Combined (d)SPE-QuEChERS Extraction of Mycotoxins in Mixed Feed Rations and Analysis by High Performance Liquid Chromatography-High-Resolution Mass Spectrometry

Abstract
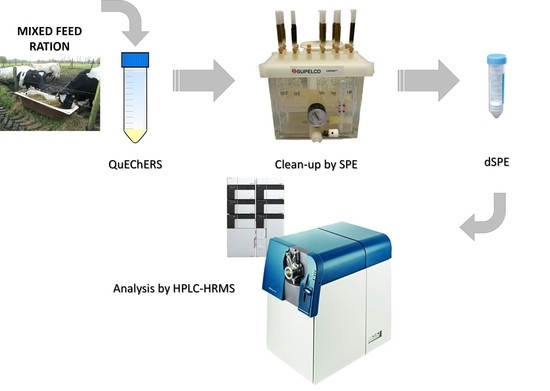
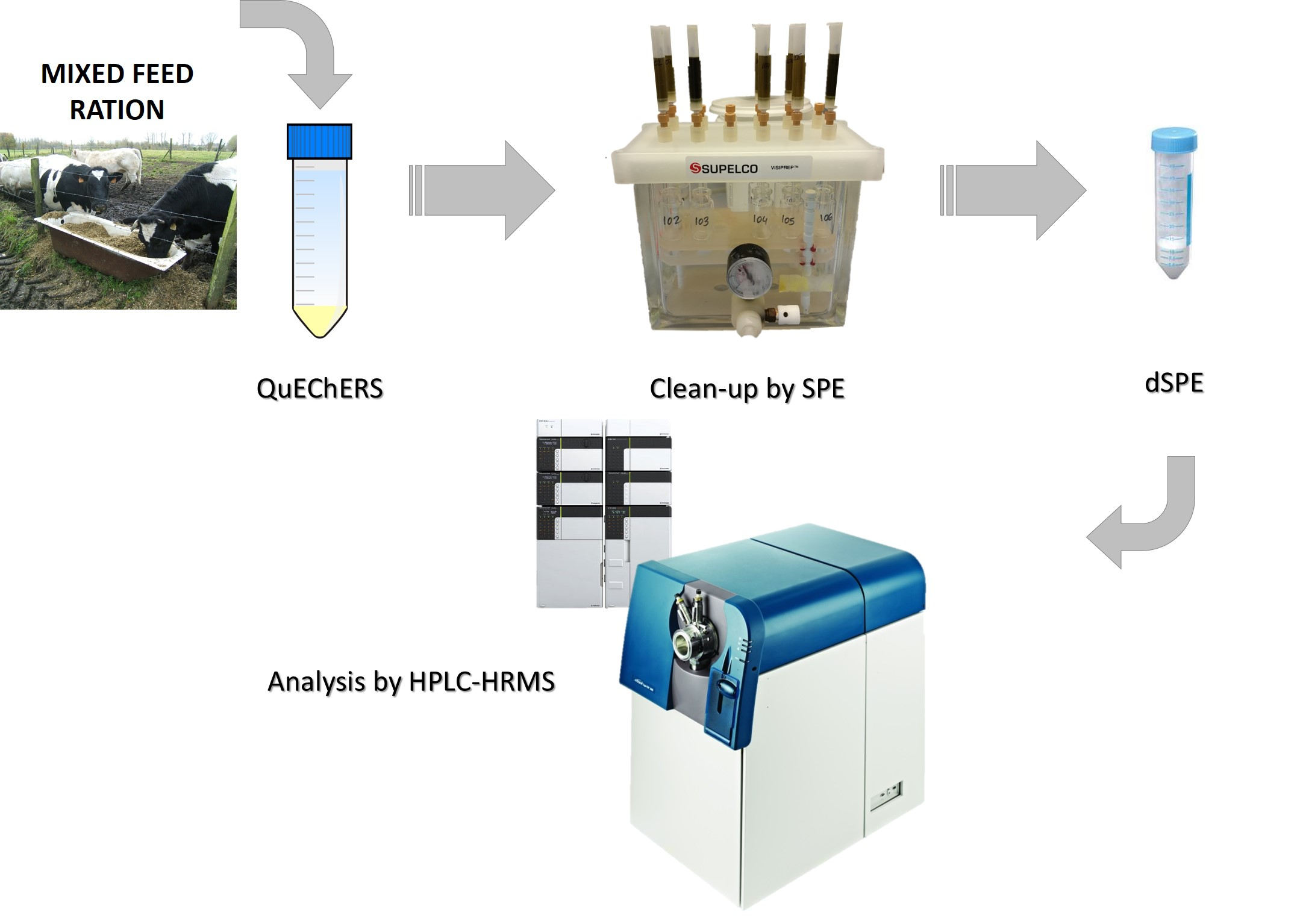
:
1. Introduction
2. Results and Discussion
2.1. Chromatographic Analysis
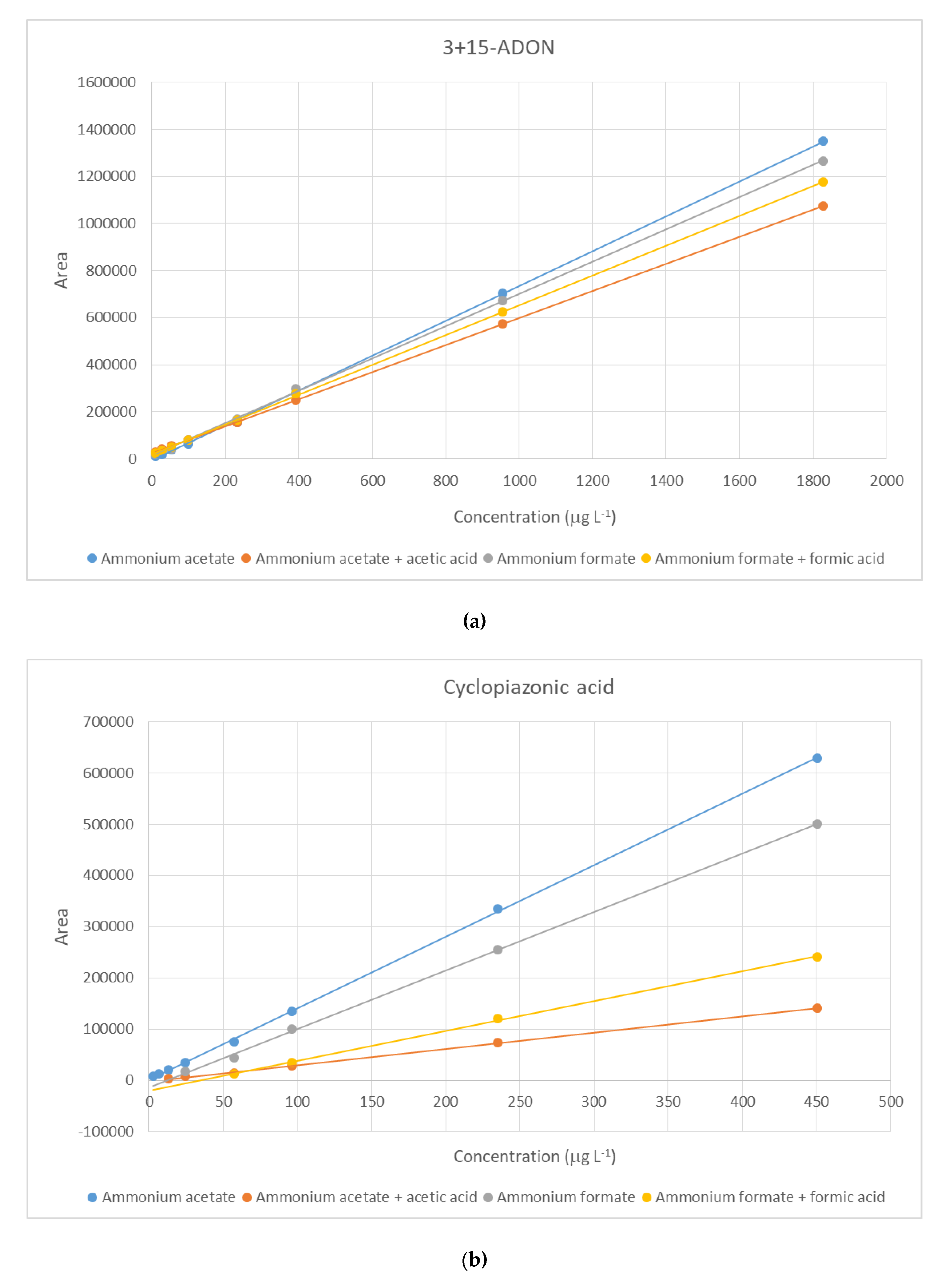
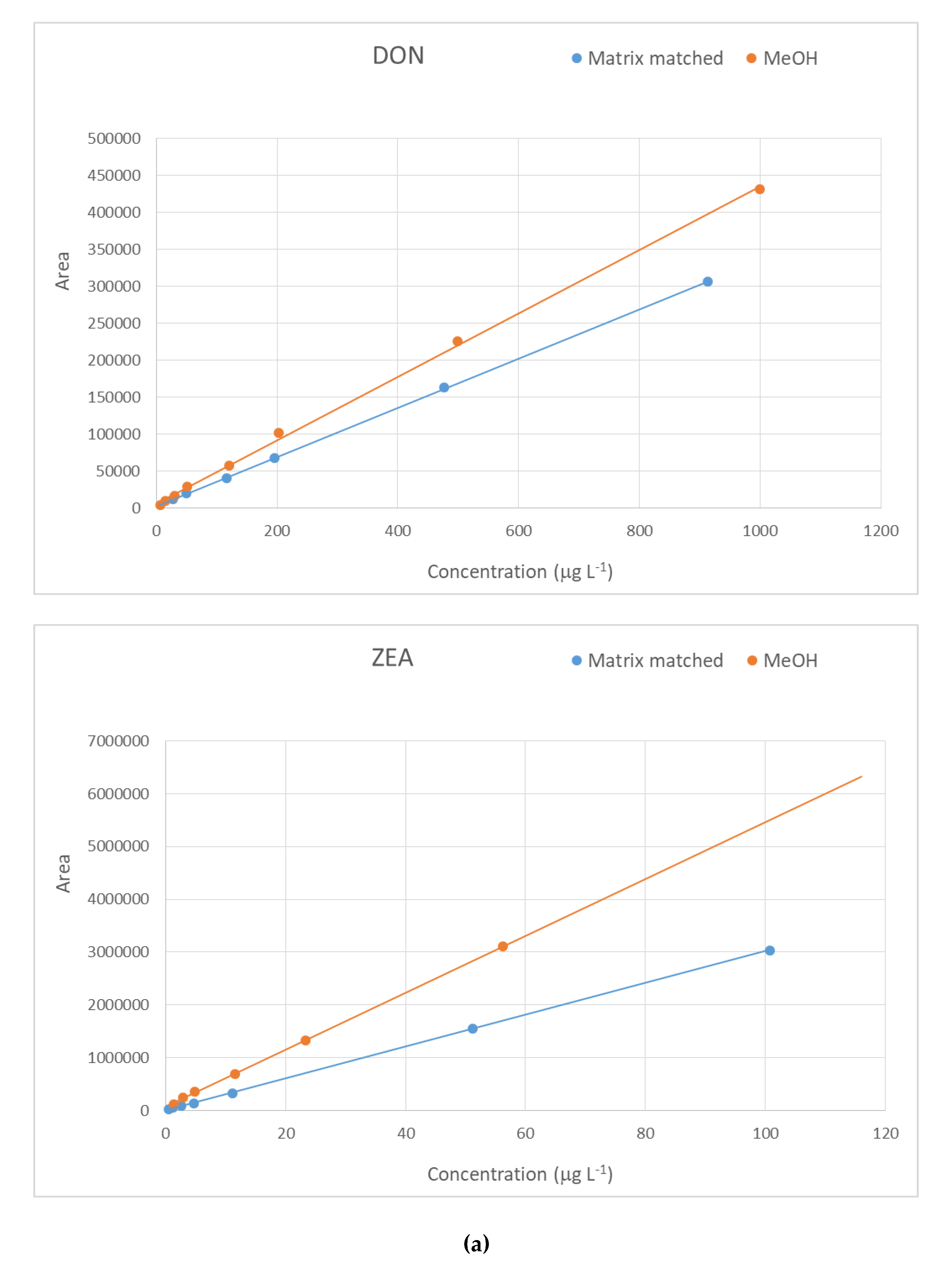
2.2. Method Performance Evaluation
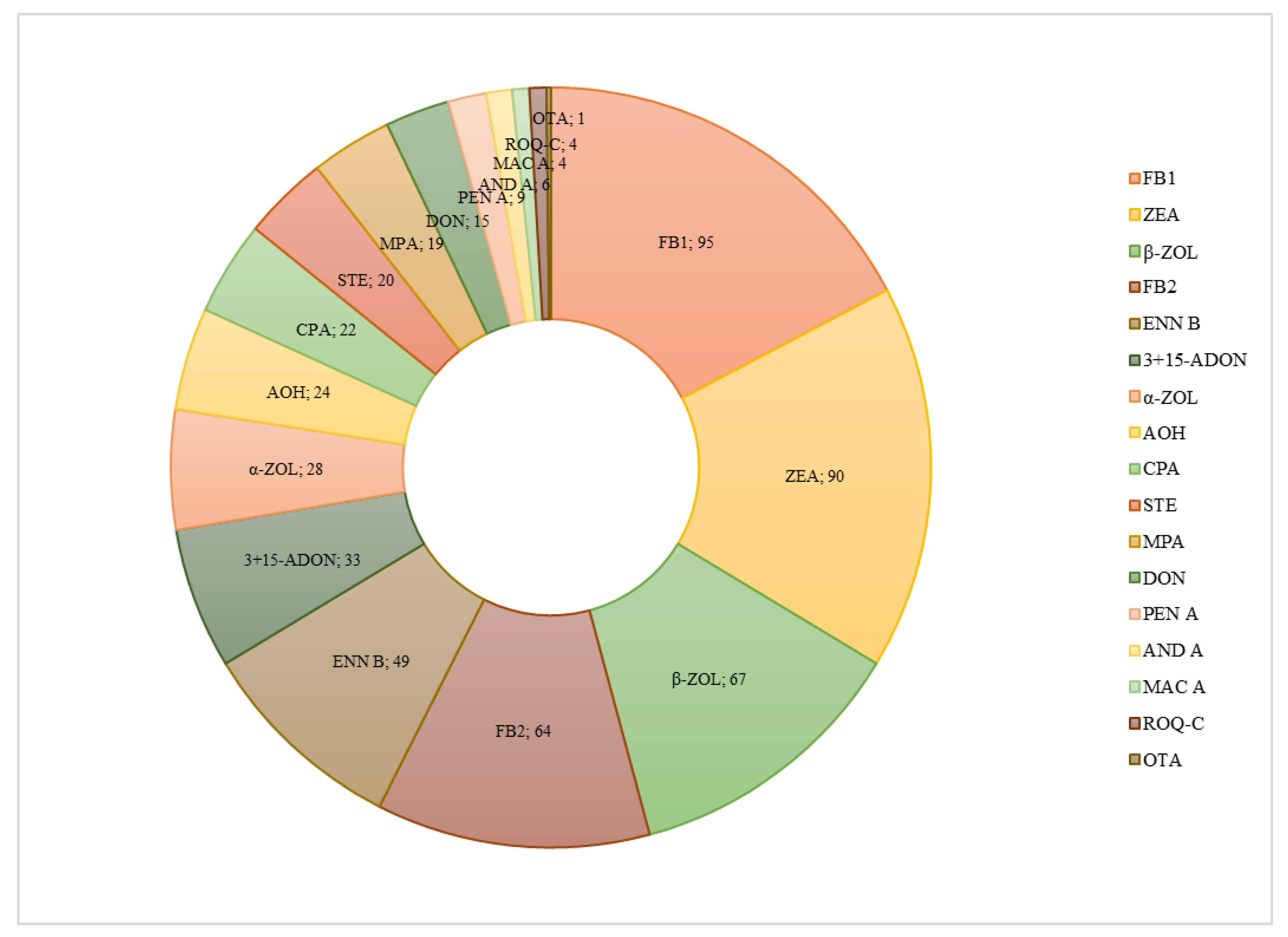
2.3. Application to Real Samples
3. Conclusions
4. Materials and Methods
4.1. Reagents and Standards
4.2. Sampling and Sample Treatment
4.3. QuEChERS Extraction
4.4. Clean-Up Procedure
4.5. HPLC-QTOF Analysis
4.6. Method Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gómez, J.V.; Gimeno-Adelantado, J.V.; Jiménez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef]
- Pitt, J.I.; Taniwaki, M.H.; Cole, M.B. Mycotoxin production in major crops as influenced by growing, harvesting, storage and processing, with emphasis on the achievement of Food Safety Objectives. Food Control 2013, 32, 205–215. [Google Scholar] [CrossRef]
- Lee, H.J.; Ryu, D. Worldwide Occurrence of Mycotoxins in Cereals and Cereal-Derived Food Products: Public Health Perspectives of Their Co-occurrence. J. Agric. Food Chem. 2017, 65, 7034–7051. [Google Scholar] [CrossRef]
- Escrivá, L.; Font, G.; Manyes, L. In vivo toxicity studies of fusarium mycotoxins in the last decade: A review. Food Chem. Toxicol. 2015, 78, 185–206. [Google Scholar] [CrossRef]
- Arroyo-Manzanares, N.; De Ruyck, K.; Uka, V.; Gámiz-Gracia, L.; García-Campaña, A.M.; De Saeger, S.; Di Mavungu, J.D. In-house validation of a rapid and efficient procedure for simultaneous determination of ergot alkaloids and other mycotoxins in wheat and maize. Anal. Bioanal. Chem. 2018, 410, 5567–5581. [Google Scholar] [CrossRef] [PubMed]
