Gymnodimine A and 13-desMethyl Spirolide C Alter Intracellular Calcium Levels via Acetylcholine Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
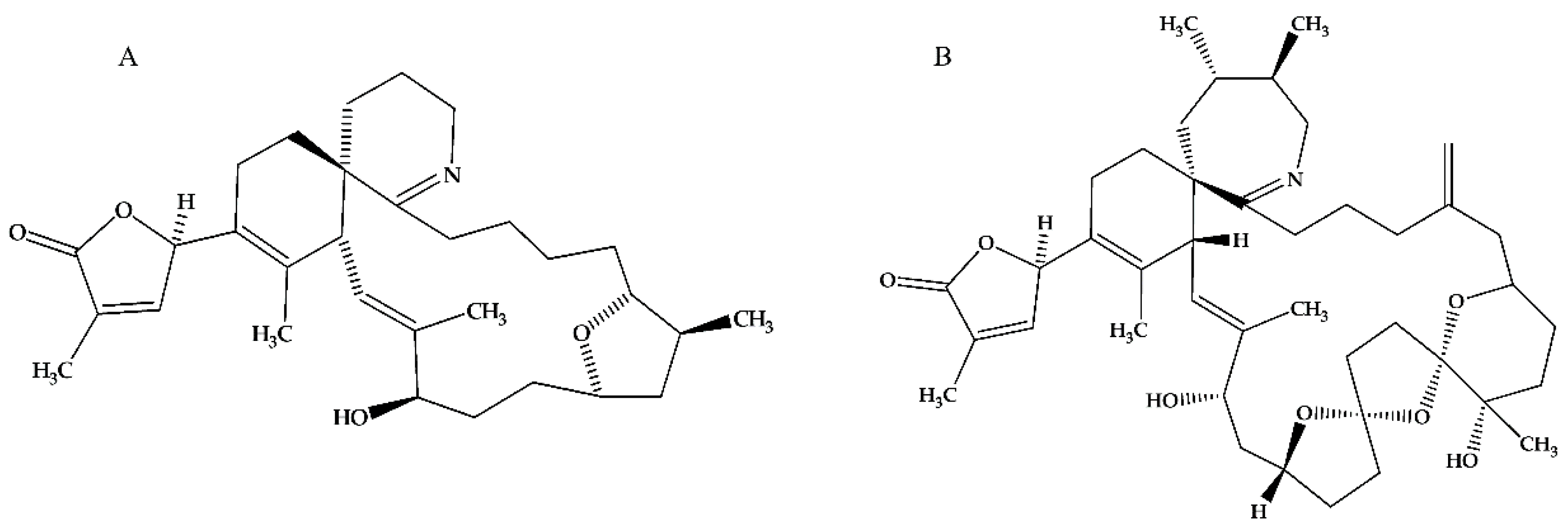
2. Results
2.1. Depolarization of Ca Channels Using K+
2.2. [Ca]i Changes Induced by ACh
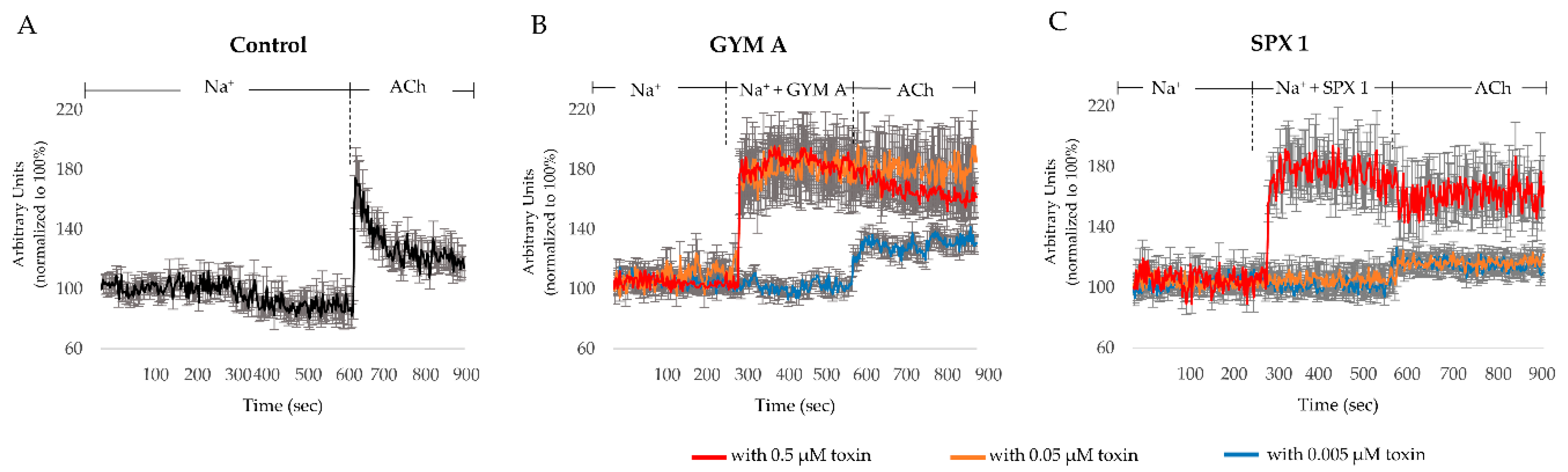
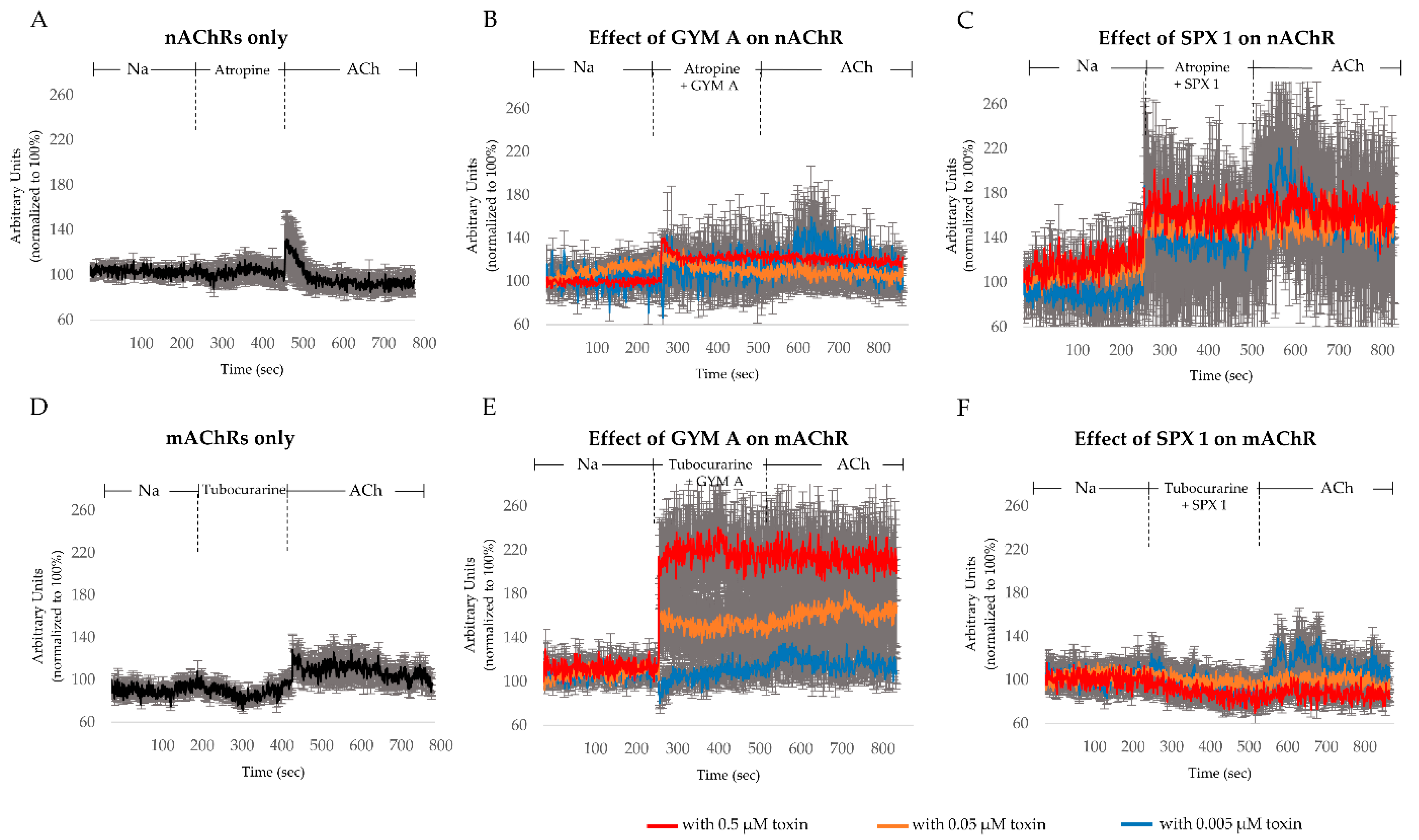
3. Discussion
3.1. GYM A and SPX 1 Do not Inhibit the Influx of [Ca]i through Voltage-Gated Ca Channels
3.2. GYM A and SPX 1 Alter [Ca]i
3.3. GYM A and SPX 1 Show a Similar Effect on nAChRs and a Differential Response to mAChRs
3.4. GYM A and SPX Alter [Ca]i through nAChRs and mAChRs
4. Materials and Methods
4.1. Extraction and Purification of Gymnodimine A and 13-Desmethyl Spirolide C
4.2. PC 12 Culture Methods
4.3. Fluorimetric Measurements of Intracellular Calcium Levels
4.4. [Ca]i Measurements Using K+ Depolarization
4.5. [Ca]i Measurements with Application of Acetylcholine
Author Contributions
Funding
Conflicts of Interest
References
- Gobler, C.J. Climate change and harmful algal blooms: Insights and perspective. Harmful Algae 2020, 91. [Google Scholar] [CrossRef] [PubMed]
- Guéret, S.M.; Brimble, M.A. Synthetic studies toward the spiroimine unit of the spirolides. Pure Appl. Chem. 2011, 83, 425–433. [Google Scholar] [CrossRef]
- Cembella, A.D.; Lewis, N.I.; Quilliam, M.A. The marine dinoflagellate Alexandrium ostenfeldii (Dinophyceae) as the causative organism of spirolide shellfish toxins. Phycologia 2000, 39, 67–74. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Misner, I.; Tomas, C.R.; Wright, J.L.C. Occurrence of 12-methylgymnodimine in a spirolide-producing dinoflagellate Alexandrium peruvianum and the biogenetic implications. Tetrahedron Lett. 2011, 52, 4243–4246. [Google Scholar] [CrossRef]
