Analysis of Covalently Bound Microcystins in Sediments and Clam Tissue in the Sacramento–San Joaquin River Delta, California, USA

 ,
,
Abstract
:1. Introduction
2. Results
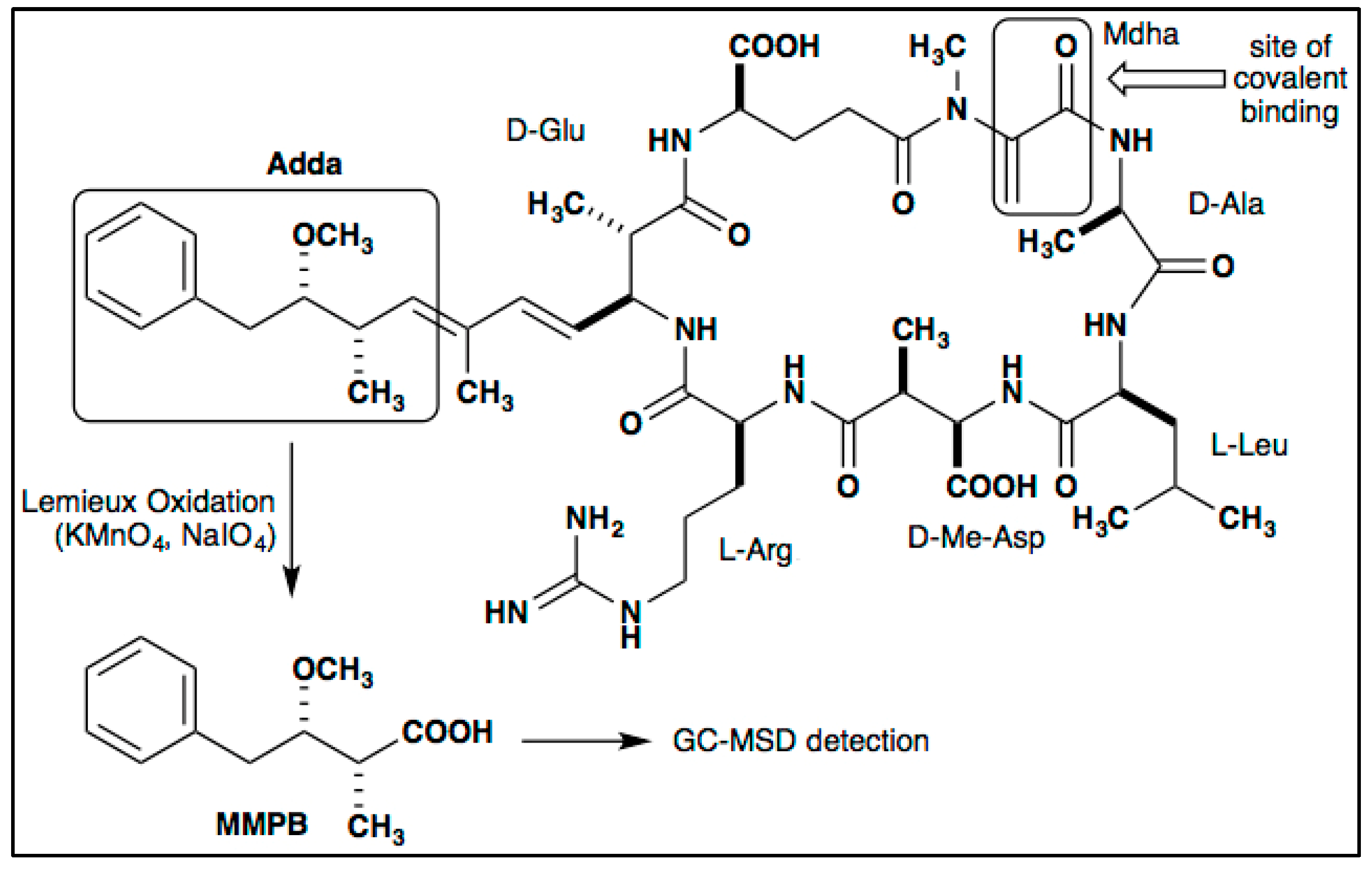
2.1. Optimization of Total MC Quantification (Bound and Unbound)
2.1.1. Hexane Extraction
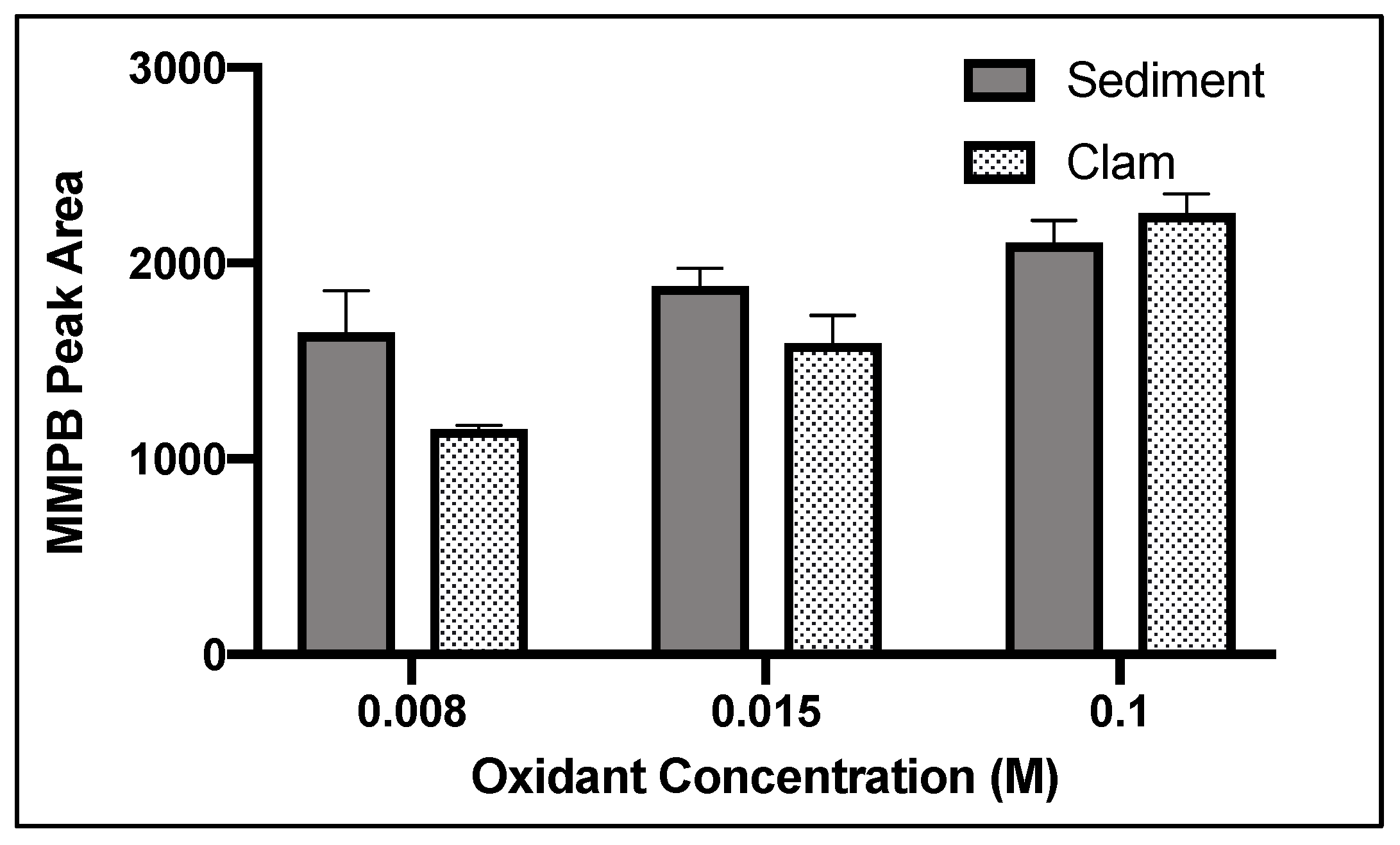
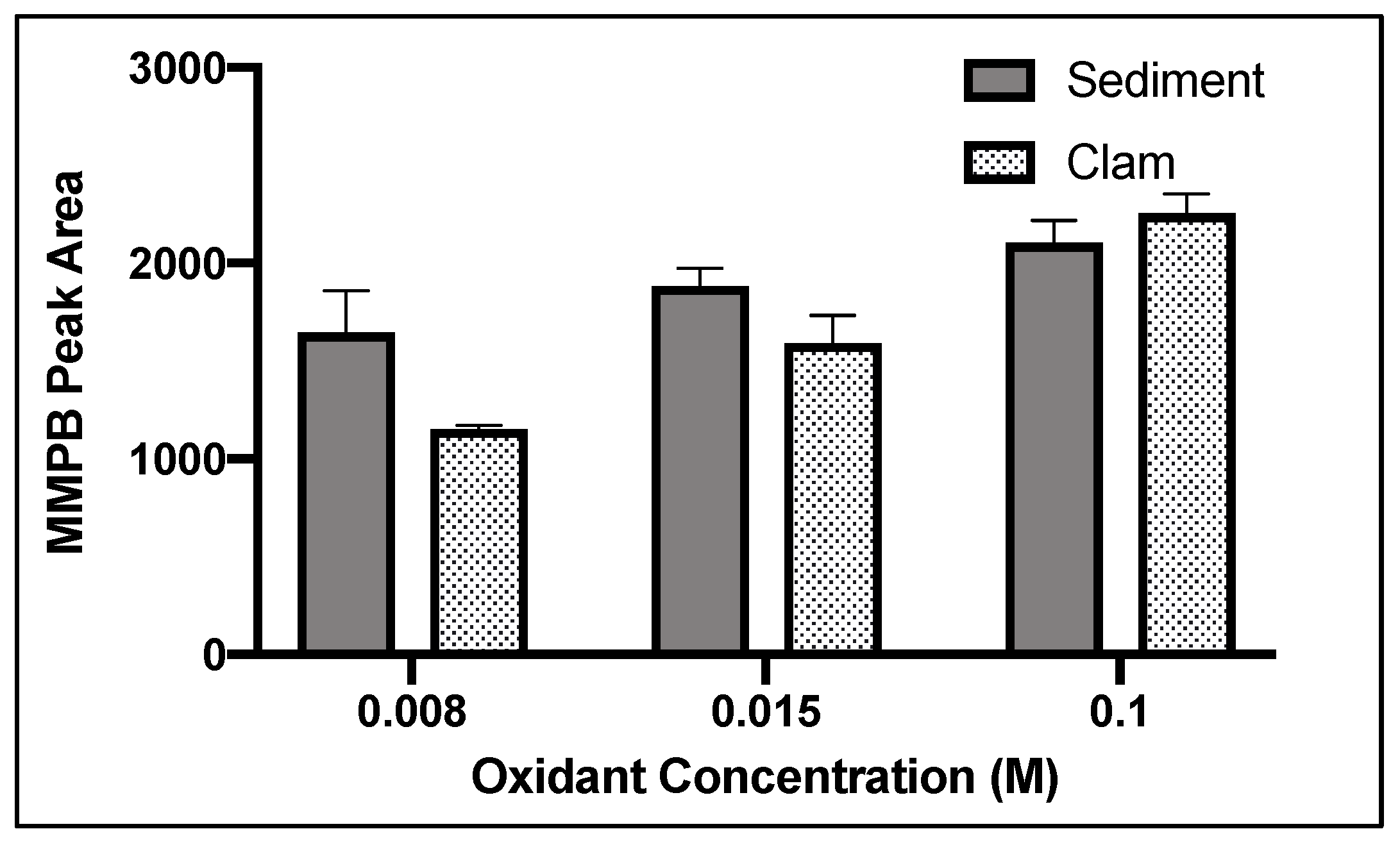
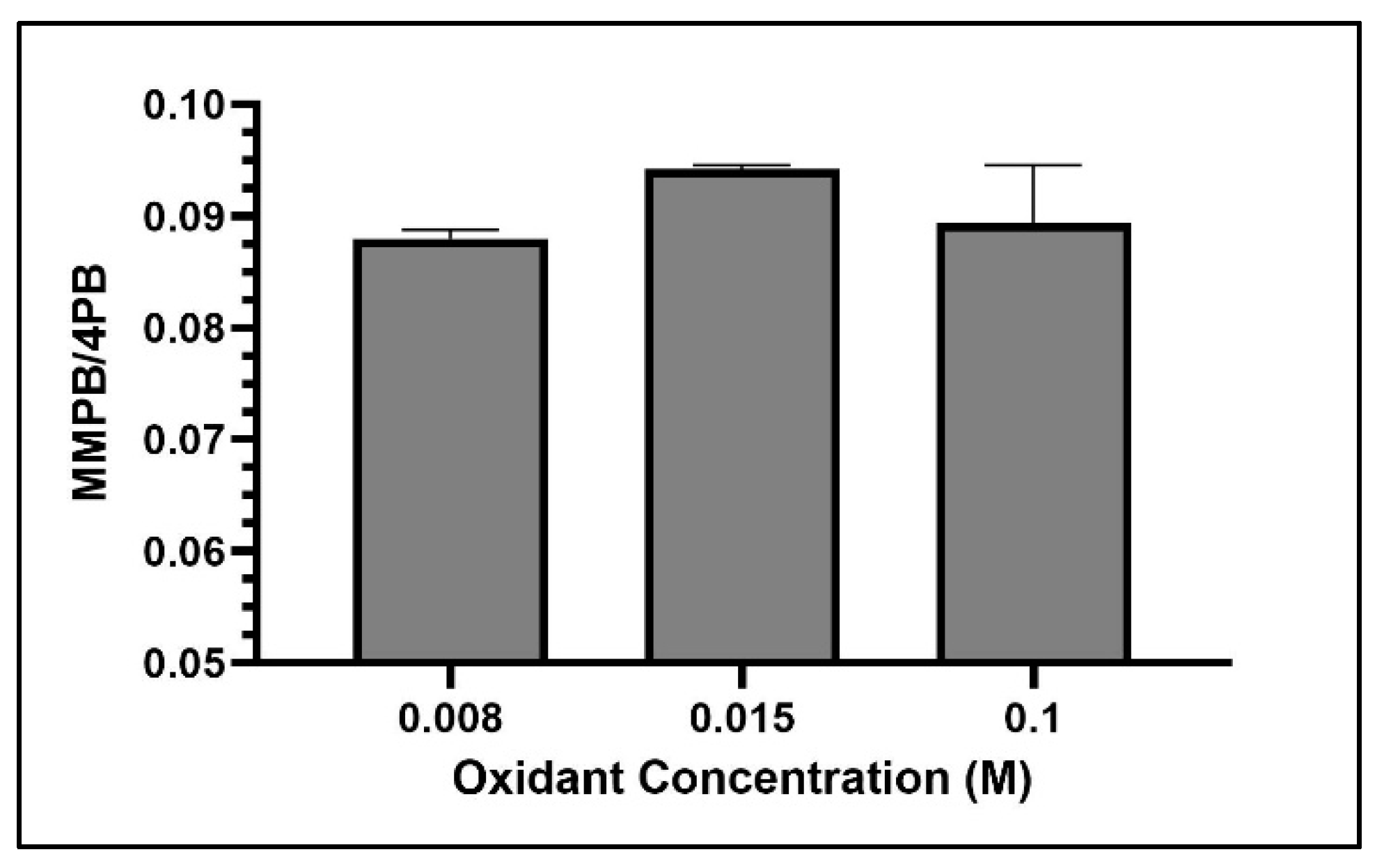
2.1.2. Oxidant Concentration
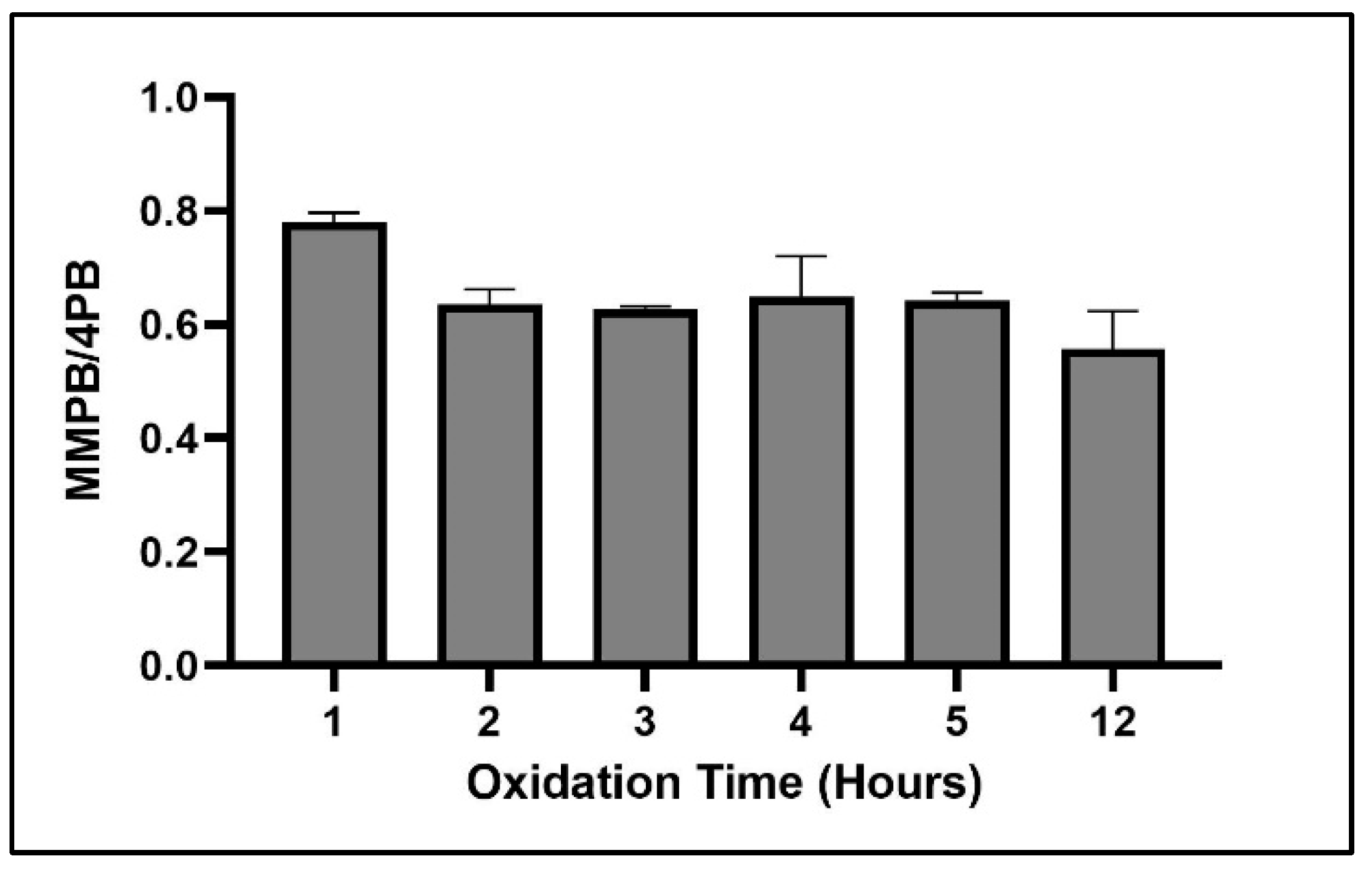
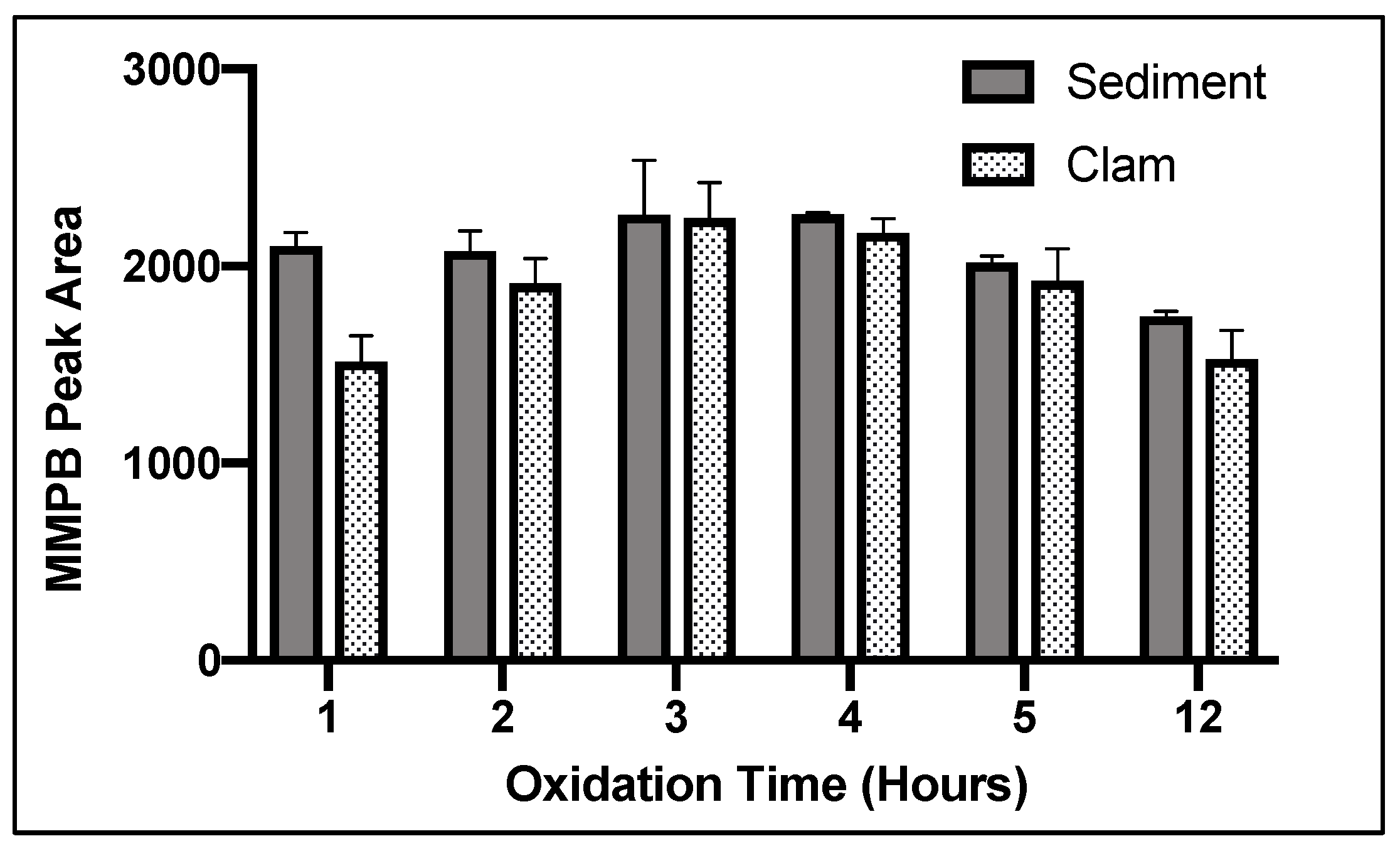
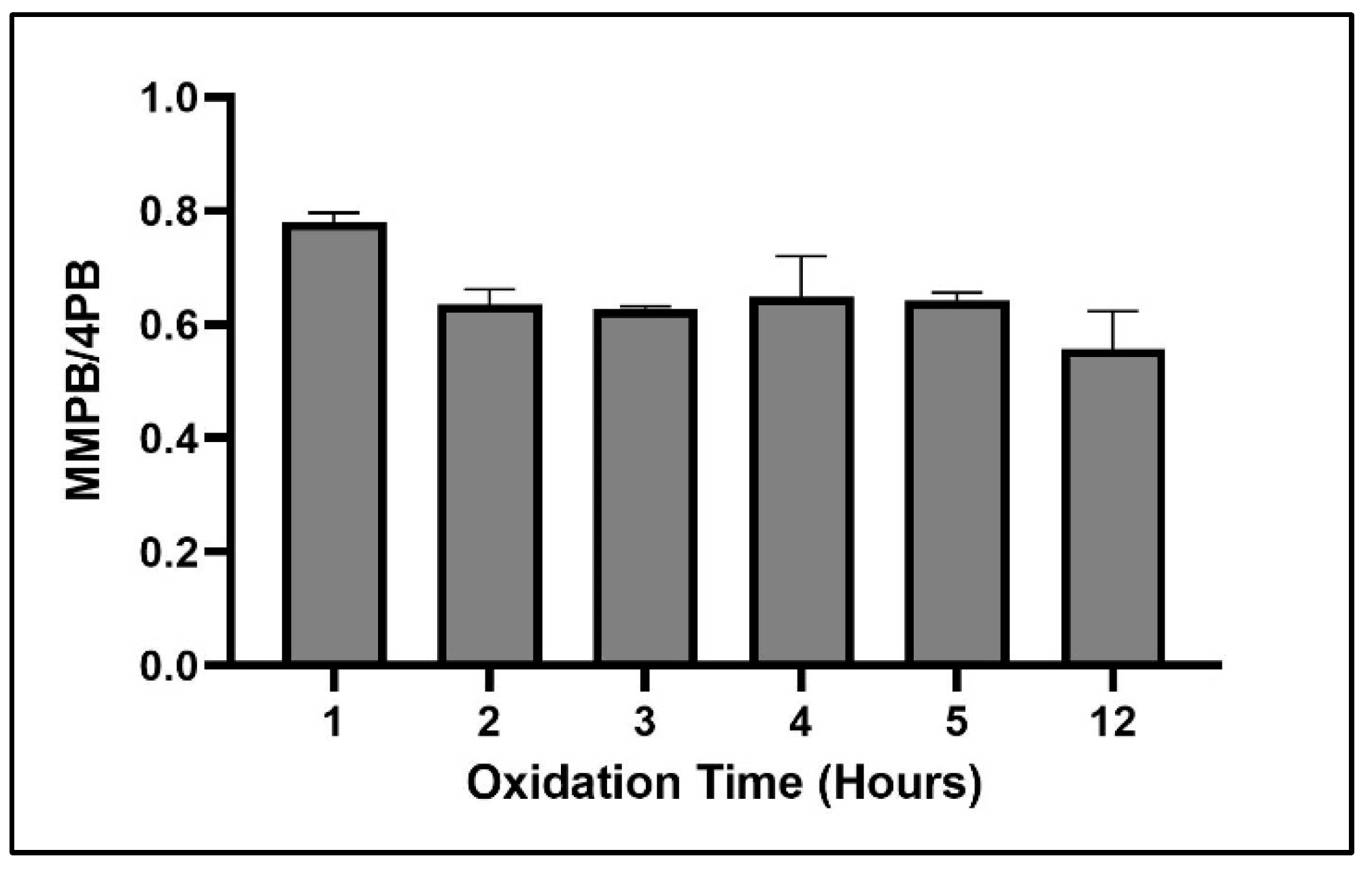
2.1.3. Oxidation Time
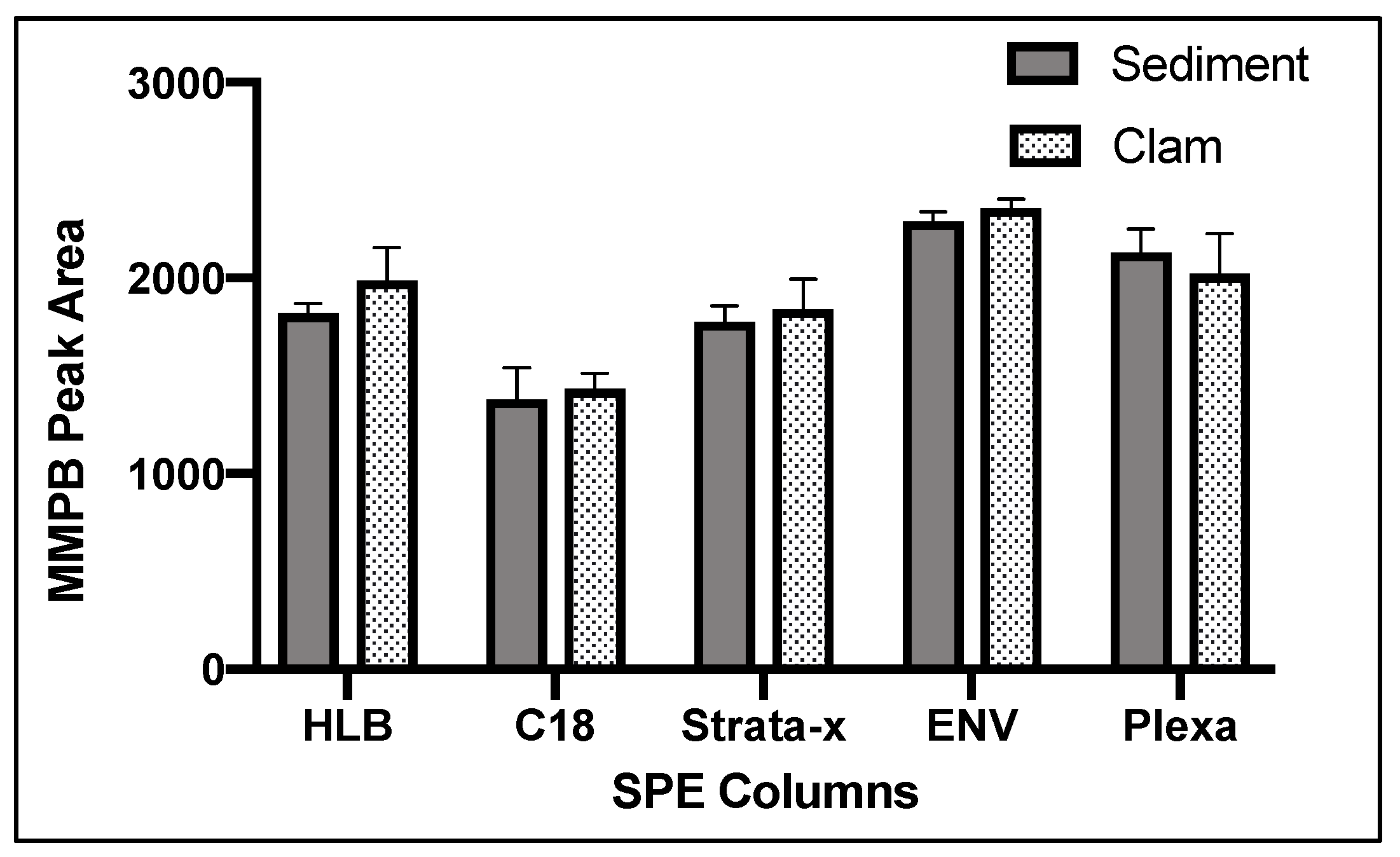
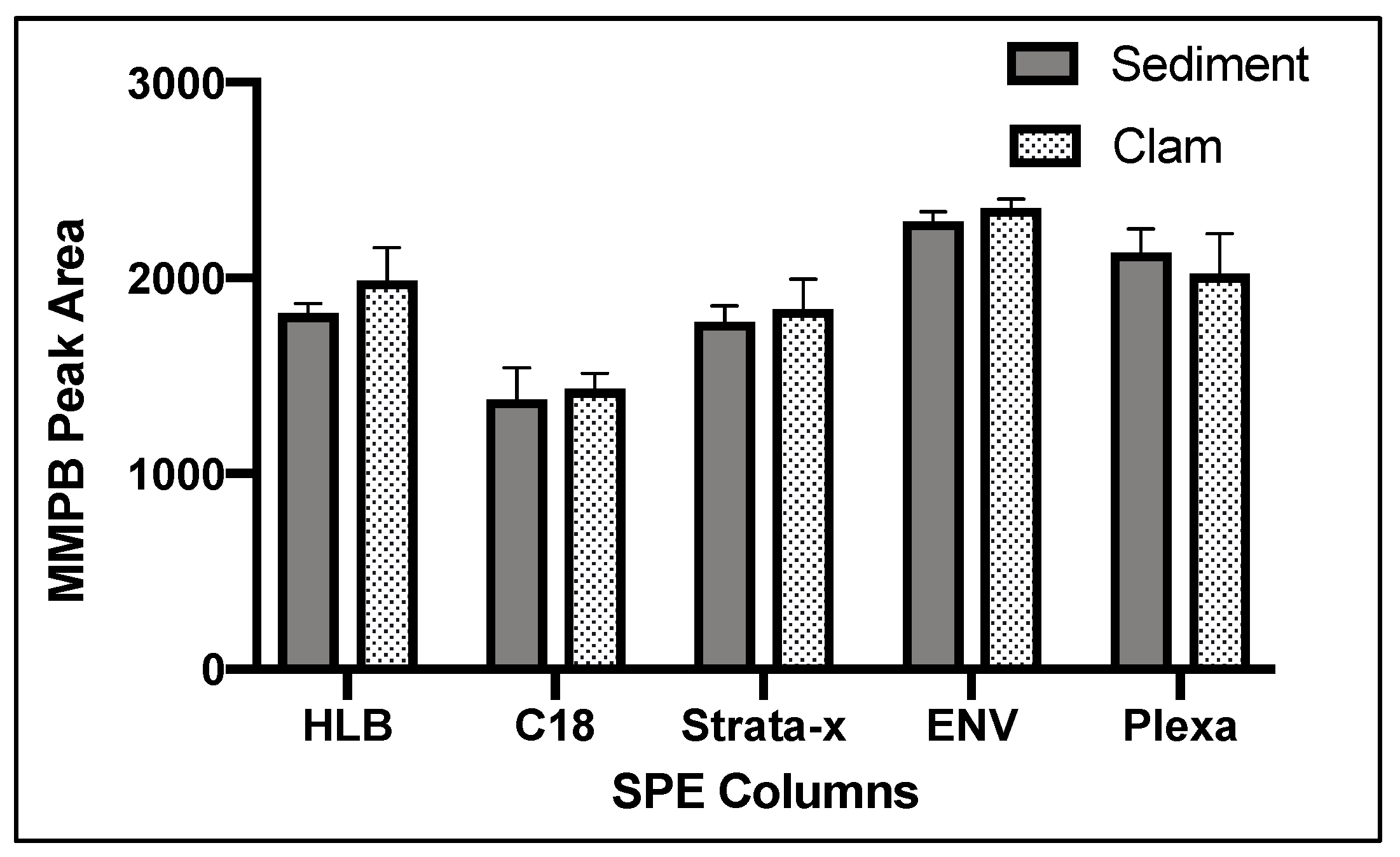
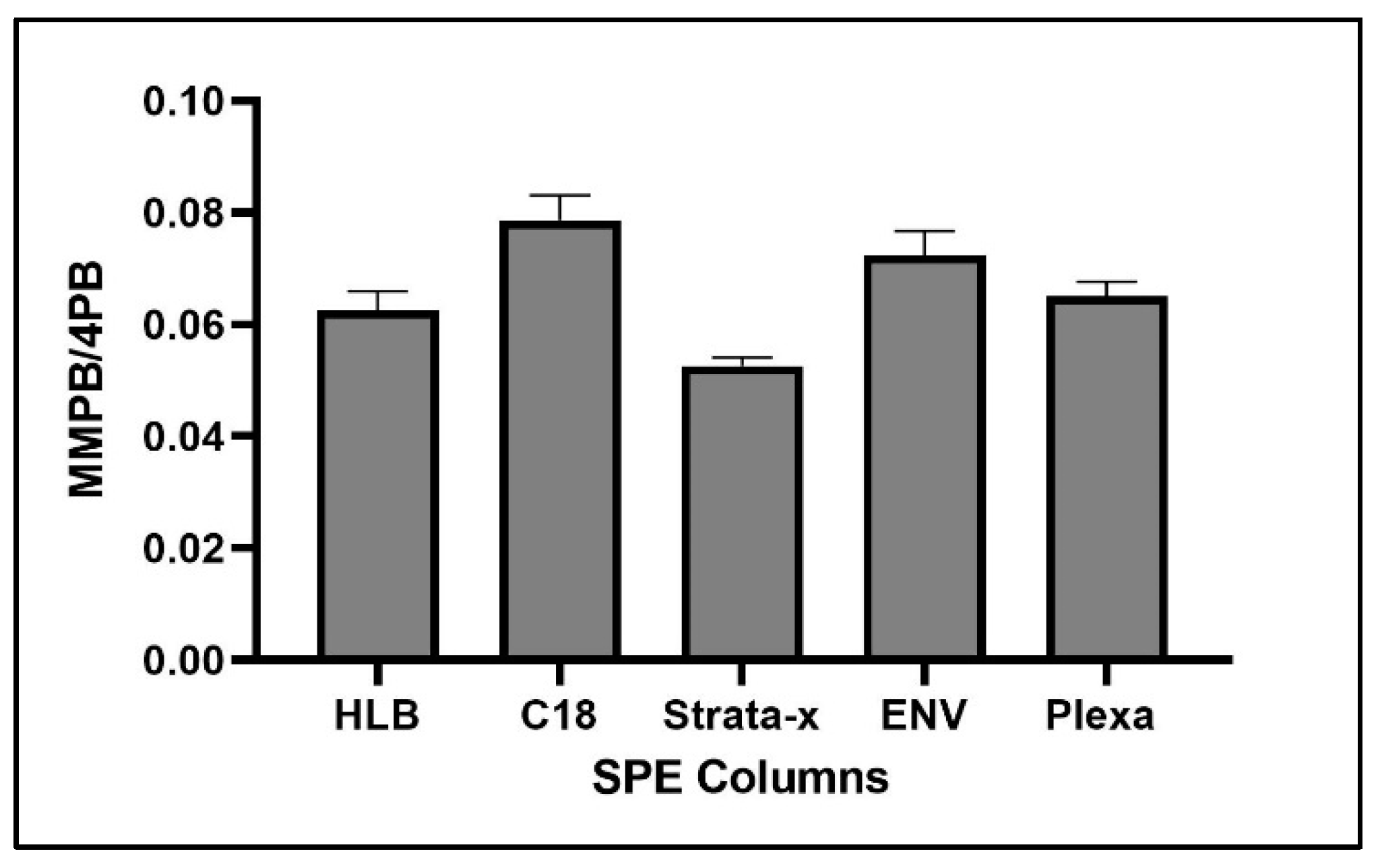
2.1.4. Solid-Phase Extraction (SPE)
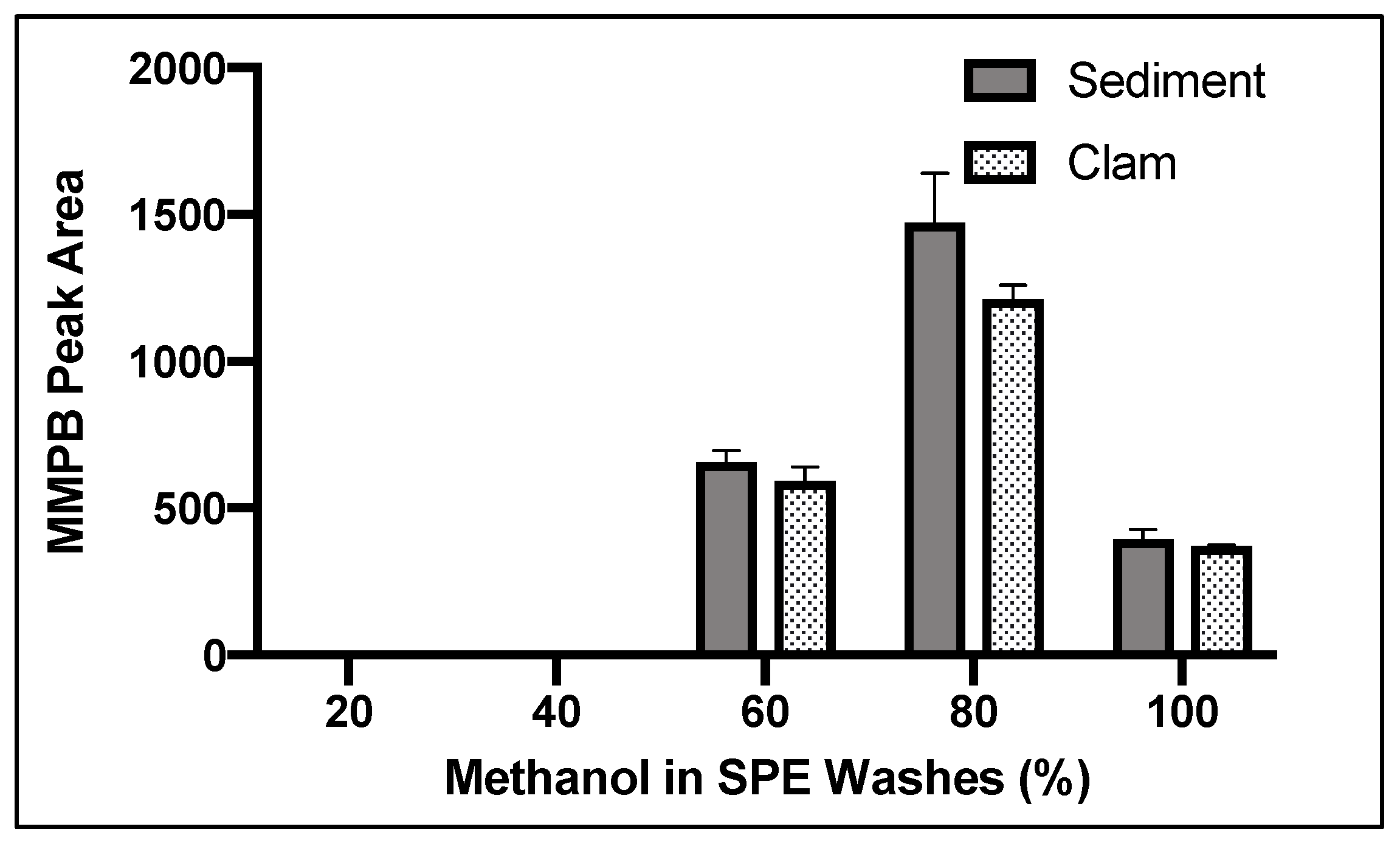
2.1.5. SPE Washes
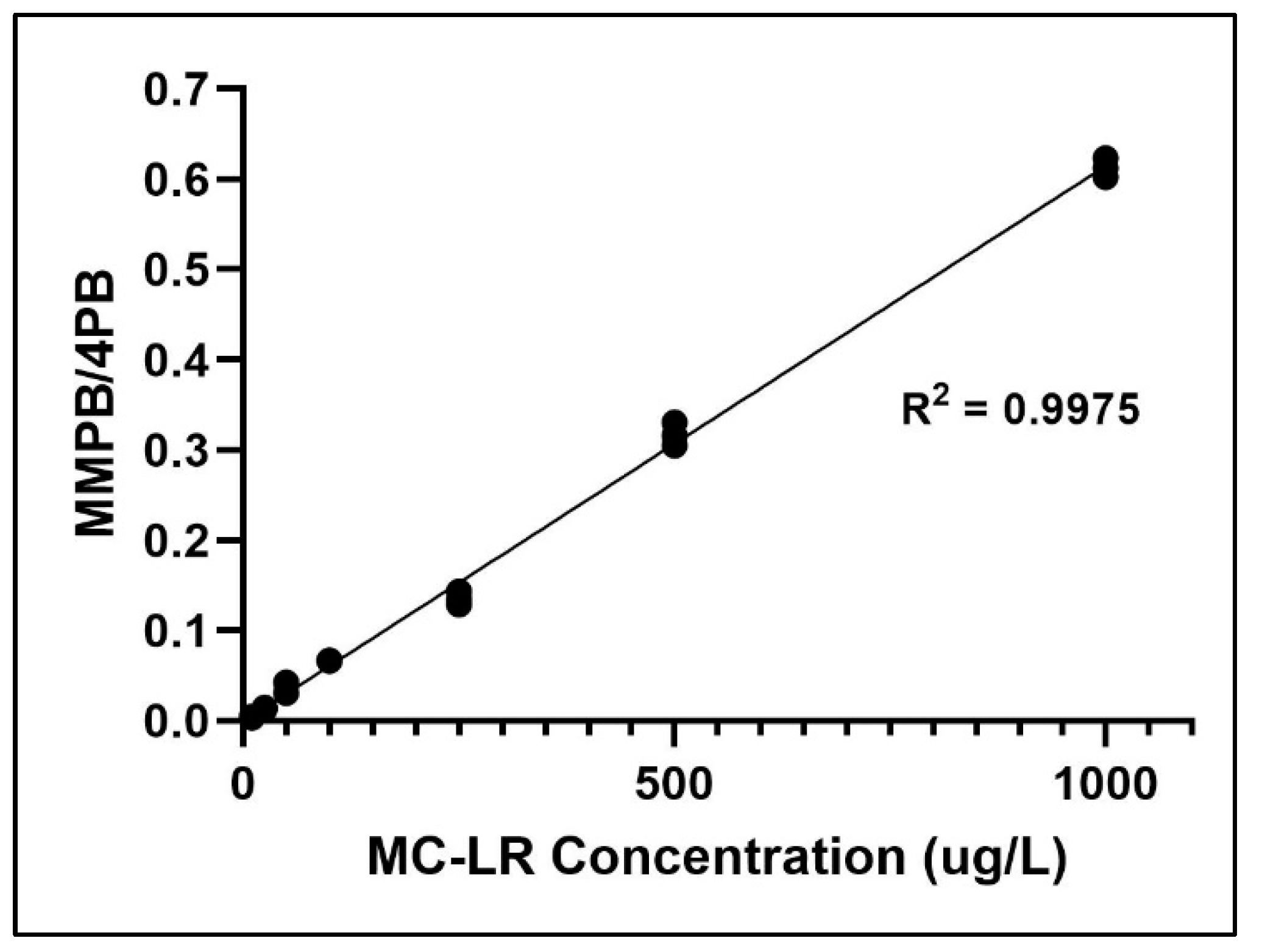
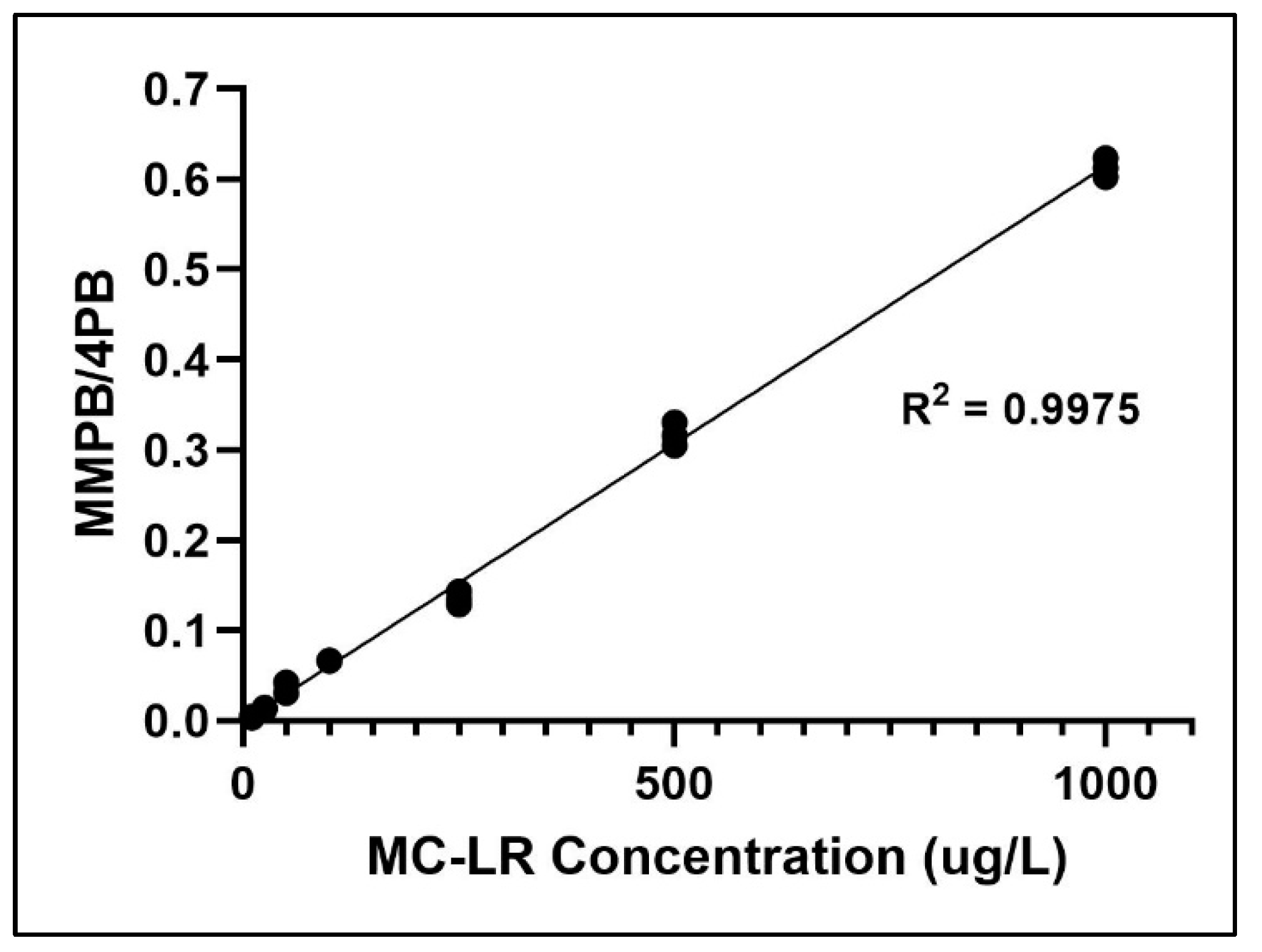
2.2. Method Validation
2.3. Loss of Product
2.4. Optimization of Unbound MC Quantification
2.4.1. Oxidant Concentration
2.4.2. Oxidation Time
2.4.3. SPE
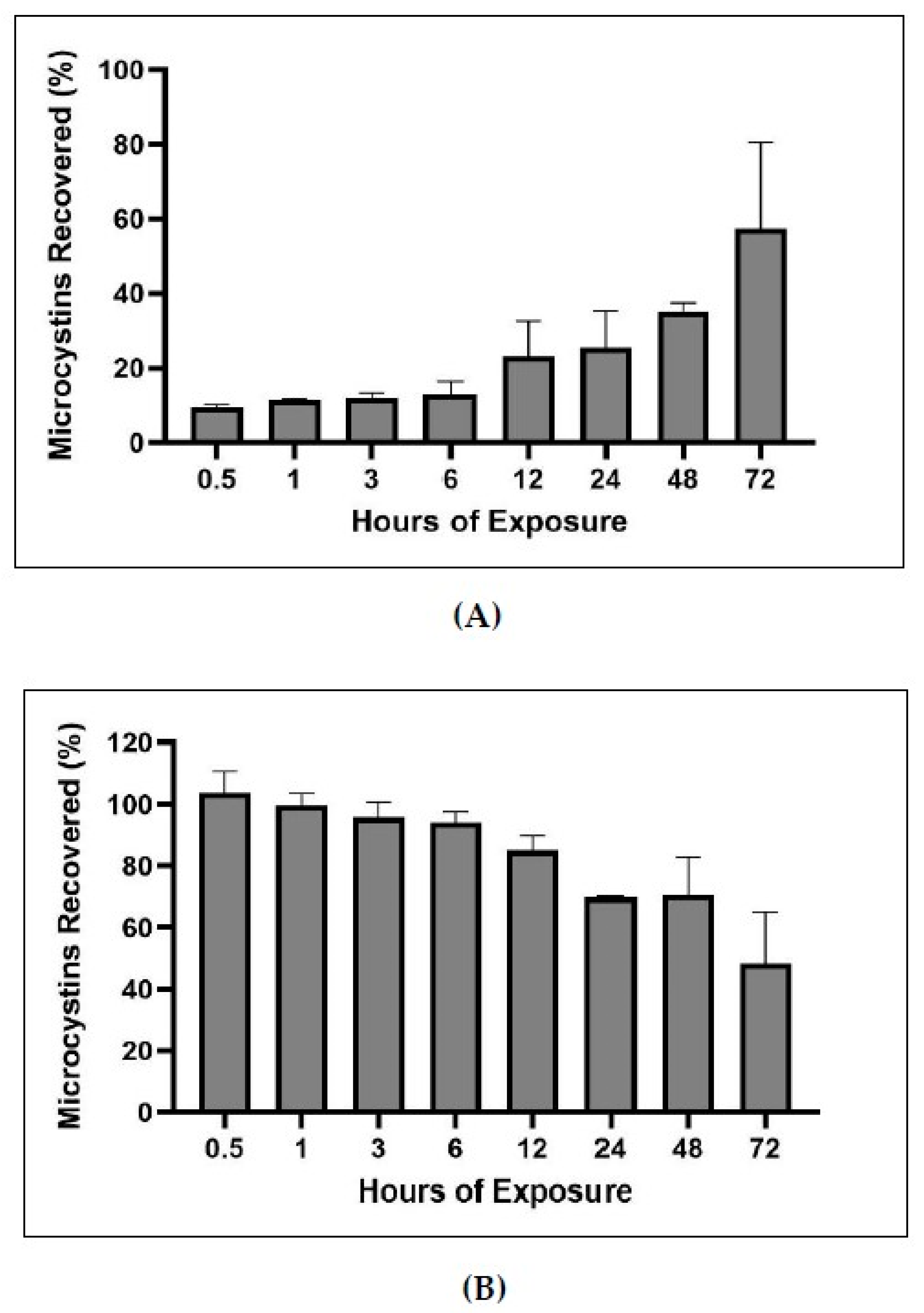
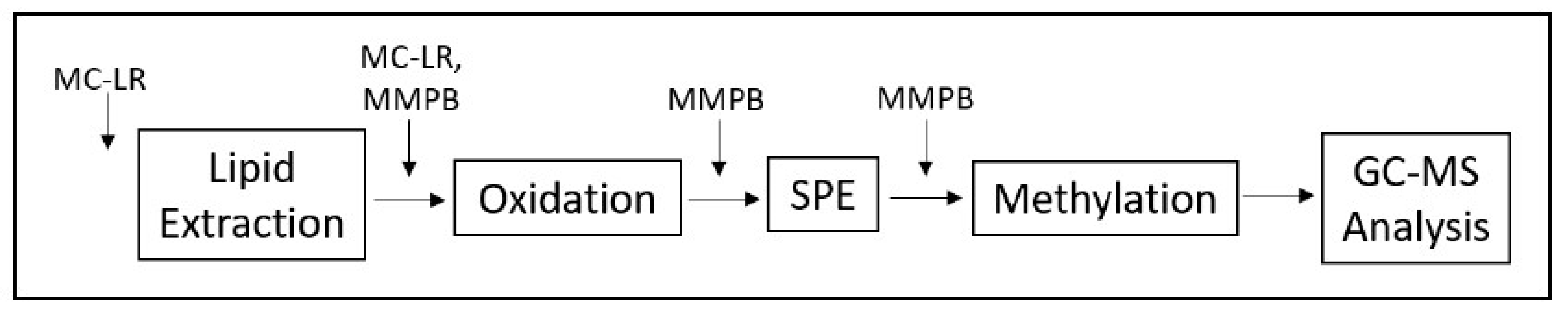
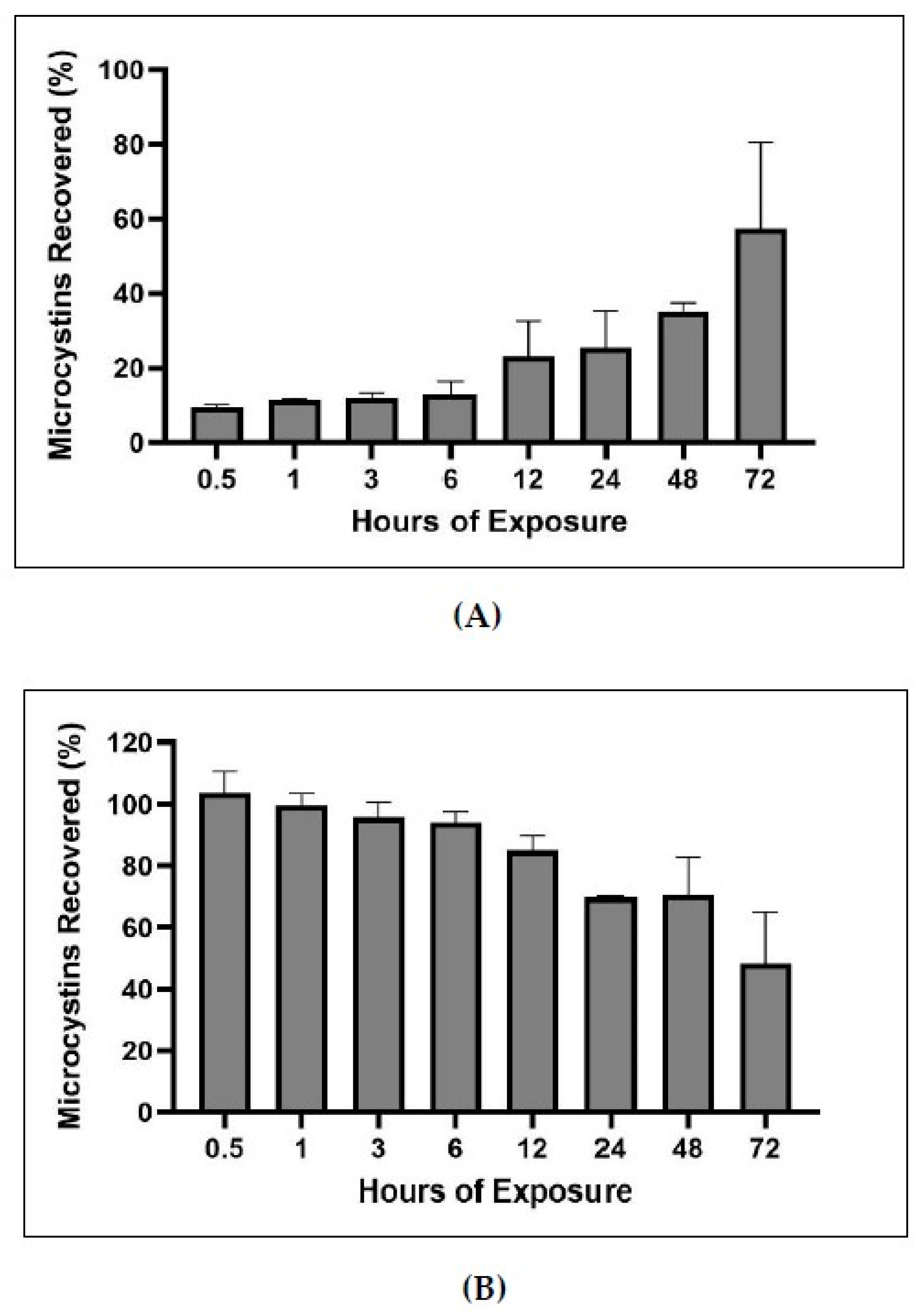
2.5. Bound and Unbound Time Series
2.6. MC Quantification in Environmental Samples
3. Discussion
3.1. Optimization of Total MC Quantification
3.2. Product Loss
3.3. Time Series of Bound and Unbound MC Fractions
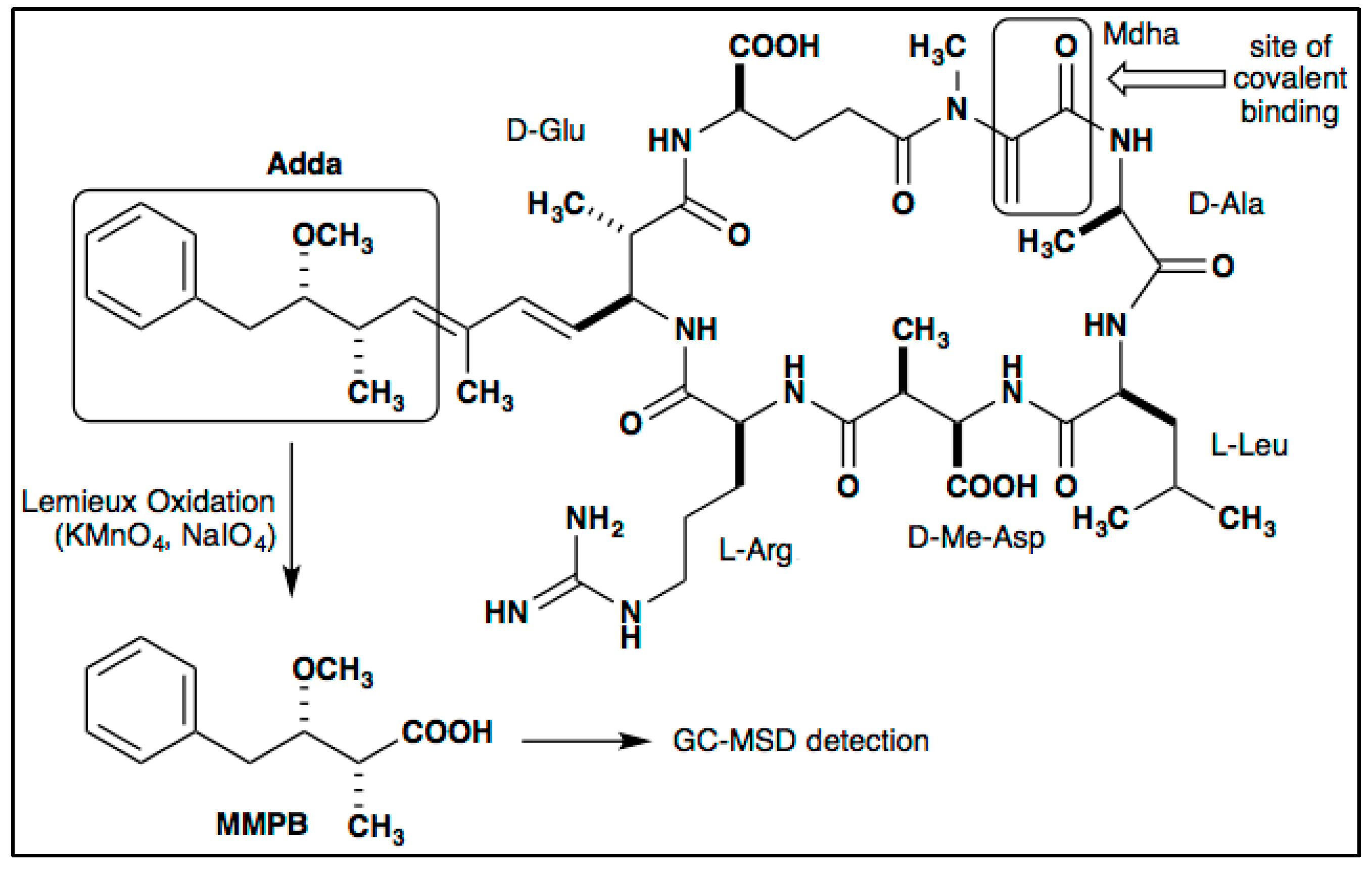
3.4. MC Detection
3.5. Contribution to Current Understanding of MC Detection and Distribution
4. Materials and Methods
4.1. Materials and Standards
4.1.1. Materials
4.1.2. Standards
4.2. Methods
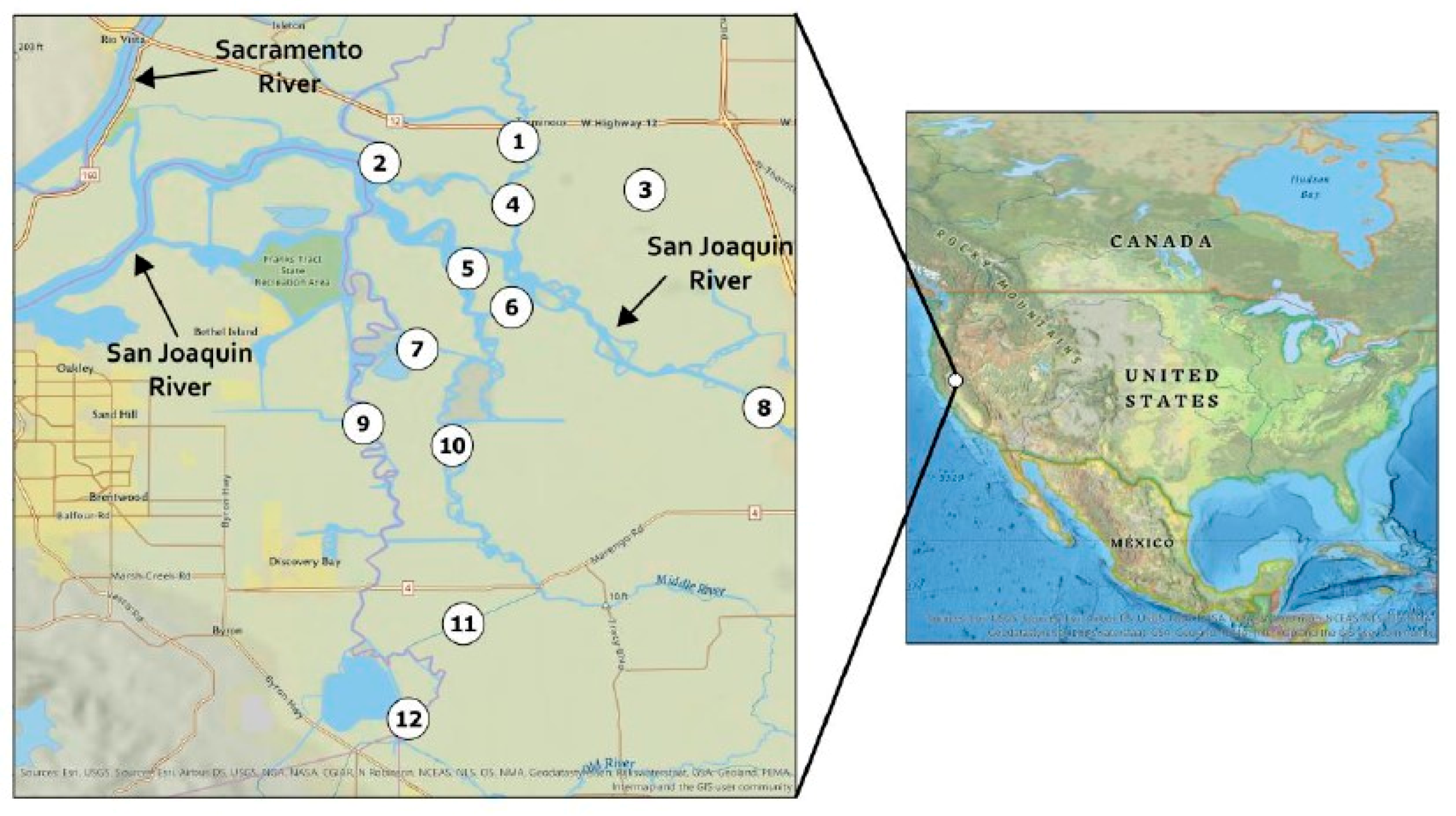
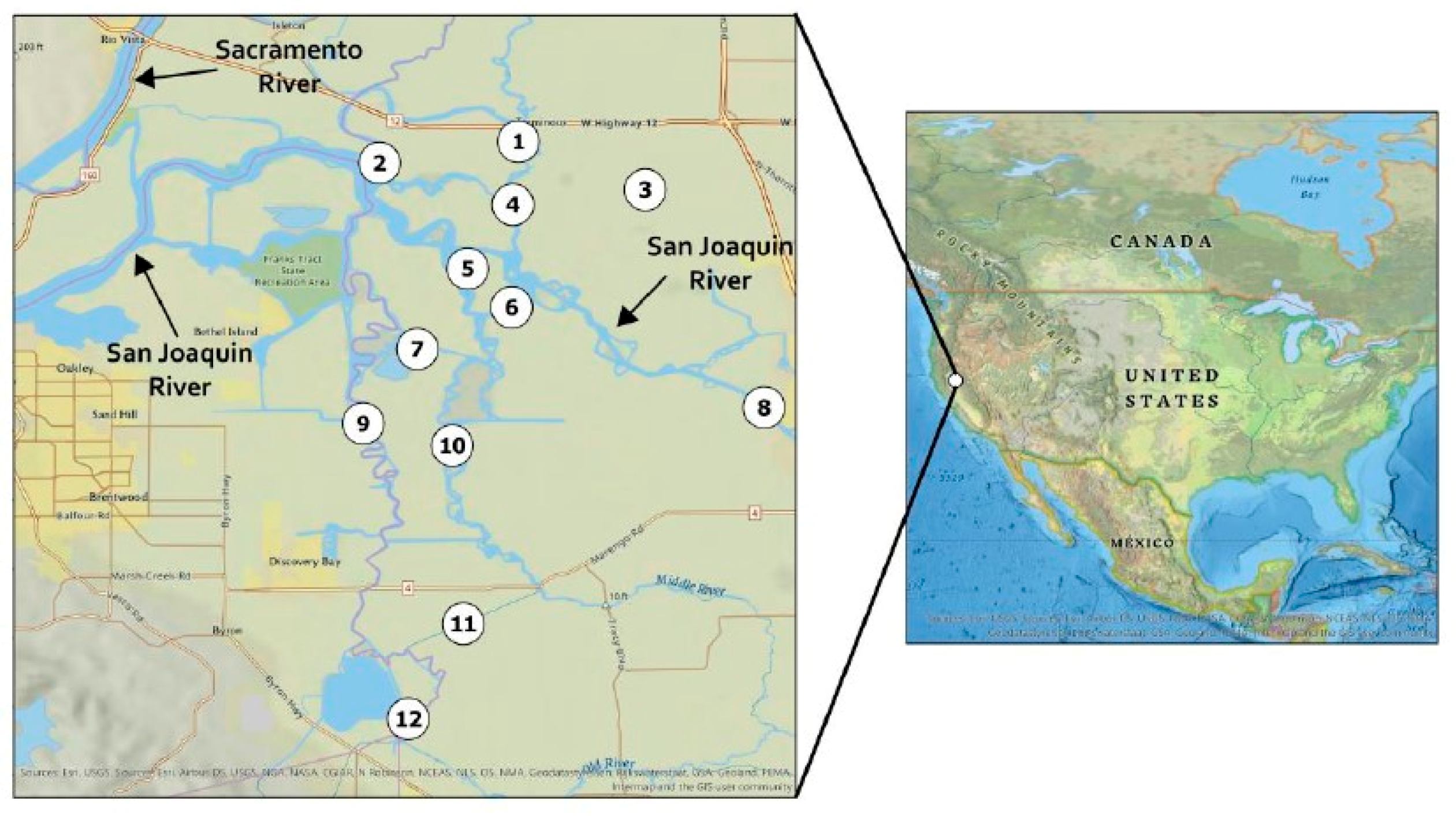
4.2.1. Site Description
4.2.2. Field Sampling
4.2.3. Analysis of MCs in Water