- Paschoal, F.N.; de Azevedo Silva, D.; de Souza, R.V.S.; de Oliveira, M.S.; Pereira, D.A.A.; de Souza, S.V.C. A Rapid Single-Extraction Method for the Simultaneous Determination of Aflatoxins B1, B2, G1, G2, Fumonisin B1, and Zearalenone in Corn Meal by Ultra Performance Liquid Chromatography Tandem Mass Spectrometry. Food Anal. Methods 2017, 10, 1631–1644. [Google Scholar] [CrossRef]
- Nathanail, A.V.; Syvähuoko, J.; Malachová, A.; Jestoi, M.; Varga, E.; Michlmayr, H.; Adam, G.; Sieviläinen, E.; Berthiller, F.; Peltonen, K. Simultaneous determination of major type A and B trichothecenes, zearalenone and certain modified metabolites in Finnish cereal grains with a novel liquid chromatography-tandem mass spectrometric method. Anal. Bioanal. Chem. 2015, 407, 4745–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolechová, M.; Benešová, K.; Běláková, S.; Čáslavský, J.; Pospíchalová, M.; Mikulíková, R. Determination of seventeen mycotoxins in barley and malt in the Czech Republic. Food Control 2015, 47, 108–113. [Google Scholar] [CrossRef]
- Qian, M.; Yang, H.; Li, Z.; Liu, Y.; Wang, J.; Wu, H.; Ji, X.; Xu, J. Detection of 13 mycotoxins in feed using modified QuEChERS with dispersive magnetic materials and UHPLC–MS/MS. J. Sep. Sci. 2018, 41, 756–764. [Google Scholar] [CrossRef]
- Wang, R.G.; Su, X.O.; Cheng, F.F.; Wang, P.L.; Fan, X.; Zhang, W. Determination of 26 mycotoxins in feedstuffs by multifunctional clean-up column and liquid chromatography-tandem mass spectrometry. Chin. J. Anal. Chem. 2015, 43, 264–270. [Google Scholar] [CrossRef]
- Miró-Abella, E.; Herrero, P.; Canela, N.; Arola, L.; Borrull, F.; Ras, R.; Fontanals, N. Determination of mycotoxins in plant-based beverages using QuEChERS and liquid chromatography–tandem mass spectrometry. Food Chem. 2017, 229, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Tsiplakou, E.; Anagnostopoulos, C.; Liapis, K.; Haroutounian, S.A.; Zervas, G. Determination of mycotoxins in feedstuffs and ruminant’s milk using an easy and simple LC-MS/MS multiresidue method. Talanta 2014, 130, 8–19. [Google Scholar] [CrossRef]