- Van de Waal, D.B.; Tillmann, U.; Martens, H.; Krock, B.; van Scheppingen, Y.; John, U. Characterization of multiple isolates from an Alexandrium ostenfeldii bloom in The Netherlands. Harmful Algae 2015, 49, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Harju, K.; Koskela, H.; Kremp, A.; Suikkanen, S.; de la Iglesia, P.; Miles, C.O.; Krock, B.; Vanninen, P. Identification of gymnodimine D and presence of gymnodimine variants in the dinoflagellate Alexandrium ostenfeldii from the Baltic Sea. Toxicon 2016, 112, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Martens, H.; Tillmann, U.; Harju, K.; Dell’Aversano, C.; Tartaglione, L.; Krock, B. Toxin Variability Estimations of 68 Alexandrium ostenfeldii (Dinophyceae) Strains from The Netherlands Reveal a Novel Abundant Gymnodimine. Microorganisms 2017, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Zurhelle, C.; Nieva, J.; Tillmann, U.; Harder, T.; Krock, B.; Tebben, J. Identification of Novel Gymnodimines and Spirolides from the Marine Dinoflagellate Alexandrium ostenfeldii. Mar. Drugs 2018, 16, 446. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Satake, M.; MacKenzie, L.; Kaspar, H.F.; Yasumoto, T. Gymnodimine, a new marine toxin of unprecedented structure isolated from New Zealand oysters and the dinoflagellate, Gymnodinium sp. Tetrahedron Lett. 1995, 36, 7093–7096. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Satake, M.; Wright, J.L.C. Polyketide biosynthesis in dinoflagellate: What makes it different? Nat. Prod. Rep. 2014, 31, 1101. [Google Scholar] [CrossRef]
- Otero, P.; Alfonso, A.; Rodríguez, P.; Rubiolo, J.A.; Cifuentes, J.M.; Bermúdez, R.; Vieytes, M.R.; Botana, L.M. Pharmacokinetic and toxicological data of spirolides after oral and intraperitoneal administration. Food Chem. Toxicol. 