4.2.4. Treatment of Sediment and Clam Tissue Samples
4.2.5. Quantification of Total MCs (Bound and Unbound)
4.2.6. Quantification of Unbound MCs
4.2.7. GC–MS Analysis
4.2.8. Loss of Product
4.2.9. Bound and Unbound MCs Time Series
4.2.10. Analysis of Environmental Samples
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paerl, H. Nutrient and other environmental controls of harmful cyanobacterial blooms along the freshwater–marine continuum. In Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs; Hudnell, H.K., Ed.; Springer: New York, NY, USA, 2008; pp. 217–237. [Google Scholar]
- Cheung, M.Y.; Liang, S.; Lee, J. Toxin-producing cyanobacteria in freshwater: A review of the problems, impact on drinking water safety, and efforts for protecting public health. J. Microbiol. 2013, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Carmichael, W.W.; Hilborn, E.D. Microcystin analysis in human sera and liver from human fatalities in Caruaru, Brazil 1996. Toxicon 2006, 48, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.; Mazur-Marzec, H. The effect of cyanobacterial blooms in the siemianówka dam reservoir on the phytoplankton structure in the Narew River. Oceanol. Hydrobiol. Stud. 2011, 40, 19–26. [Google Scholar] [CrossRef]
- Chellappa, N.T.; Chellappa, S.L.; Chellappa, S. Harmful phytoplankton blooms and fish mortality in a eutrophicated reservoir of Northeast Brazil. Braz. Arch. Biol. Technol. 2008, 51, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Giannuzzi, L.; Sedan, D.; Echenique, R.; Andrinolo, D. An acute case of intoxication with cyanobacteria and cyanotoxins in recreational water in Salto Grande Dam, Argentina. Mar. Drugs 2011, 9, 2164–2175. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Loiselle, S.A.; Zhu, L.; Feng, L.; Zhang, Y.; Ma, R. Distribution and incidence of algal blooms in Lake Taihu. Aquat. Sci. 2014, 77, 9–16. [Google Scholar] [CrossRef]
