Simultaneous Determination of Multiple Mycotoxins in Swine, Poultry and Dairy Feeds Using Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry




Abstract
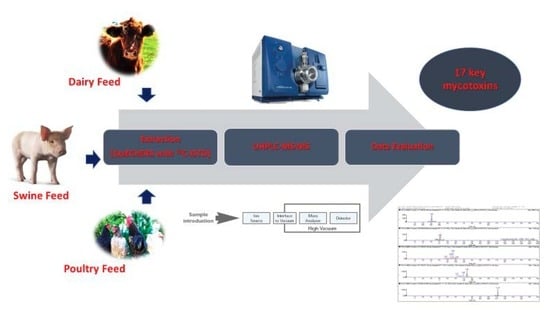
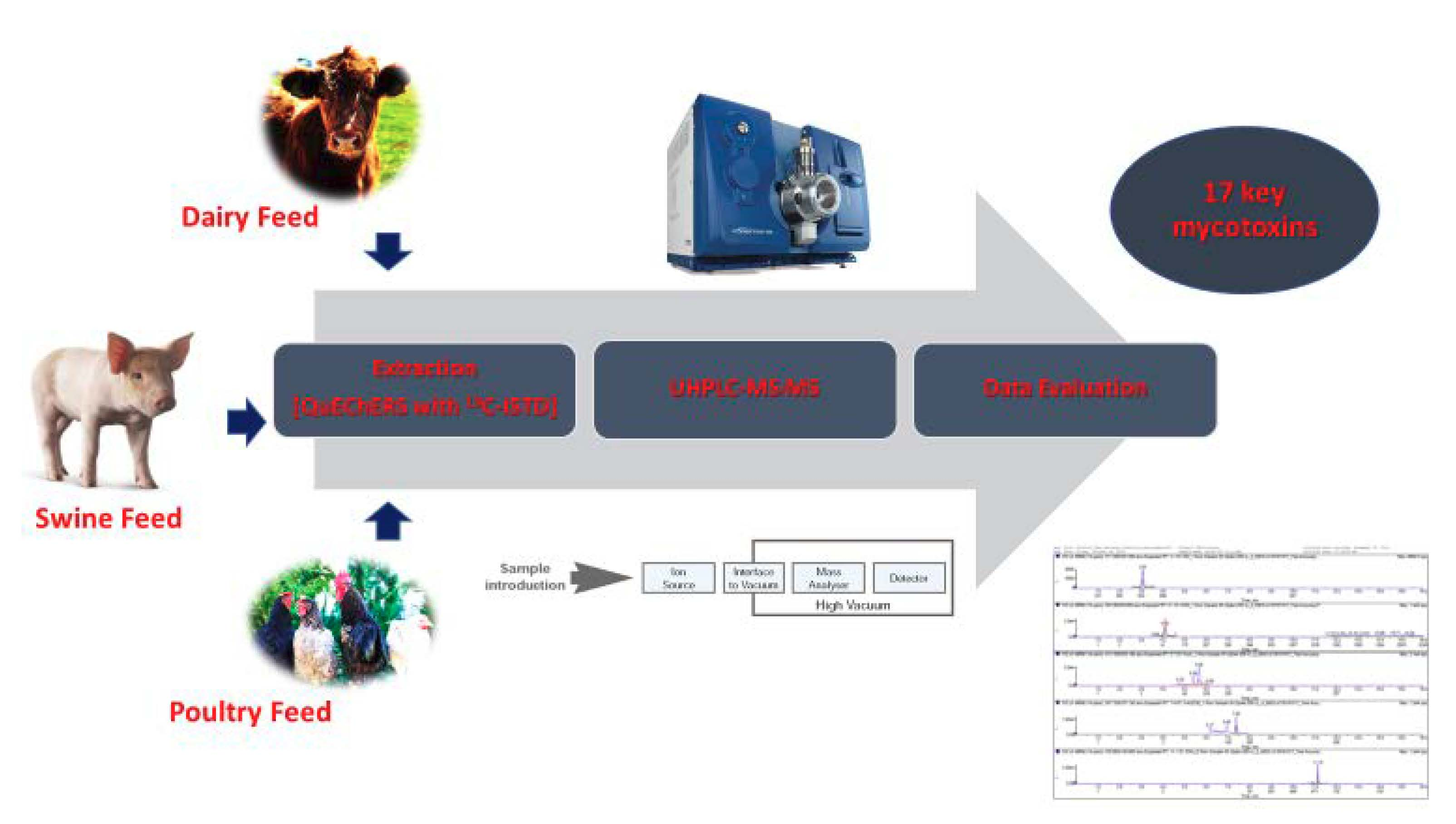
:
1. Introduction
2. Results and Discussion
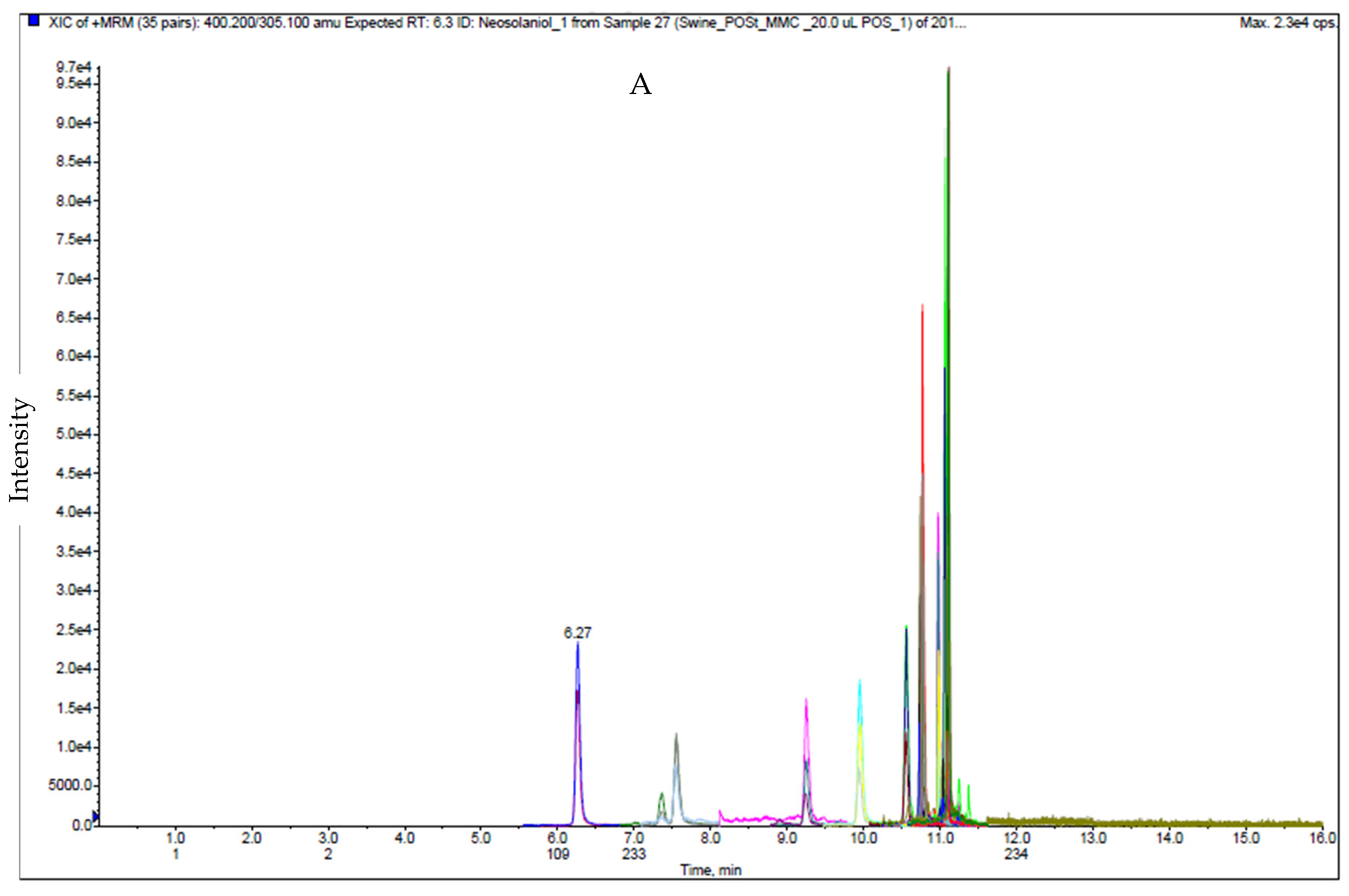
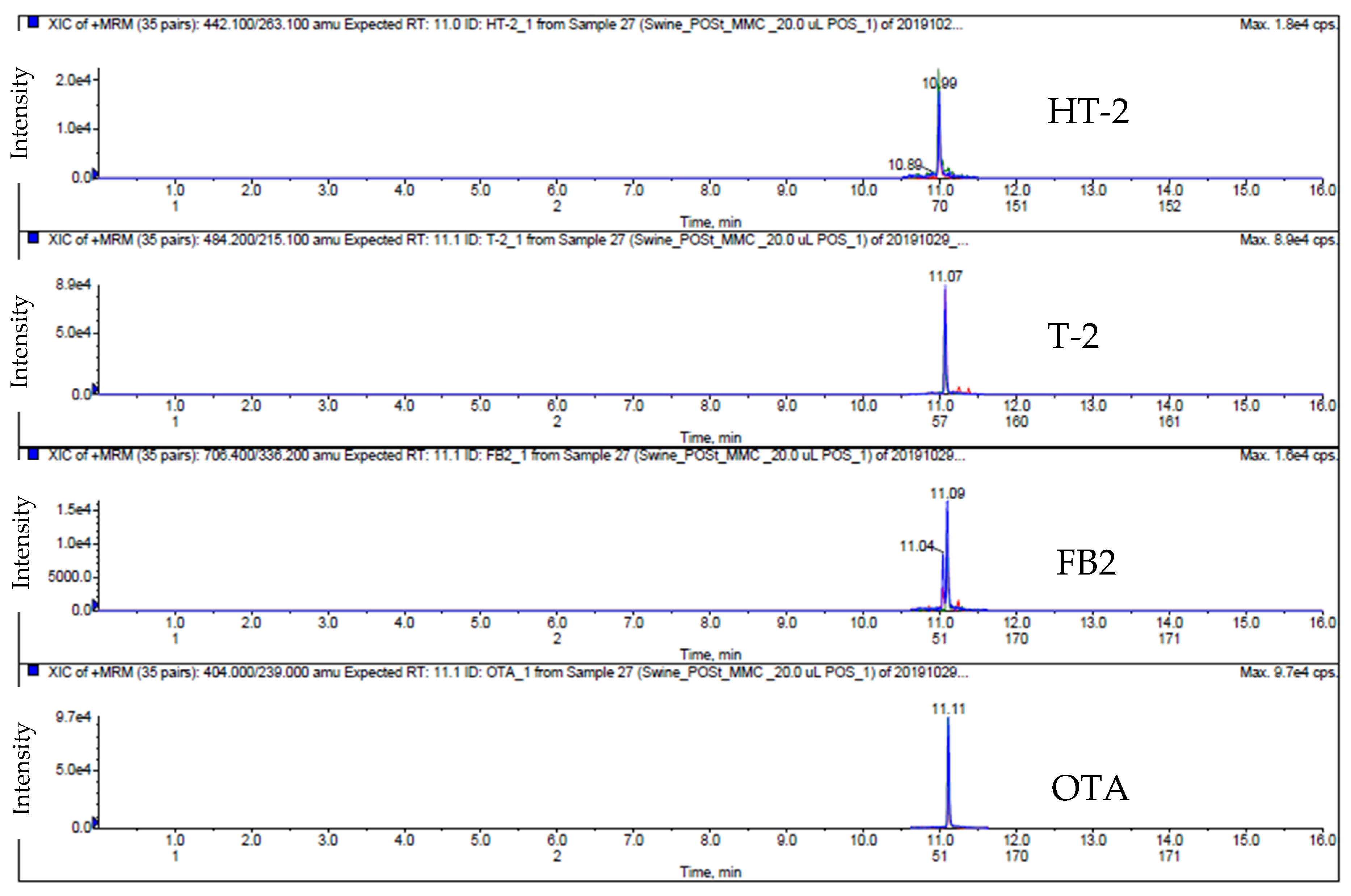
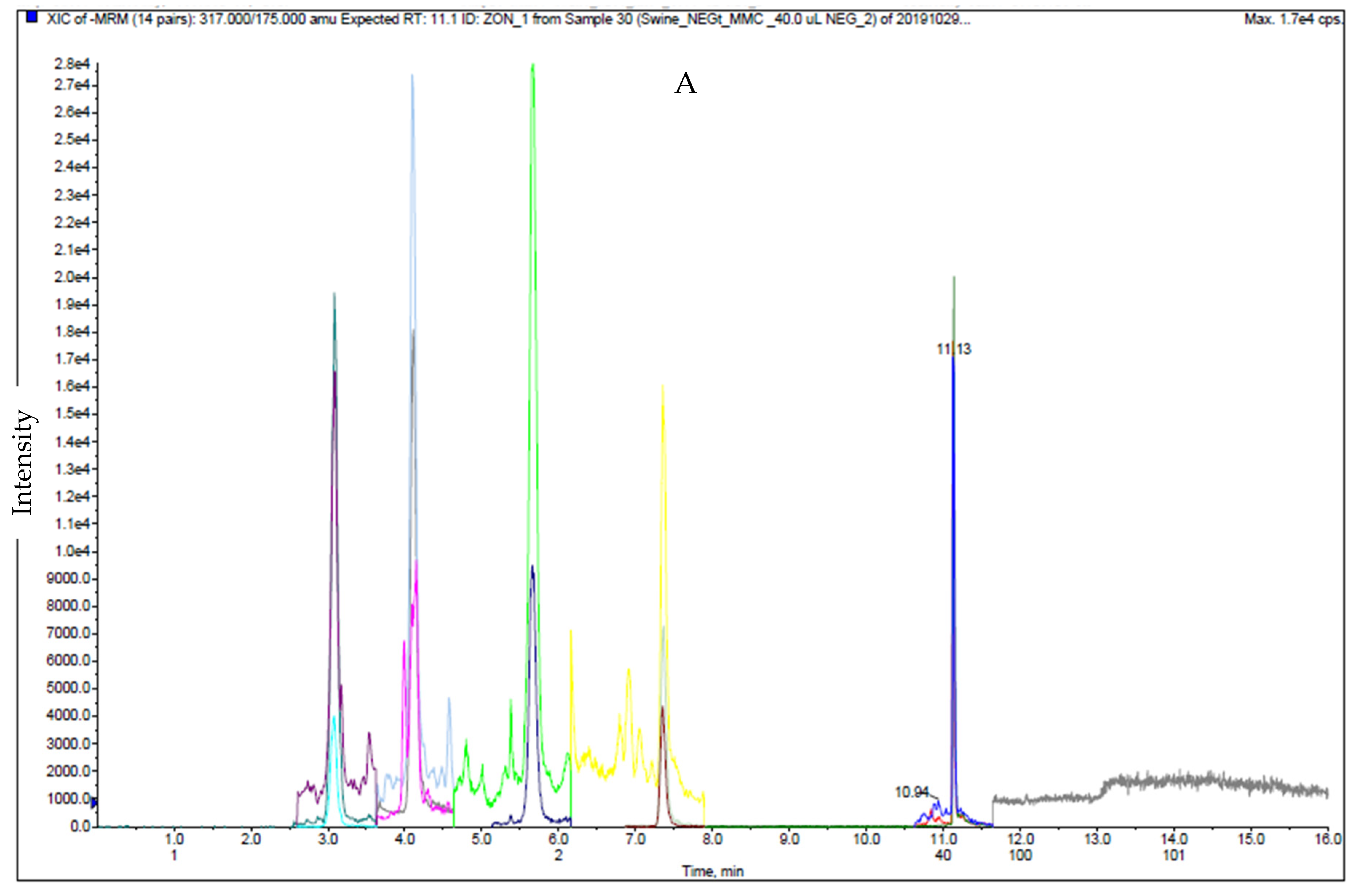
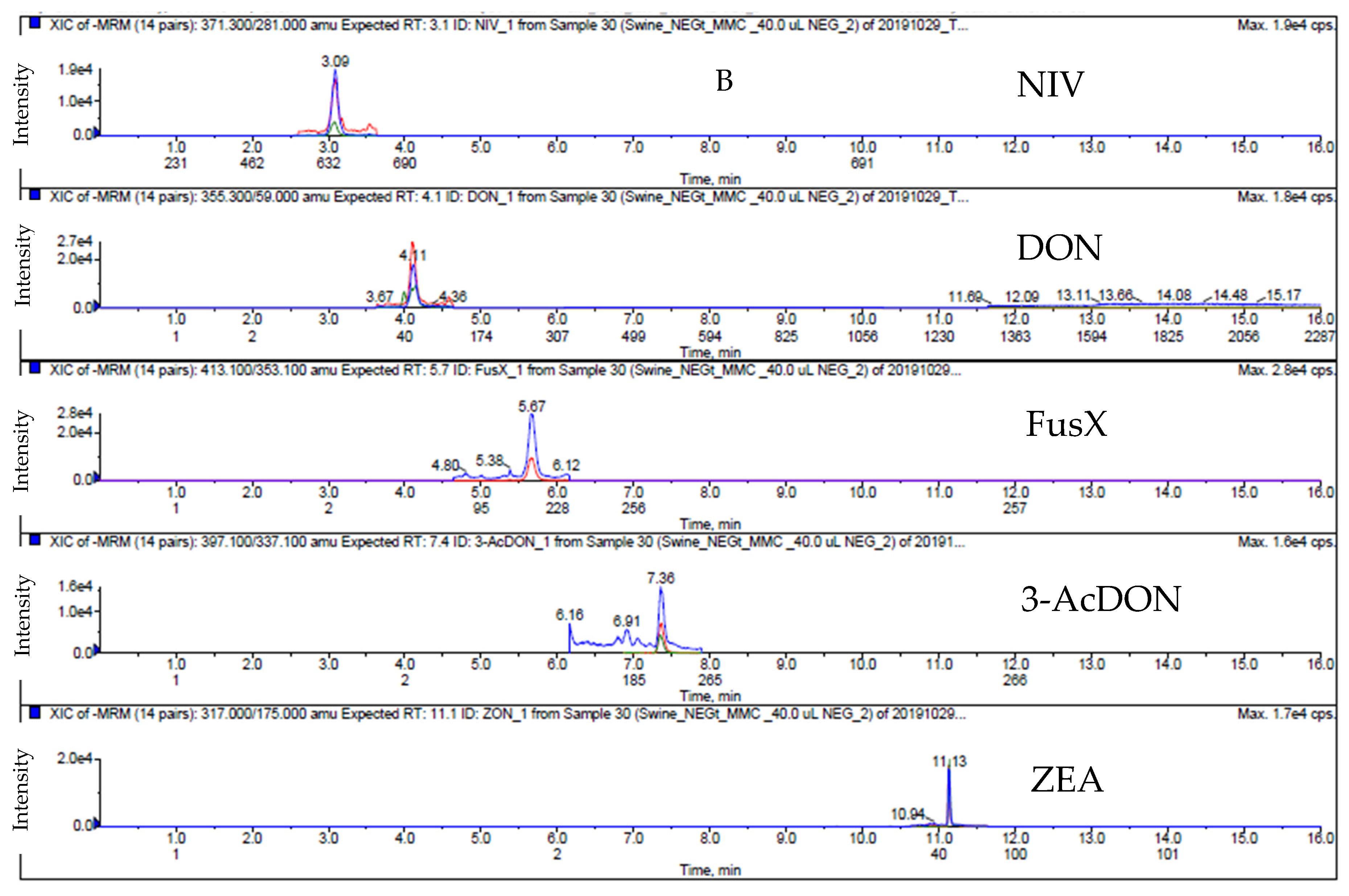
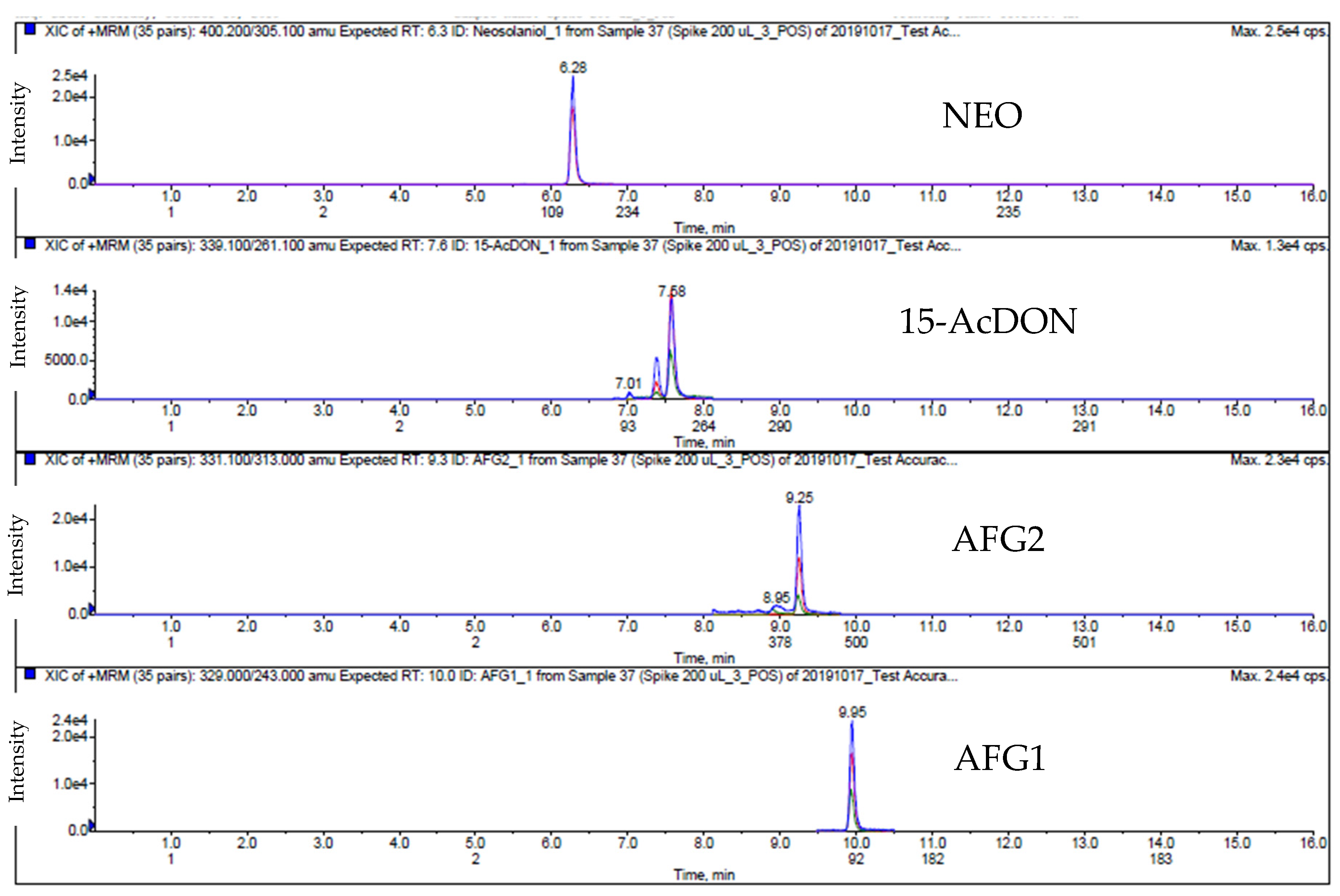