2012, 50, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Murphy, M.; Clausen, J.; Richard, D.; Quilliam, M.; MacKinnon, S.; LaBlanc, P.; Mueller, R.; Pulido, O. Neural Injury Biomarkers of Novel Shellfish Toxins, Spirolides: A Pilot Study Using Immunochemical and Transcriptional Analysis. Neurotoxicology 2003, 24, 593–604. [Google Scholar] [CrossRef]
- Munday, R.; Towers, N.R.; Mackenzie, L.; Beuzenberg, V.; Hollan, P.T.; Miles, C.O. Acute toxicity of gymnodimine to mice. Toxicon 2004, 44, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, R.; Servent, D.; Girard, E.; Ouanounou, G.; Amar, M.; Marrouchi, R.; Benoit, E.; Molgo, J. The marine phycotoxin gymnodimin targets muscular and neuronal nicotinic acetylcholine receptor subtypes with high affinity. J. Neurochem. 2008, 107, 952–963. [Google Scholar] [CrossRef]
- Aráoz, R.; Ouanounou, G.; Iorga, B.I.; Goudet, A.; Alili, D.; Amar, M.; Benoit, E.; Molgó, J.; Servent, D. The Neurotoxic Effect of 13,19-Didesmethyl and 13-Desmethyl Spirolide C Phycotoxins Is Mainly Mediated by Nicotinic Rather Than Muscarinic Acetylcholine Receptors. Toxicol. Sci. 2015, 147, 156–157. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Radić, Z.; Aráoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgó, J.; Marchot, P. Structural determinants in phycotoxins an AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef] [Green Version]
- Hauser, T.A.; Hepler, C.D.; Kombo, D.C.; Grinevich, V.P.; Kiser, M.N.; Hooker, D.N.; Zhang, J.; Mountfort, D.; Selwood, A.; Akireddy, S.R.; et al. Comparison of acetylcholine receptor interactions of the marine toxins, 13-desmethylspirolide C and gymnodimine. Neuropharmacology 2012, 62, 2239–2250. [Google Scholar] [CrossRef]
- Wandscheer, C.B.; Vilariño, N.; Espiña, B.; Louzao, M.C.; Botana, L.M. Human muscarinic acetylholine receptors are a target of the marine toxin 13-desmethyl C spirolide. Chem. Res. Toxicol. 2010, 11, 1753–1761. [Google Scholar] [CrossRef]