- Michalak, A.M.; Anderson, E.J.; Beletsky, D.; Boland, S.; Bosch, N.S.; Bridgeman, T.B.; Chaffin, J.D.; Cho, K.; Confesor, R.; Daloglu, I.; et al. Record-setting algal bloom in Lake Erie caused by agricultural and meteorological trends consistent with expected future conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6448–6452. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xie, P.; Li, L.; Xu, J. First identification of the hepatotoxic microcystins in the serum of a chronically exposed human population together with indication of hepatocellular damage. Toxicol. Sci. 2009, 108, 81–89. [Google Scholar] [CrossRef]
- Jochimsen, E.; Carmichael, W.W.; An, J.; Cardo, D.M.; Cookson, S.T.; Holmes, C.E.M.; Antunes, B.C.; Melo Filho, D.A.; Lyra, T.M.; Barreto, V.S.T.; et al. Liver failure and death after exposure to microcystins. N. Engl. J. Med. 1998, 338, 873–878. [Google Scholar] [CrossRef]
- Davis, T.W.; Berry, D.L.; Boyer, G.L.; Gobler, C.J. The effects of temperature and nutrients on the growth and dynamics of toxic and non-toxic strains of Microcystis during cyanobacteria blooms. Harmful Algae 2009, 8, 715–725. [Google Scholar] [CrossRef]
- Paerl, H.W.; Huisman, J. Climate change: A catalyst for global expansion of harmful cyanobacterial blooms. Environ. Microbiol. Rep. 2009, 1, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.J.; Altheimer, S.; Cattori, V.; Meier, P.J.; Dietrich, D.R.; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKintosh, R.W.; Dalby, K.N.; Campbell, D.G.; Cohen, P.T.W.; Cohen, P.; MacKintosh, C. The cyanobacterial toxin microcystin binds covalently to cysteine-273 on protein phosphatase 1. FEBS Lett. 1995, 371, 236–240. [Google Scholar] [PubMed] [Green Version]