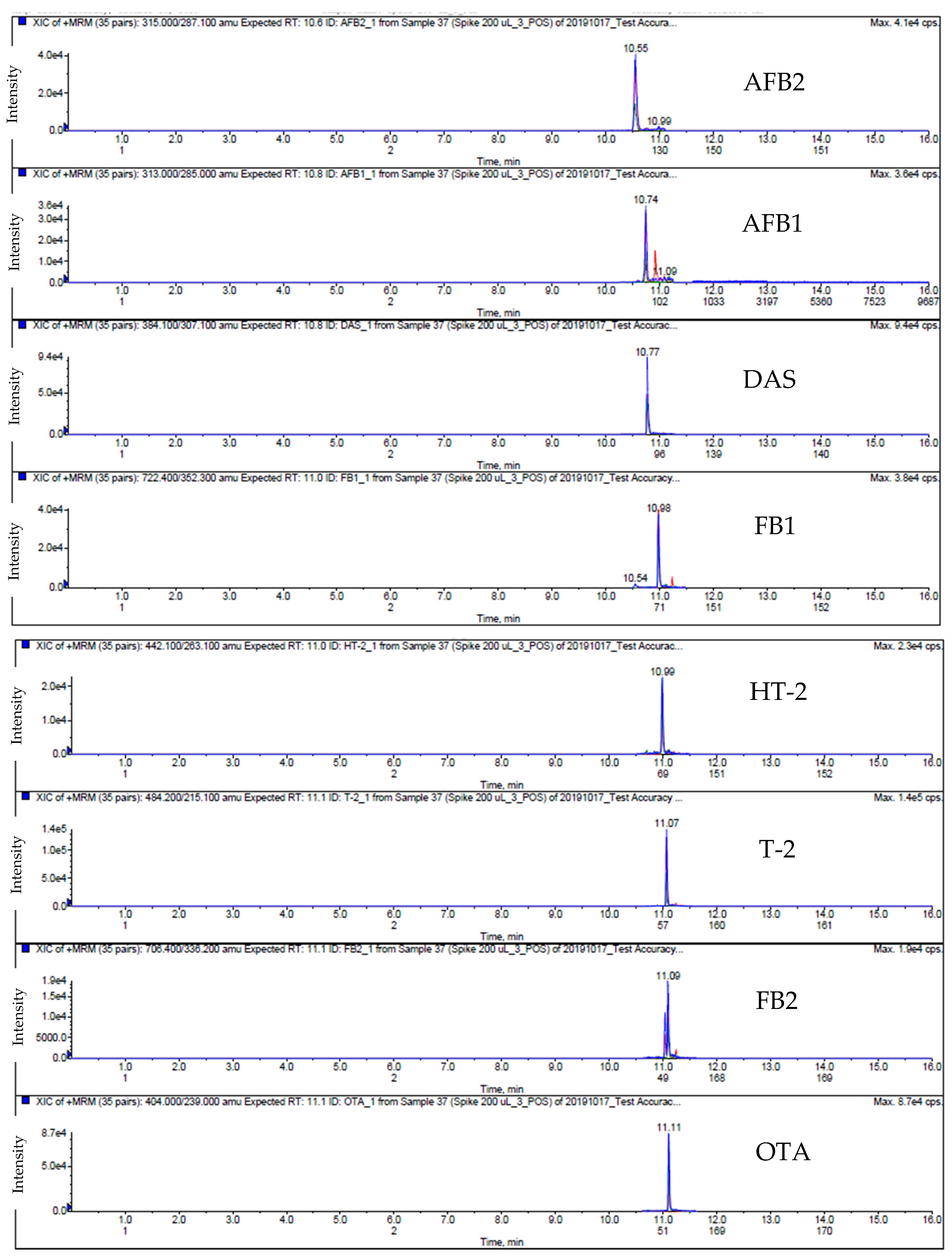
2.1. LC-MS/MS Analysis
2.2. QuEChERS-Based Procedure
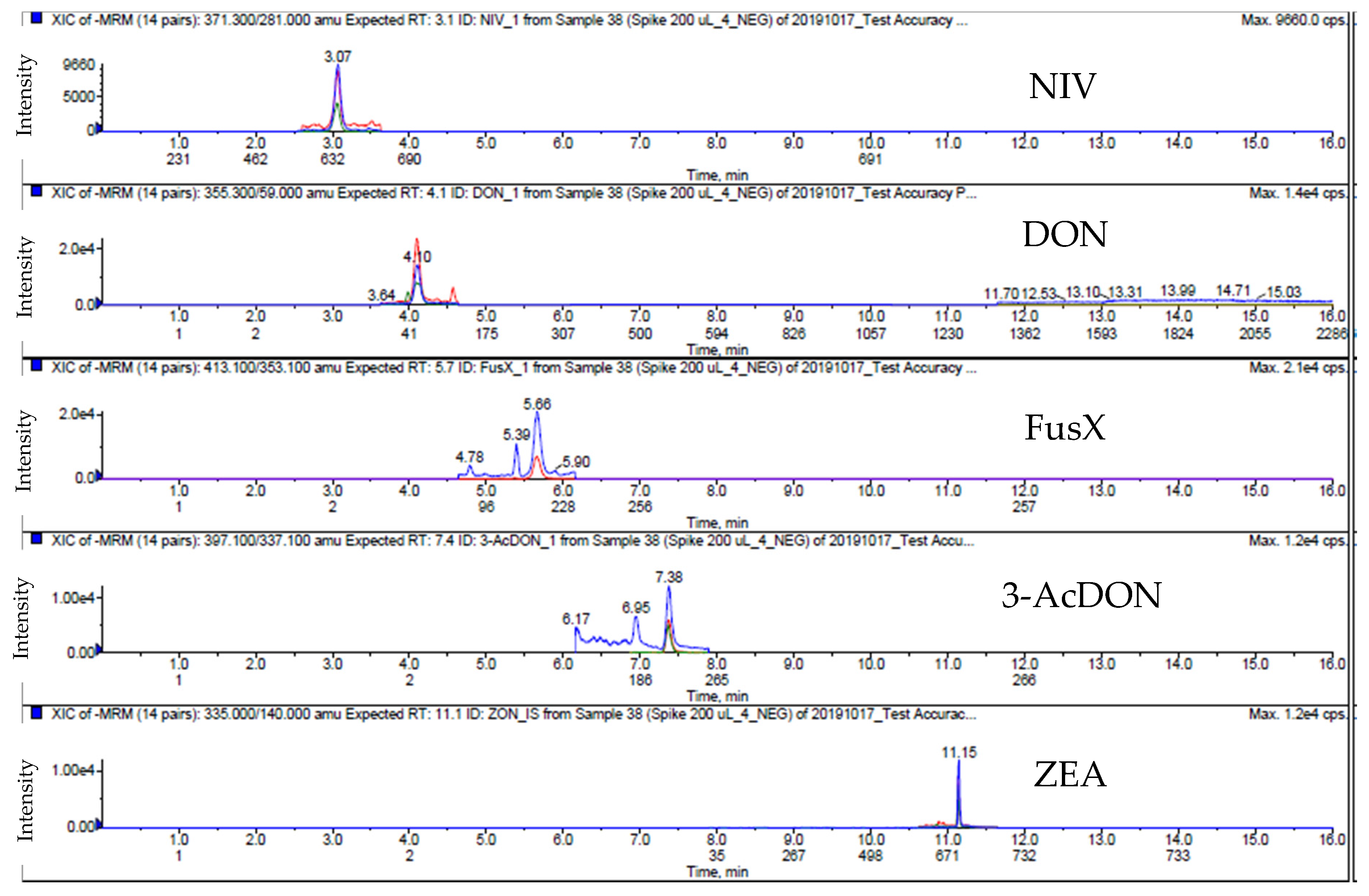
2.3. Method Validation
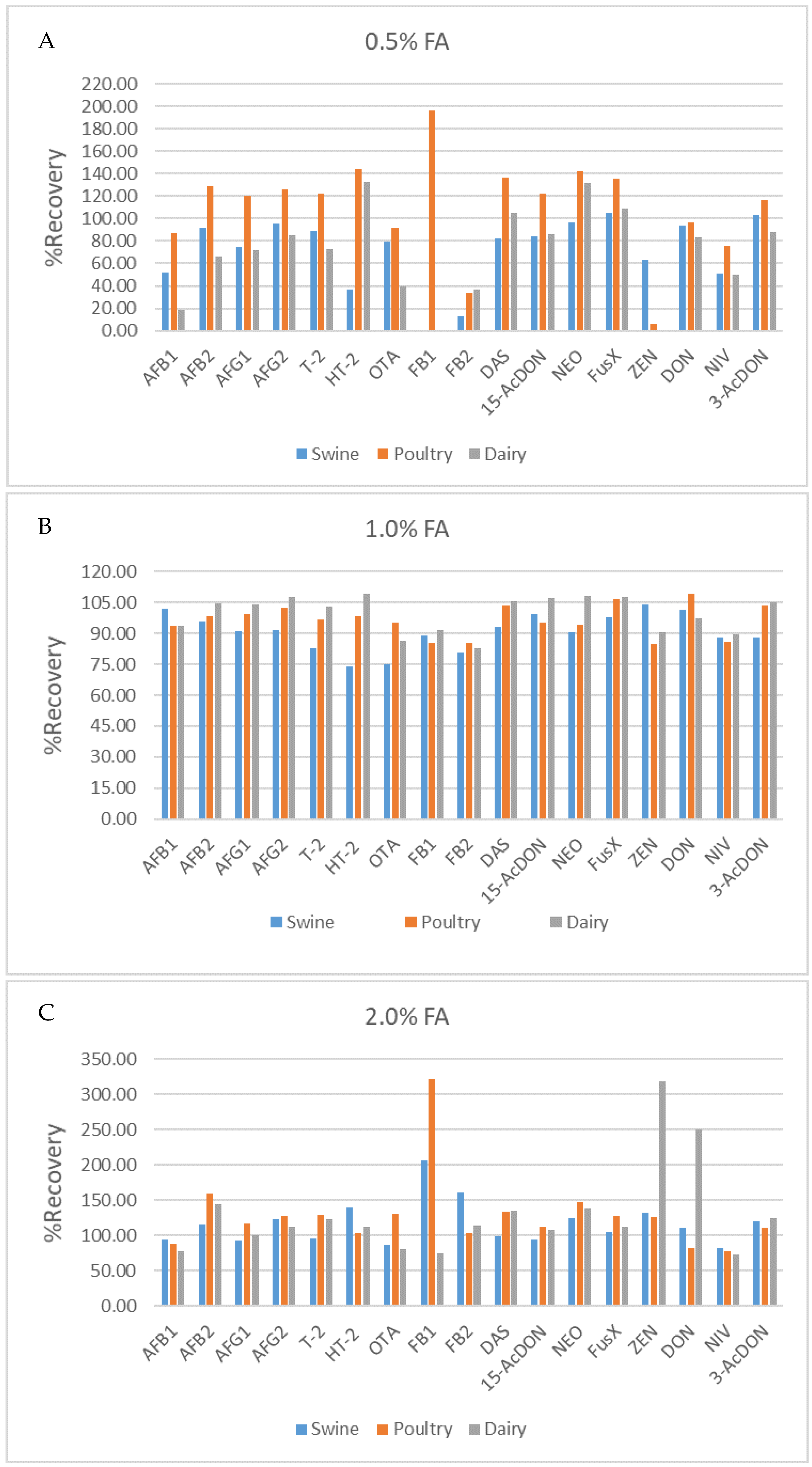
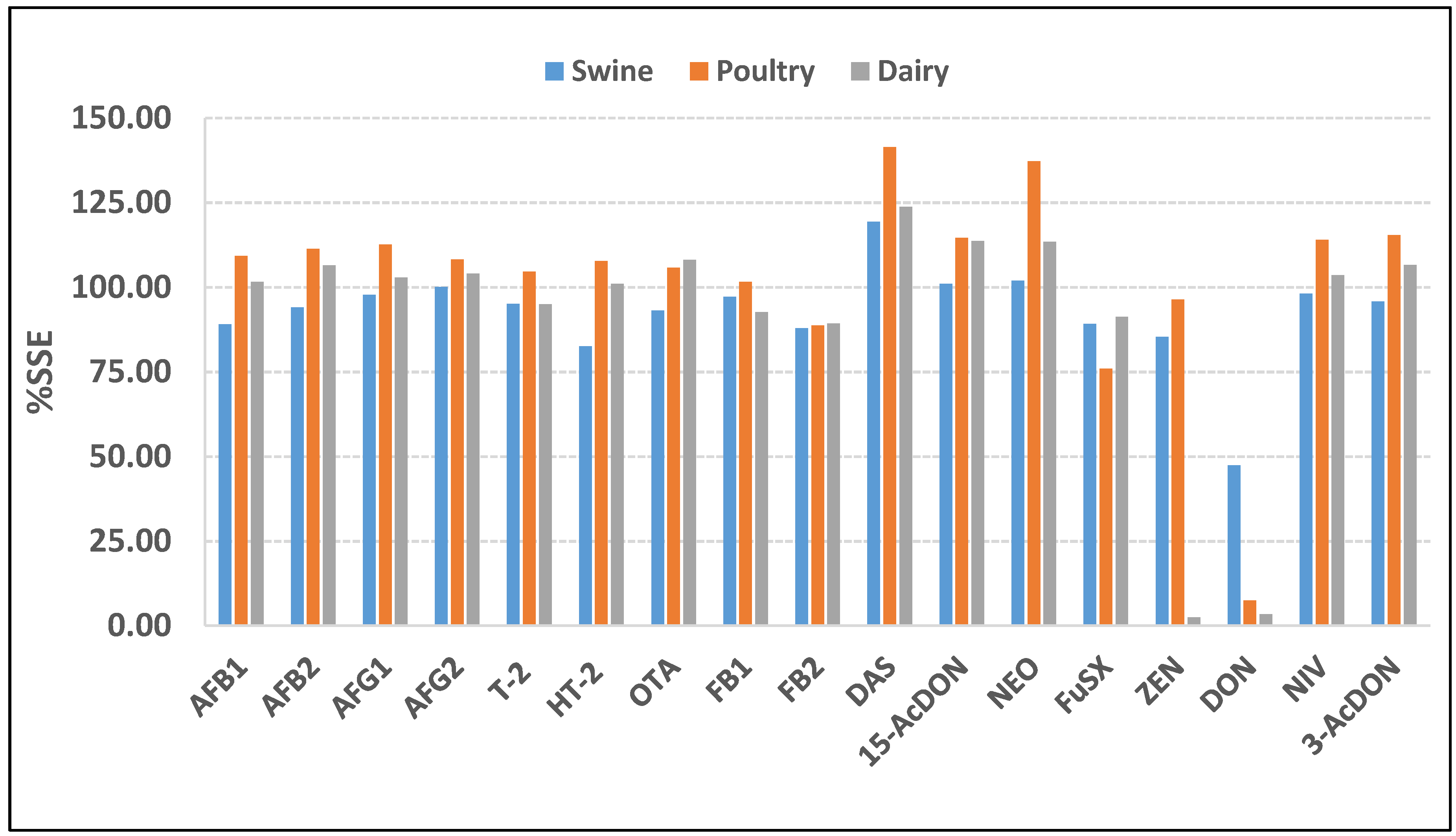
2.4. Matrix Effect Study
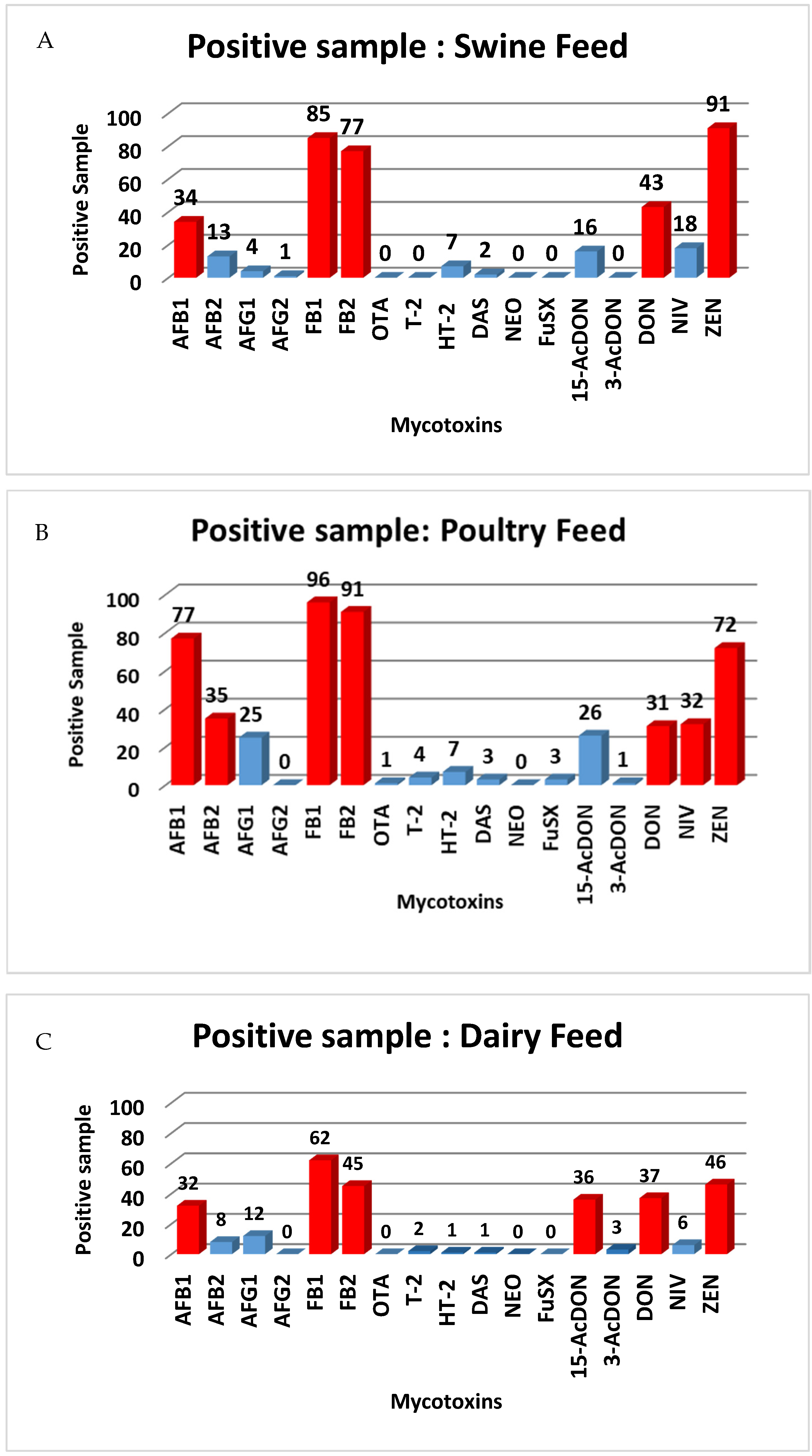
2.5. Occurrence of the Mycotoxins in Animal Feed
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Unlabeled Analytical Standards
4.3. Isotopically Internal Standards