- Friis, S.; Mathes, C.; Sunesen, M.; Bowlby, M.R.; Dunlop, J. Characterization of compounds on nicotinic acetylcholine receptor alpha7 channels using high throughput electrophysiology. J. Neurosci. Methods 2009, 177, 142–148. [Google Scholar] [CrossRef]
- Shafer, T.J.; Atchison, W.D. Transmitter, ion channel and receptor properties of pheochromocytoma (PC12) cells: A model for neurotoxicological studies. Neurotoxicology 1991, 12, 473–492. [Google Scholar]
- Seta, K.; Kim, H.W.; Ferguson, T.; Kim, R.; Pathrose, P.; Yuan, Y.; Lu, G.; Spicer, Z.; Millhorn, D.E. Genomic and Physiological Analysis of Oxygen Sensitivity and Hypoxia Tolerance in PC12 Cells. Ann. N. Y. Acad. Sci. 2002, 971, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995, 64, 493–531. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Therapeut. 2013, 1, 22–54. [Google Scholar] [CrossRef]
- Brann, M.R.; Ellis, J.; Jørgensen, H.; Hill-Eubanks, D.; Jones, S.V.P. Chapter 12: Muscarinic acetylcholine receptor subtypes: Localization and structure/function. Prog. Brain Res. 1993, 98, 121–127. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamashita, H.; Nakamura, S.; Ishiguro, H.; Nagatsu, T.; Kawakami, H. Effects of nerve growth factor and nicotine on the expression of nicotinic acetylcholine receptor subunits in PC12 cells. J. Neurosci. Sci. 1999, 35, 175–181. [Google Scholar] [CrossRef]
- Michel, A.D.; Stefanich, E.; Whiting, R.L. PC12 phaeochromocytoma cells contain an atypical muscarinic receptor binding site. Br. J. Pharmacol. 1989, 97, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility of calcium signaling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bickmeyer, U.; Grube, A.; Klings, K.W.; Pawlick, J.R.; Köck, M. Siphonodictyal B1 from a Marine Spong Increases Intracellular Calcium Levels Comparable to Ca2+-ATPase (SERCA) Inhibitor Thapsigargin. Mar. Biotechnol. 