- Craig, M.; Luu, H.A.; McCready, T.L.; Holmes, C.F.B.; Williams, D.; Andersen, R.J. Molecular mechanisms underlying the interaction of motuporin and microcystins with type-1 and type-2A protein phosphatases. Biochem. Cell Biol. 1996, 74, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Toivola, D.M.; Eriksson, J.E. Toxins affecting cell signalling and alteration of cytoskeletal structure. Toxicol. In Vitro 1999, 13, 521–530. [Google Scholar] [CrossRef]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, F.; Ikai, Y.; Oka, H.; Okumura, M.; Ishikawa, N.; Harada, K.; Matsuura, K.; Murata, H.; Suzuki, M. Formation, characterization, and toxicity of the glutathione and cysteine conjugates of toxic heptapeptide Microcystins. Chem. Res. Toxicol. 1992, 5, 591–596. [Google Scholar] [CrossRef]
- Williams, D.E.; Craig, M.; Dawe, S.C.; Kent, M.L.; Holmes, C.F.B.; Andersen, R.J. Evidence for a covalently bound form of microcystin-LR in salmon liver and dungeness crab larvae. Chem. Res. Toxicol. 1997, 10, 463–469. [Google Scholar] [CrossRef]
- Cazenave, J.; Wunderlin, D.A.; Bistoni, M.D.L.Á.; Amé, M.V.; Krause, E.; Pflugmacher, S.; Wiegand, C. Uptake, tissue distribution and accumulation of microcystin-RR in Corydoras paleatus, Jenynsia multidentata and Odontesthes bonariensis: A field and laboratory study. Aquat. Toxicol. 2005, 75, 178–190. [Google Scholar] [CrossRef]
- Miller, M.A.; Kudela, R.M.; Mekebri, A.; Crane, D.; Oates, S.C.; Tinker, M.T.; Staedler, M.; Miller, W.A.; Toy-Choutka, S.; Dominik, C.; et al. Evidence for a novel marine harmful algal bloom: Cyanotoxin (microcystin) transfer from land to sea otters. PLoS ONE 2010, 5, e12576. [Google Scholar] [CrossRef]