4.4. Preparation of Standards Solution
4.5. Preparation of Isotopically Internal Standards
4.6. Feed Samples
4.7. QuEChERS-Based Procedure
4.8. LC-MS/MS Analysis
4.9. Method Validation
4.10. Matrix Effect Study
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kongkapan, J.; Poapolathep, S.; Isariyodom, S.; Kumagai, S.; Poapolathep, A. Simultaneous detection of multiple mycotoxins in broiler feeds using a liquid chromatography tandem-mass spectrometry. J. Veter-Med. Sci. 2016, 78, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Logrieco, A.; Miller, J.D.; Eskola, M.; Krska, R.; Ayalew, A.; Bandyopadhyay, R.; Battilani, P.; Bhatnagar, D.; Chulze, S.N.; De Saeger, S.; et al. The Mycotox Charter: Increasing Awareness of, and Concerted Action for, Minimizing Mycotoxin Exposure Worldwide. Toxins 2018, 10, 149. [Google Scholar] [CrossRef] [Green Version]
- Marín, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.G.; Ross, P.F. Methods for Detection and Quantitation of Fumonisins in Corn, Cereal Products and Animal Excreta. J. Food Prot. 1994, 57, 536–540. [Google Scholar] [CrossRef]
- Eskola, M.; Elliott, C.T.; HajšLová, J.; Steiner, D.; Krska, R. Towards a dietary-exposome assessment of chemicals in food: An update on the chronic health risks for the European consumer. Crit. Rev. Food Sci. Nutr. 2019, 1–22. [Google Scholar] [CrossRef]
- European Commission. Directive 2002/32/EC of the European Parliament and of the Council of 7 May 2002 on undesirable substances in animal feed. Off. J. Eur. Commun. 