2010, 12, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Spalding, T.A.; Burstein, E.S. Constitutive activity of muscarinic acetylcholine receptors. J. Recept. Sig. Transd. 2006, 26, 61–65. [Google Scholar] [CrossRef]
- Zwart, R.; Vijverberg, H.P.M. Potentiation and inhibition of neuronal nicotinic receptors by atropine: Competitive and non-competitive effects. Mol. Pharmacol. 1997, 52, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.C.; Sarkar, D.; Quick, M.W.; Lester, R.A.J. Interactions of atropine with heterogously expressed and native α3 subunit-containing nicotine acetylcholine receptors. Br. J. Pharmacol. 2003, 138, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2008, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eglen, R.M. Muscarinic Receptor Subtype Pharmacology and Physiology. Prog. Med. Chem. 2005, 43, 105–136. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieva, J.A.; Krock, B.; Tillmann, U.; Tebben, J.; Zurhelle, C.; Bickmeyer, U. Gymnodimine A and 13-desMethyl Spirolide C Alter Intracellular Calcium Levels via Acetylcholine Receptors. Toxins 2020, 12, 751. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120751
Nieva JA, Krock B, Tillmann U, Tebben J, Zurhelle C, Bickmeyer U. Gymnodimine A and 13-desMethyl Spirolide C Alter Intracellular Calcium Levels via Acetylcholine Receptors. Toxins. 2020; 12(12):751. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120751
Chicago/Turabian StyleNieva, Joyce A., Bernd Krock, Urban Tillmann, Jan Tebben, Christian Zurhelle, and Ulf Bickmeyer. 2020. "Gymnodimine A and 13-desMethyl Spirolide C Alter Intracellular Calcium Levels via Acetylcholine Receptors" Toxins 12, no. 12: 751. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120751