- Vasconcelos, J.F.; Barbosa, J.E.L.; Lira, W.; Azevedo, S.M.F.O. Microcystin bioaccumulation can cause potential mutagenic effects in farm fish. Egypt J. Aquat. Res. 2013, 39, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.R.; Shaskus, M.; Estenik, J.F.; Oesch, C.; Khidekel, R.; Boyer, G.L. Variations in the microcystin content of different fish species collected from a Eutrophic Lake. Toxins 2013, 5, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Magonono, M.; Oberholster, P.; Addmore, S.; Stanley, M.; Gumbo, J. The presence of toxic and non-toxic cyanobacteria in the sediments of the Limpopo River Basin: Implications for human health. Toxins 2018, 10, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, K.; Nalto, S.; Kondo, F.; Ishiawa, N.; Watanabe, M.F.; Suzuki, M.; Harada, K.-I. Stability of microcystins from cyanobacteria: Effect of light on decomposition and isomerization. Environ. Sci. Technol. 1994, 28, 173–177. [Google Scholar] [CrossRef]
- Harada, K.; Tsuji, K.; Watanabe, M.F.; Kondo, F. Stability of microcystins from cyanobacteria—III. Effect of pH and temperature. Phycologia 1996, 35, 83–88. [Google Scholar] [CrossRef]
- Wu, X.; Wang, C.; Xiao, B.; Wang, Y.; Zheng, N.; Liu, J. Optimal strategies for determination of free/extractable and total microcystins in lake sediment. Anal. Chim. Acta 2012, 709, 66–72. [Google Scholar] [CrossRef]
- Smith, J.L.; Schulz, K.L.; Zimba, P.V.; Boyer, G.L. Possible mechanism for the foodweb transfer of covalently bound microcystins. Ecotoxicol. Environ. Saf. 2010, 73, 757–761. [Google Scholar] [CrossRef]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Ballot, A.; Wilkins, A.L.; Rise, F.; Jaabaek, J.A.H.; Loader, J.I. Conjugation of Microcystins with thiols is reversible: Base-catalyzed deconjugation for chemical analysis. Chem. Res. Toxicol. 2016, 29, 860–870. [Google Scholar] [CrossRef]