- Rubert, J.; León, N.; Sáez, C.; Martins, C.P.B.; Godula, M.; Yusà, V.; Mañes, J.; Soriano, J.M.; Soler, C. Evaluation of mycotoxins and their metabolites in human breast milk using liquid chromatography coupled to high resolution mass spectrometry. Anal. Chim. Acta 2014, 820, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Cherkani-Hassani, A.; Mojemmi, B.; Mouane, N. Occurrence and levels of mycotoxins and their metabolites in human breast milk associated to dietary habits and other factors: A systematic literature review, 1984–2015. Trends Food Sci. Technol. 2016, 50, 56–69. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Directive 2002/32/EC of the European Parliament and of the Council of 7 May 2002 on Undesirable Substances in Animal Feed and its Amendments; European Parliament and of the Council: Brussels, Belgium, 2002.
- Commission Recommendation of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding) (2006/576/EC); The Commission of The European Communities: Brussels, Belgium, 2006.
- 2013/165/EU, C.R. Recomendations on the presence of T-2 and HT-2 Toxin in Cereals and Cereal Products; Official Journal of the European Union: Brussels, Belgium, 2013.
- Turner, N.W.; Bramhmbhatt, H.; Szabo-Vezse, M.; Poma, A.; Coker, R.; Piletsky, S.A. Analytical methods for determination of mycotoxins: An update (2009–2014). Anal. Chim. Acta 2015, 901, 12–33. [Google Scholar] [CrossRef]
- Rubert, J.; Dzuman, Z.; Vaclavikova, M.; Zachariasova, M.; Soler, C.; Hajslova, J. Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: Comparison of efficiency and efficacy of different extraction procedures. Talanta 2012, 99, 712–719. [Google Scholar] [CrossRef]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, J.J.; Huang, B.F.; Cai, Z.X.; Ren, Y.P. High-performance liquid chromatographic determination of multi-mycotoxin in cereals and bean foodstuffs using interference-removal solid-phase extraction combined with optimized dispersive liquid–liquid microextraction. J. Sep. Sci. 2017, 40, 2141–2150. