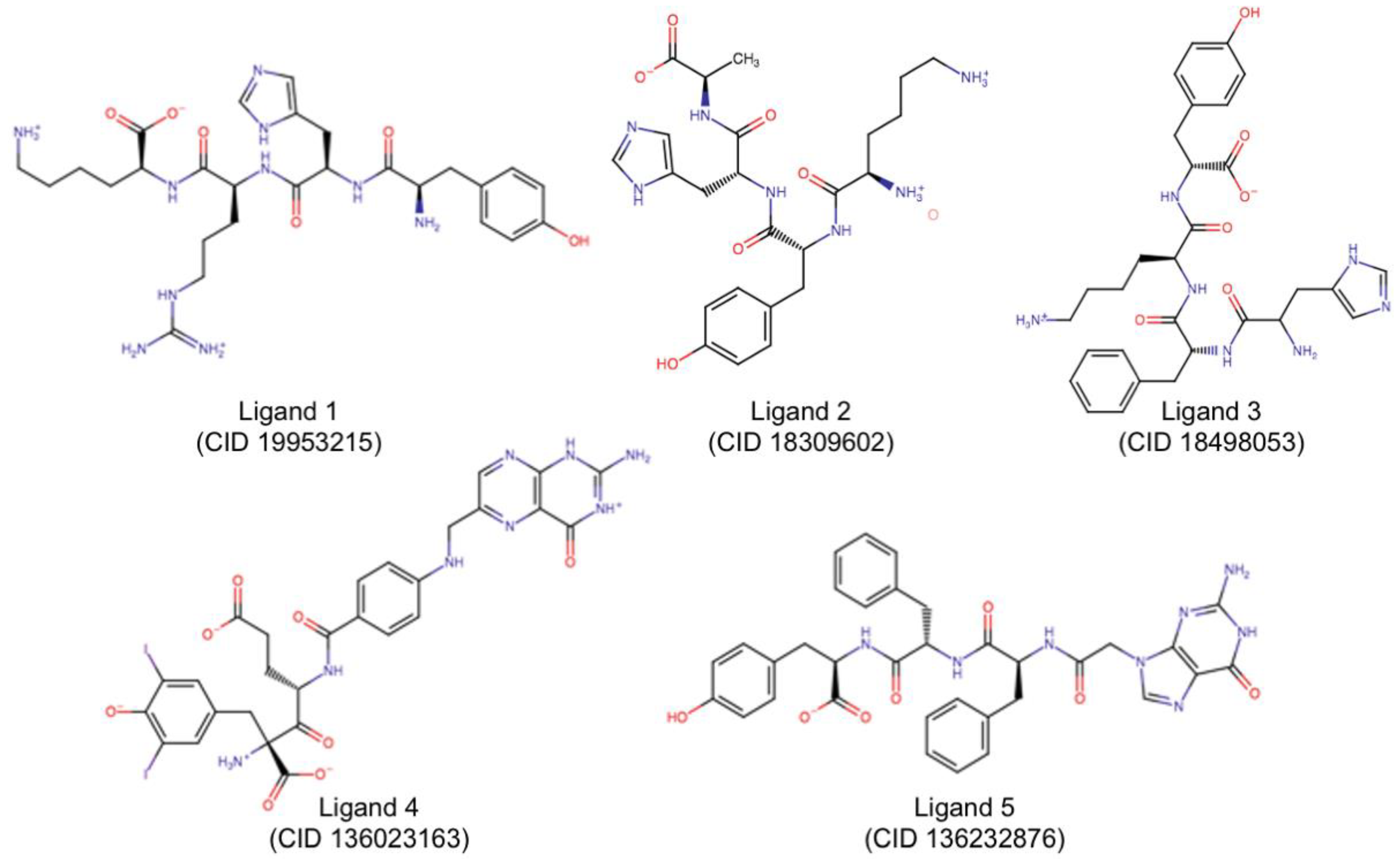
| Group 1 | 19953215 | −160.63 | Glu177Arg180Asp75Asp96Asp100Tyr123Trp211 Asn122 Gly212 Arg258 Glu208 |
| Group 2 | 18309602 | −152.14 | Glu177Arg180Asp75Asp96Asp100Tyr123Asn209 Asn122 Asp124 Glu208 |
| Group 3 | 18498053 | −161.20 | Glu177Arg180Asn78Asp96Asp100Val81 Asn122 Ser176 Glu208 Arg258 |
| Group 4 | 136023163 | −203.93 | Arg180Asn78Asp96Asp100Tyr80Val81Gly121Tyr123 Arg56 Thr77 Arg258 |
| Group 5 | 136232876 | −157.66 | Arg180Asn78Asp96Asp100Trp211 Thr77 Asn122 Glu208 Gly212 |
| Group | Molecule CID | Mut 1 | Tumor | Irr | cLogP | Sol | Mol. Weight | Drug Score | H Donor | H Acceptor |
|---|---|---|---|---|---|---|---|---|---|---|
| --- | NNPT | N | N | N | −2.16 | −2.04 | 427 | 0.42 | 6 | 9 |
| 1 | 19953215 | N | N | N | −6.85 | −2.74 | 602 | 0.29 | 10 | 10 |
| 1 | 18305509 | N | N | N | −6.85 | −2.74 | 602 | 0.29 | 10 | 10 |
| 1 | 18493267 | N | N | N | −6.85 | −2.74 | 602 | 0.29 | 10 | 10 |
| 1 | 18243472 | N | N | N | −5.52 | −1.99 | 531 | 0.34 | 9 | 9 |
| 1 | 67312445 | N | N | N | −5.52 | −1.99 | 531 | 0.34 | 9 | 9 |
| 2 | 18309602 | N | N | N | −5.77 | −2.03 | 517 | 0.35 | 8 | 9 |
| 2 | 18309609 | N | N | N | −6.13 | −1.65 | 503 | 0.37 | 8 | 9 |
| 2 | 18499956 | N | N | N | −6.13 | −1.65 | 503 | 0.37 | 8 | 9 |
| 2 | 18305842 | N | N | N | −7.46 | −2.41 | 574 | 0.31 | 9 | 10 |
| 2 | 18500025 | N | N | N | −5.77 | −2.03 | 517 | 0.35 | 8 | 9 |
| 2 | 18306834 | N | N | N | −7.46 | −2.41 | 574 | 0.31 | 9 | 10 |
| 2 | 19953410 | N | N | N | −6.13 | −1.65 | 503 | 0.37 | 8 | 9 |
| 2 | 22659428 | N | N | N | −6.99 | −1.85 | 560 | 0.33 | 9 | 10 |
| 2 | 19953311 | N | N | N | −5.20 | −1.37 | 503 | 0.37 | 8 | 9 |
| 2 | 19953235 | N | N | N | −6.99 | −1.85 | 560 | 0.33 | 9 | 10 |
| 2 | 18309613 | N | N | N | −7.46 | −2.41 | 574 | 0.31 | 9 | 10 |
| 3 | 18498053 | N | N | N | −4.33 | −3.17 | 593 | 0.29 | 8 | 9 |
| 3 | 18500076 | N | N | N | −4.33 | −3.17 | 593 | 0.29 | 8 | 9 |
| 3 | 18500176 | N | N | N | −4.67 | −2.87 | 609 | 0.29 | 9 | 10 |