- Paldavičiene, A.; Zaiko, A.; Mazur-Marzec, H.; Razinkovas-Baziukas, A. Bioaccumulation of microcystins in invasive bivalves: A case study from the boreal lagoon ecosystem. Oceanologia 2015, 57, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, V.; Soares, R.; Azevedo, S. Microcystin contamination in fish from the Jacarepagua Lagoon (Rio de Janeiro, Brazil): Ecological implication and human health risk. Toxicon 2001, 39, 1077–1085. [Google Scholar] [CrossRef]
- Prepas, E.E.; Kotak, B.G.; Campbell, L.M.; Evans, J.C.; Hrudey, S.E.; Holmes, C.F. Accumulation and elimination of Cyanobacterial hepatotoxins by the freshwater clam Anodonta grandis simpsoniana. Can. J. Fish. Aquat. Sci. 1997, 54, 41–46. [Google Scholar] [CrossRef]
- Larson, D.; Ahlgren, G.; Willén, E. Bioaccumulation of microcystins in the food web: A field study of four Swedish lakes. Inland Waters 2014, 4, 91–104. [Google Scholar] [CrossRef]
- Smith, J.L.; Haney, J.F. Foodweb transfer, accumulation, and depuration of microcystins, a cyanobacterial toxin, in pumpkinseed sunfish (Lepomis gibbosus). Toxicon 2006, 48, 580–589. [Google Scholar] [CrossRef]
- Greer, B.; Maul, R.; Campbell, K.; Elliott, C.T. Detection of freshwater cyanotoxins and measurement of masked microcystins in tilapia from Southeast Asian aquaculture farms. Anal. Bioanal. Chem. 2017, 409, 4057–4069. [Google Scholar] [CrossRef] [Green Version]
- Moreno, I.M.; Molina, R.; Jos, A.; Picó, Y.; Cameán, A.M. Determination of microcystins in fish by solvent extraction and liquid chromatography. J. Chromatogr. A 2005, 1080, 199–203. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Rudloff, E. Periodate-permanganate oxidations. Can. J. Chem. 1955, 33, 1701–1709. [Google Scholar] [CrossRef]