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Garrido Frenich, A. Determination of toxic substances, pesticides and mycotoxins, in ginkgo biloba nutraceutical products by liquid chromatography Orbitrap-mass spectrometry. Microchem. J. 2015, 118, 124–130. [Google Scholar] [CrossRef]
- Bueno, D.; Istamboulie, G.; Muñoz, R.; Marty, J.L. Determination of Mycotoxins in Food: A Review of Bioanalytical to Analytical Methods. Appl. Spectrosc. Rev. 2015, 50, 728–774. [Google Scholar] [CrossRef]
- Rahmani, A.; Jinap, S.; Soleimany, F. Qualitative and quantitative analysis of mycotoxins. Compr. Rev. Food Sci. Food Saf. 2009, 8, 202–251. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, N.; Yang, L.; Deng, Y.; Wang, J.; Song, S.; Lin, S.; Wu, A.; Zhou, Z.; Hou, J. Multi-mycotoxin analysis of animal feed and animal-derived food using LC-MS/MS system with timed and highly selective reaction monitoring. Anal. Bioanal. Chem. 2015, 407, 7359–7368. [Google Scholar] [CrossRef] [PubMed]
- Dagnac, T.; Latorre, A.; Lorenzo, B.F.; Llompart, M. Validation and application of a liquid chromatography-tandem mass spectrometry based method for the assessment of the co-occurrence of mycotoxins in maize silages from dairy farms in NW Spain. Food Addit. Contam. Part A 2016, 33, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Veprikova, Z.; Godula, M.; Hajslova, J. Multi-analyte high performance liquid chromatography coupled to high resolution tandem mass spectrometry method for control of pesticide residues, mycotoxins, and pyrrolizidine alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; de Boevre, M.; Preußke, N.; de Saeger, S.; Birr, T.; Sönnichsen, F.D. Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices. Toxins 2019, 11, 531. [Google Scholar] [CrossRef] [Green Version]
- ISO 21748. Guidance for the Use of Repeatability, Reproducibility and Trueness Estimates in Measurement Uncertainty Estimation; International Organization for Standardization: Geneva, Switzerland, 2017. [Google Scholar]