| 3 | 20044260 | N | N | N | −4.33 | −3.17 | 593 | 0.29 | 8 | 9 |
| 3 | 18492007 | N | N | N | −3.00 | −2.41 | 522 | 0.35 | 7 | 8 |
| 3 | 18500043 | N | N | N | −4.67 | −2.87 | 609 | 0.29 | 9 | 10 |
| 3 | 18499958 | N | N | N | −3.00 | −2.41 | 522 | 0.35 | 7 | 8 |
| 4 | 136023163 | N | N | N | −2.59 | −5.69 | 856 | 0.27 | 8 | 13 |
| 4 | 135977982 | N | N | N | −3.36 | −3.97 | 622 | 0.30 | 8 | 14 |
| 4 | 136149436 | N | N | N | −4.02 | −4.67 | 730 | 0.32 | 8 | 13 |
| 4 | 136132835 | N | N | N | −2.92 | −4.98 | 748 | 0.23 | 8 | 14 |
| 5 | 136232876 | S | N | N | 0.09 | −4.35 | 666 | 0.14 | 7 | 9 |
| Mut 1 | Tumor | Irr | cLogP | Sol | Mol. Weight | Drug Score | H Donor | H Acceptor | |
|---|---|---|---|---|---|---|---|---|---|
| Reference values | N | N | N | <5 | >−4 | <500 | Close to 1 | <5 | <10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botelho, F.D.; dos Santos, M.C.; Gonçalves, A.d.S.; Kuca, K.; Valis, M.; LaPlante, S.R.; França, T.C.C.; de Almeida, J.S.F.D. Ligand-Based Virtual Screening, Molecular Docking, Molecular Dynamics, and MM-PBSA Calculations towards the Identification of Potential Novel Ricin Inhibitors. Toxins 2020, 12, 746. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120746
Botelho FD, dos Santos MC, Gonçalves AdS, Kuca K, Valis M, LaPlante SR, França TCC, de Almeida JSFD. Ligand-Based Virtual Screening, Molecular Docking, Molecular Dynamics, and MM-PBSA Calculations towards the Identification of Potential Novel Ricin Inhibitors. Toxins. 2020; 12(12):746. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120746
Chicago/Turabian StyleBotelho, Fernanda D., Marcelo C. dos Santos, Arlan da S. Gonçalves, Kamil Kuca, Martin Valis, Steven R. LaPlante, Tanos C. C. França, and Joyce S. F. D. de Almeida. 2020. "Ligand-Based Virtual Screening, Molecular Docking, Molecular Dynamics, and MM-PBSA Calculations towards the Identification of Potential Novel Ricin Inhibitors" Toxins 12, no. 12: 746. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120746