- Xu, X.; Yu, R.; Wang, L.P.; Wu, S.; Song, Q. Sensitive determination of total microcystins with GC-MS method by using methylchloroformate as a derivatizing reagent. Anal. Methods 2013, 5, 1799–1805. [Google Scholar] [CrossRef]
- Ortiz-Rodríguez, R.; Wiegand, C. Age related acute effects of microcystin-LR on Daphnia magna biotransformation and oxidative stress. Toxicon 2010, 56, 1342–1349. [Google Scholar] [CrossRef]
- Lehman, P.W.; Boyer, G.; Hall, C.; Waller, S.; Gehrts, K. Distribution and toxicity of a new colonial Microcystis aeruginosa bloom in the San Francisco Bay Estuary, California. Hydrobiologia 2005, 541, 87–99. [Google Scholar] [CrossRef]
- Sommer, T.; Mejia, F. A place to call home: A synthesis of delta smelt habitat in the upper San Francisco estuary. San Franc. Estuary Watershed Sci. 2013, 11. [Google Scholar] [CrossRef]
- Frank, W. Fisher past and present status of central valley chinook salmon. Conserv. Biol. 1994, 8, 870–873. [Google Scholar]
- Lehman, P.W.; Boyer, G.; Satchwell, M.; Waller, S. The influence of environmental conditions on the seasonal variation of Microcystis cell density and microcystins concentration in San Francisco Estuary. Hydrobiologia 2008, 600, 187–204. [Google Scholar] [CrossRef]
- Lehman, P.W.; Kurobe, T.; Lesmeister, S.; Baxa, D.; Tung, A.; Teh, S.J. Impacts of the 2014 severe drought on the Microcystis bloom in San Francisco Estuary. Harmful Algae 2017, 63, 94–108. [Google Scholar] [CrossRef]
- Sanan, T.T.; Lazorchak, J. EPA Current Research on Cyanotoxins in Fish Tissue. New Jersey Water Monitoring Council, Cincinnati, OH. 2017. Available online: https://cfpub.epa.gov/si/si_public_record_report.cfm?Lab=NRMRL&dirEntryId=339470 (accessed on 4 July 2018).