- Ediage, E.N.; Van Poucke, C.; De Saeger, S. A multi-analyte LC-MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chem. 2015, 177, 397–404. [Google Scholar] [CrossRef]
- Franco, L.T.; Petta, T.; Rottinghaus, G.E.; Bordin, K.; Gomes, G.A.; Oliveira, C.A.F. Co-occurrence of mycotoxins in maize food and maize-based feed from small-scale farms in Brazil: A pilot study. Mycotoxin Res. 2019, 35, 65–73. [Google Scholar] [CrossRef]
- European Food Safety Authority. Deoxynivalenol in food and feed: Occurrence and exposure 1. EFSA J. 2013, 11, 3379. [Google Scholar]
- Istituto Superiore di Sanità (ISS); Italian National Agency for New Technologies, Energy and Sustainable Economic Development (ENEA); French Agency for Food, Environmental and Occupational Health & Safety (ANSES). In vivo Toxicity and Genotoxicity of Auvericin and Enniatins. Combined Approach to Study In Vivo Toxicity and Genotoxicity of Mycotoxins Beauvericin (BEA) and Enniatin B (ENNB). EFSA Supporting Publ. 2018, 15, 1406E. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R2 | Linear Range (ng mL−1) | Intra-Day Precision (%) (n = 3) | Inter-Day Precision (%) (n = 3) | LOQs (ng mL−1) | Recoveries | Matrix Effect (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (ng mL−1) | Recovery (%) (% RSD) | Concentration (ng mL−1) | Recovery (%) (% RSD) | |||||||
| 3+15-ADON | 0.9998 | 10.7–1828.3 | 2.4 | 5.8 | 24 | 339.6 | 85.8 (3.9) | 95.4 | 90.5 (3.0) | −0.6 |
| DON | 0.9999 | 26.7–912.8 | 2.4 | 6.2 | 41.1 | 169.6 | 114.2 (0.8) | 47.7 | 121.9 (8.7) | −22.4 |
| ENN B | 0.9999 | 40.6–905.4 | 2.5 | 5.9 | 46.8 | 168.2 | 88.8 (4.1) | 47.6 | 132.6 (5.8) | 33.9 |
| ENN B1 | 1.0000 | 13.5–479.7 | 1.1 | 1.4 | 24 | 170.6 | 58.3 (1.2) | 47.9 | 67.4 (3.5) | −29.6 |
| FB1 | 0.9996 | 2.7–92.6 | 1.7 | 5.2 | 2.9 | 17.2 | 92.7 (2.2) | 4.8 | 29.6 (6.6) | 195.3 |
| FB2 | 0.9998 | 1.3–85.8 | 0.2 | 0.4 | 2.4 | 15.9 | 118.1 (0.3) | 4.5 | 98.7 (4.2) | 197.8 |
| HT-2 | 0.9998 | 1.1–190.7 | 2.3 | 2.2 | 3.3 | 35.4 | 98.9 (0.4) | 10.0 | 61.3 (2.0) | −69.8 |
| MAC A | 0.9998 | 2.6–87.9 | 3.1 | 6.0 | 4.2 | 16.3 | 104.0 (5.5) | 4.6 | 70.4 (2.9) | −51.2 |
| OTA | 0.9991 | 1.6–36.4 | 4.8 | 5.2 | 2.0 | 6.8 | 71.4 (5.2) | 1.9 | 54.8 (1.1) | −49.9 |
| ROQ-C | 0.9998 | 0.2–36.1 | 2.5 | 2.9 | 0.6 | 6.7 | 32.2 (2.5) | 1.9 | 87.0 (8.0) | −31.0 |
| STE | 0.9996 | 0.5–35.9 | 4.7 | 5.1 | 1.2 | 6.7 | 80.9 (3.0) | 1.9 | 76.3 (9.1) | −56.3 |
| T-2 | 0.9999 | 0.4–9.1 | 0.9 | 1.1 | 0.5 | 1.7 | 155.2 (1.9) | 0.6 | 85.7 (0.3) | −28.8 |
| CPA | 0.9998 | 2.6–450.5 | 1.1 | 3.8 | 7.0 | 83.7 | 53.7 (0.3) | 23.5 | 83.4 (5.5) | −25.9 |
| AND A1 | 0.9999 | 0.1–19.1 | 1.2 | 1.3 | 0.3 | 3.5 | 78.2 (2.5) | 0.9 | 97.6 (3.0) | −53.0 |
| AOH1 | 0.9996 | 2.0–394.7 | 3.7 | 5.6 | 3.5 | 73.1 | 66.7 (2.1) | 18.6 | 84.2 (3.0) | −89.6 |
| MPA1 | 0.9999 | 1.4–33.2 | 4.1 | 3.6 | 1.6 | 6.1 | 81.4 (1.8) | 1.7 | 101.0 (1.1) | −34.6 |
| PEN A | 0.9993 | 1.0–191.3 | 1.4 | 4.0 | 2.1 | 35.4 | 37.8 (0.5) | 9.0 | 56.5 (0.2) | −11.4 |
| α-ZOL1 | 0.9999 | 2.5–249.2 | 4.9 | 9.4 | 5.7 | 90.8 | 95.8 (6.5) | 23.2 | 90.5 (1.7) | −3.0 |
| β-ZOL1 | 0.9999 | 2.6–256.4.4 | 4.8 | 5.2 | 5.7 | 93.4 | 77.9 (5.2) | 23.8 | 108.2 (4.2) | −50.0 |
| ZEA1 | 0.9999 | 0.5–100.7 | 4.6 | 4.8 | 1.2 | 18.7 | 83.4 (5.9) | 4.8 | 103.7 (5.9) | −44.0 |
| FUS X | 0.9995 | 2.0–395.3 | 0.6 | 0.6 | 6 | 73.2 | 96.4 (0.7) | 18.7 | 109.6 (0.9) | 25.0 |
| AF B1 | 0.9998 | 0.1–103.5 | 4.4 | 10.4 | 0.3 | 21.3 | 103.3 (5.2) | 11.4 | 57.8 (8.9) | −65.0 |
| AF B2 | 1.0000 | 0.4–25.9 | 4.7 | 4.4 | 1.2 | 5.3 | 105.2 (2.9) | 2.9 | 62.5 (7.6) | −43.2 |
| AF G1 | 0.9998 | 0.1–103.5 | 6.8 | 10.1 | 0.3 | 21.3 | 96.5 (1.9) | 11.4 | 49.2 (6.2) | −36.3 |
| AF G2 | 0.9998 | 0.4–25.9 | 4.6 | 4.3 | 1.2 | 5.3 | 118.3 (0.6) | 2.9 | 135.5 (0.4) | −30.3 |
| Compounds | Concentration (ng g−1) | N° Positive | Occurrence Frequency (%) | |||
|---|---|---|---|---|---|---|
| Average | Max | Min | U (%) | |||