- Ott, J.L.; Carmichael, W.W. LC/ESI/MS method development for the analysis of hepatotoxic cyclic peptide microcystins in animal tissues. Toxicon 2006, 47, 734–741. [Google Scholar] [CrossRef]
- Brown, A.; Foss, A.; Miller, M.A.; Gibson, Q. Detection of cyanotoxins (microcystins/nodularins) in livers from estuarine and coastal bottlenose dolphins (Tursiops truncatus) from Northeast Florida. Harmful Algae 2018. [Google Scholar] [CrossRef]
- Neffling, M.-R.; Lance, E.; Meriluoto, J. Detection of free and covalently bound microcystins in animal tissues by liquid chromatography-tandem mass spectrometry. Environ. Pollut. 2010, 158, 948–952. [Google Scholar] [CrossRef]
- Umehara, A.; Takahashi, T.; Komorita, T.; Orita, R.; Choi, J. Widespread dispersal and bio-accumulation of toxic microcystins in benthic marine ecosystems. Chemosphere 2017, 167, 492–500. [Google Scholar] [CrossRef]
- Chen, W.; Song, L.; Peng, L.; Wan, N.; Zhang, X.; Gan, N. Reduction in microcystin concentrations in large and shallow lakes: Water and sediment-interface contributions. Water Res. 2008, 42, 763–773. [Google Scholar] [CrossRef]
- Zastepa, A.; Taranu, Z.E.; Kimpe, L.E.; Blais, J.M.; Gregory-Eaves, I.; Zurawell, R.W.; Pick, F.R. Science of the total environment reconstructing a long-term record of microcystins from the analysis of lake sediments. Sci. Total Environ. 2017, 579, 893–901. [Google Scholar] [CrossRef]
- Pavagadhi, S.; Gong, Z.; Balasubramanian, R. Toxicological implications of microcystins for zebrafish embryos in the presence of other environmental pollutants. Environ. Toxicol. Chem. 2013, 32, 1574–1581. [Google Scholar] [CrossRef]
- Wu, Q.; Cheng, H.; Liu, C.; Li, G.; Yan, W.; Hung, T.-C.; Guo, X. Parental transfer of microcystin-LR induced transgenerational effects of developmental neurotoxicity in zebrafish offspring. Environ. Pollut. 2017, 231, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Lehman, P.W.; Marr, K.; Boyer, G.L.; Acuna, S.; Teh, S.J. Long-term trends and causal factors associated with Microcystis abundance and toxicity in San Francisco Estuary and implications for climate change impacts. Hydrobiologia 2013, 718, 141–158. [Google Scholar] [CrossRef]
- Kurobe, T.; Lehman, P.W.; Hammock, B.G.; Bolotaolo, M.B.; Lesmeister, S.; Teh, S.J. Biodiversity of cyanobacteria and other aquatic microorganisms across a freshwater to brackish water gradient determined by shotgun metagenomic sequencing analysis in the San Francisco Estuary, USA. PLoS ONE 2018, 13, e0203953. [Google Scholar] [CrossRef] [PubMed]
- Orihel, D.M.; Hadas, O.; Pinkas, R.; Viner-Mozzini, Y.; Sukenik, A. Internal nutrient loading may increase microcystin concentrations in freshwater lakes by promoting growth of Microcystis populations. Ann. Limnol. Int. J. Limnol. 2013, 49, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Takamura, N.; Yasuno, M.; Sugahara, K. Overwintering of Microcystis aeruginosa Kütz. in a shallow lake. J. Plankton Res. 1984, 6, 1019–1029. [Google Scholar] [CrossRef]
- Robarts, R.D.; Zohary, T. Temperature effects on photosynthetic capacity, respiration, and growth rates of bloom-forming cyanobacteria. N. Z. J. Mar. Freshw. Res. 1987, 21, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.S. Growth and buoyancy of Microcystis aeruginosa Kutz. emend. Elenkin in a shallow eutrophic lake. Proc. R. Soc. Lond. Biol. Sci. 1973, 184, 29–50. [Google Scholar]
- Preston, T.; Stewart, W.D.; Reynolds, C.S. Bloom-forming cyanobacterium Microcystis aeruginosa overwinters on sediment surface. Nature 1980, 288, 365–367. [Google Scholar] [CrossRef]
- Bronzi, B.P.; Rosenthal, H.; Gessner, J. Global sturgeon aquaculture production: An overview. Appl. Ichthyol. 2011, 27, 169–175. [Google Scholar] [CrossRef]
- Wijsman, J.W.M.; Troost, K.; Fang, J.; Roncarati, A. Global production of marine bivalves. Trends and challenges. In Goods and Services of Marine Bivalves; Smaal, A.C., Ferreira, J.G., Grant, J., Petersen, J.K., Strand, Ø., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 7–26. [Google Scholar]
- McCabe, G.; Emmett, R.L.; Hinton, S.A. Feeding ecology of juvenile white sturgeon (Acipenser transmontanus) in the Lower Columbia river. Northwest Sci. Am. 1993, 67. [Google Scholar]
- Carlton, J.T.; Thompson, J.K.; Schemel, L.E.; Nichols, F.H. Remarkable invasion of San Francisco Bay (CA, USA), by the Asian clam Potamocorbula amurensis. I. Introduction and dispersal. Mar. Ecol. Prog. Ser. 1990, 66, 81–94. [Google Scholar] [CrossRef]
- Niedermeyer, T. Microcystin Congeners Described in the Literature, Version 5. Available online: http://0-dx-doi-org.brum.beds.ac.uk/10.6084/m9.figshare.880756 (accessed on 1 September 2018).
- Baxa, D.V.; Kurobe, T.; Ger, K.A.; Lehman, P.W.; Teh, S.J. Estimating the abundance of toxic Microcystis in the San Francisco Estuary using quantitative real-time PCR. Harmful Algae 2010, 9, 342–349. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike (ng gDW −1) | Sediment | Clam Tissue | ||
|---|---|---|---|---|
| Yield (%) | RSD (%) | Yield (%) | RSD (%) | |
| 50 | 44–53 | 9 | 41–56 | 10 |
| 100 | 50–55 | 3 | 44–54 | 13 |
| 500 | 47–53 | 4 | 39–42 | 3 |
| Matrix | Spike (µg/L) | Lipid Extraction (%) | MC-LR to MMPB Conversion (%) | Oxidation (%) | SPE (%) | Overall Loss (%) |
|---|---|---|---|---|---|---|
| Sediment | 100 | 3.3 | 20.6 | 11.2 | 13.3 | 48.5 |
| Sediment | 500 | 5 | 29.5 | 7.3 | 5.5 | 47.3 |
| Clam | 100 | 4.5 | 39.5 | 5.7 | 5 | 54.6 |
| Clam | 500 | 4 | 32.3 | 11.6 | 12.9 | 60.9 |
| Sample Site | Total MC Concentration (ng gDW −1) ± SD | Unbound MC Concentration (ng gDW −1) ± SD | ||
|---|---|---|---|---|
| May 2017 | October 2017 | May 2017 | October 2017 | |
| 1 | <LOD | <LOD | <LOD | <LOD |
| 2 | 59.3 ± 3.8 | <LOD | <LOD | <LOD |
| 3 | 20.4 ± 4.4 * | <LOD | <LOD | <LOD |
| 4 | <LOD | <LOD | <LOD | <LOD |
| 5 | 161.5 ± 21.6 | 27.6 ± 5.0 * | <LOD | 33.3 ± 0.2 * |
| 6 | 114.9 ± 4.9 | 81.8 ± 6.9 | <LOD | <LOD |
| 7 | <LOD | <LOD | <LOD | <LOD |
| 8 | N/A | 23.4 ± 6.1 * | <LOD | <LOD |
| 9 | <LOD | N/A | <LOD | <LOD |
| 10 | 40.6 ± 5.2 * | <LOD | <LOD | <LOD |
| 11 | N/A | <LOD | <LOD | <LOD |
| 12 | <LOD | <LOD | <LOD | <LOD |
| Collection Date: May 2017 | Collection Date: October 2017 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Day Sampled | Sample Site | Water | Clam | Sediment | Day Sampled | Sample Site | Water | Clam | Sediment |
| May 10 | 1 | x | x | Oct 2 | 1 | x | x | ||
| May 10 | 2 | x | x | Oct 2 | 2 | x | x | x | |
| May 10 | 3 | x | x | x | Oct 2 | 3 | x | x | |
| May 10 | 4 | x | x | x | Oct 2 | 4 | x | x | x |
| May 10 | 5 | x | x | x | Oct 2 | 5 | x | x | |
| May 10 | 6 | x | x | Oct 2 | 6 | x | x | ||
| May 10 | 7 | x | x | Oct 3 | 7 | ||||
| May 10 | 8 | Oct 2 | 8 | x | x | x | |||
| May 10 | 9 | x | x | x | Oct 3 | 9 | |||
| May 10 | 10 | x | x | x | Oct 3 | 10 | x | x | x |
| May 10 | 11 | Oct 3 | 11 | x | x | x | |||
| May 10 | 12 | x | x | Oct 3 | 12 | x | x | ||
| May 10 | FC * | Oct 2 | FC * | x | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolotaolo, M.; Kurobe, T.; Puschner, B.; Hammock, B.G.; Hengel, M.J.; Lesmeister, S.; Teh, S.J. Analysis of Covalently Bound Microcystins in Sediments and Clam Tissue in the Sacramento–San Joaquin River Delta, California, USA. Toxins 2020, 12, 178. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030178
Bolotaolo M, Kurobe T, Puschner B, Hammock BG, Hengel MJ, Lesmeister S, Teh SJ. Analysis of Covalently Bound Microcystins in Sediments and Clam Tissue in the Sacramento–San Joaquin River Delta, California, USA. Toxins. 2020; 12(3):178. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030178
Chicago/Turabian StyleBolotaolo, Melissa, Tomofumi Kurobe, Birgit Puschner, Bruce G Hammock, Matt J. Hengel, Sarah Lesmeister, and Swee J. Teh. 2020. "Analysis of Covalently Bound Microcystins in Sediments and Clam Tissue in the Sacramento–San Joaquin River Delta, California, USA" Toxins 12, no. 3: 178. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030178