| 3+15-ADON | 426.9 | 770.0 | 214.1 | 13.2 | 33 | 33.7 |
| DON | 52.8 | 81.3 | 36.5 | 14.1 | 15 | 16.3 |
| ENN B | 322.0 | 1305.5 | 53.1 | 13.4 | 49 | 50.0 |
| FB1 | 578.8 | 1453.9 | 14.0 | 11.6 | 95 | 96.9 |
| FB2 | 160.6 | 481.1 | 13.3 | 1.1 | 64 | 65.3 |
| MAC A | 124.3 | 209.9 | 46.1 | 13.6 | 4 | 4.1 |
| OTA | 5.1 | 5.1 | 5.1 | 11.0 | 1 | 1.0 |
| ROQ-C | 36.9 | 59.3 | 19.6 | 6.3 | 4 | 4.1 |
| STER | 8.7 | 15.3 | 2.6 | 10.9 | 20 | 20.4 |
| CPA | 96.5 | 257.0 | 2.6 | 8.7 | 22 | 22.4 |
| AND A | 33.3 | 91.0 | 8.8 | 2.8 | 6 | 6.1 |
| AOH | 126.3 | 295.4 | 12.7 | 12.4 | 24 | 24.5 |
| MPA | 262.2 | 3151.4 | 8.2 | 7.4 | 19 | 19.4 |
| PEN A | 159.7 | 234.5 | 63.7 | 9.1 | 9 | 9.2 |
| α-ZOL | 32.5 | 50.3 | 25.3 | 21.1 | 28 | 28.6 |
| β-ZOL | 35.3 | 78.9 | 8.1 | 11.0 | 67 | 68.4 |
| ZEA | 10.2 | 23.2 | 3.5 | 10.2 | 90 | 91.8 |
| Mycotoxins | Acronym | Molecular Formula | Exact Mass (m/z) | CAS Number | Molecular Ion | RT (min) | Structure |
|---|---|---|---|---|---|---|---|
| 3-Acetyl deoxynivalenol | 3-ADON | C17H22O7 | 338.1365 | 50722-38-8 | [M+H]+ | 3.83 |  |
| 15-Acetyl deoxynivalenol | 15-ADON | C17H22O7 | 338.1365 | 88337-96-6 | [M+H]+ | 3.83 |  |
| Deoxynivalenol | DON | C15H20O6 | 296.1259 | 51481-10-8 | [M+H]+ | 2.24 |  |
| Enniatin B | ENN B | C33H57N3O9 | 639.4094 | 917-13-5 | [M+H]+ | 8.15 |  |
| Enniatin B1 | ENN B1 | C34H59N3O9 | 653.4251 | 19914-20-6 | [M+H]+ | 8.30 |  |
| Fumonisin B1 | FB1 | C34H59NO15 | 721.3884 | 116355-83-0 | [M+H]+ | 5.20 |  |
| Fumonisin B2 | FB2 | C34H59NO14 | 705.3935 | 116355-84-1 | [M+H]+ | 6.20 |  |
| HT-2 toxin | HT-2 | C22H32O8 | 424.2097 | 26934-87-2 | [M+NH4]+ | 5.70 |  |
| Marcfortine A | MAC A | C28H35N3O4 | 477.2627 | 75731-43-0 | [M+H]+ | 6.97 |  |
| Ochratoxin A | OTA | C20H18ClNO6 | 403.0822 | 303-47-9 | [M+H]+ | 5.36 |  |
| Roquefortine C | ROQ-C | C22H23N5O2 | 389.1851 | 58735-64-1 | [M+H]+ | 6.50 |  |
| Sterigmatocystin | STE | C18H12O6 | 324.0633 | 10048-13-2 | [M+H]+ | 6.62 |  |
| T-2 toxin | T-2 | C24H34O9 | 466.2202 | 21259-20-1 | [M+NH4]+ | 6.16 |  |
| Cyclopiazonic acid | CPA | C20H20N2O3 | 336.1473 | 18172-33-3 | [M+H]+ | 5.00 |  |
| Andrastin A | AND A | C28H38O7 | 486.2617 | 174232-42-9 | [M-H]- | 5.95 |  |
| Alternariol | AOH | C14H10O5 | 258.0528 | 641-38-3 | [M-H]- | 5.64 |  |
| Mycophenolic acid | MPA | C17H20O6 | 320.1259 | 24280-93-1 | [M-H]- | 4.93 |  |
| Penitrem A | PEN A | C37H44ClNO6 | 633.2857 | 12627-35-9 | [M+H]+ | 7.64 |  |
| α-Zearalenol | α-ZOL | C18H24O5 | 320.1623 | 36455-72-8 | [M+H]+ | 5.95 |  |
| β-Zearalenol | β-ZOL | C18H24O5 | 320.1623 | 71030-11-0 | [M+H]+ | 6.34 |  |
| Zearalenone | ZEA | C18H22O5 | 318.1467 | 17924-92-4 | [M+H]+ | 6.48 |  |
| Fusarenon X | FUS X | C17H22O8 | 354.1314 | 23255-69-8 | [M+H]+ | 3.03 |  |
| Aflatoxin B1 | AF B1 | C17H12O6 | 312.0633 | 1162-65-8 | [M+H]+ | 4.95 |  |
| Aflatoxin B2 | AF B2 | C17H14O6 | 314.0790 | 7220-81-7 | [M+H]+ | 4.75 |  |
| Aflatoxin G1 | AF G1 | C17H12O7 | 328.0583 | 1165-39-5 | [M+H]+ | 4.54 |  |
| Aflatoxin G2 | AF G2 | C17H14O7 | 330.0739 | 7241-98-7 | [M+H]+ | 4.33 |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facorro, R.; Llompart, M.; Dagnac, T. Combined (d)SPE-QuEChERS Extraction of Mycotoxins in Mixed Feed Rations and Analysis by High Performance Liquid Chromatography-High-Resolution Mass Spectrometry. Toxins 2020, 12, 206. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030206
Facorro R, Llompart M, Dagnac T. Combined (d)SPE-QuEChERS Extraction of Mycotoxins in Mixed Feed Rations and Analysis by High Performance Liquid Chromatography-High-Resolution Mass Spectrometry. Toxins. 2020; 12(3):206. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030206
Chicago/Turabian StyleFacorro, Rocio, Maria Llompart, and Thierry Dagnac. 2020. "Combined (d)SPE-QuEChERS Extraction of Mycotoxins in Mixed Feed Rations and Analysis by High Performance Liquid Chromatography-High-Resolution Mass Spectrometry" Toxins 12, no. 3: 206. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12030206