2002, 140, 10–21.
- Commission Recommendation 2006/576/EC of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxinA, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off. J. Eur. Commun. 2006, L229, 7–9.
- Dzuman, Z.; Stranska-Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavíková, P.; HajšLová, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef]
- Rasmussen, R.R.; Storm, I.M.L.D.; Rasmussen, P.H.; Smedsgaard, J.; Nielsen, K.F. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal. Bioanal. Chem. 2010, 397, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, S.; Van Poucke, C.; Detavernier, C.; Dumoulin, F.; Van De Velde, M.; Schoeters, E.; Van Dyck, S.; Averkieva, O.; Van Peteghem, C.; De Saeger, S. Occurrence of Mycotoxins in Feed as Analyzed by a Multi-Mycotoxin LC-MS/MS Method. J. Agric. Food Chem. 2010, 58, 66–71. [Google Scholar] [CrossRef]
- Jackson, L.C.; Kudupoje, M.B.; Yiannikouris, A. Simultaneous multiple mycotoxin quantification in feed samples using three isotopically labeled internal standards applied for isotopic dilution and data normalization through ultra-performance liquid chromatography/electrospray ionization tandem mass spe. Rapid Commun. Mass Spectrom. 2012, 26, 2697–2713. [Google Scholar] [CrossRef]
- Streit, E.; Schwab, C.; Sulyok, M.; Nährer, K.; Krska, R.; Schatzmayr, G. Multi-Mycotoxin Screening Reveals the Occurrence of 139 Different Secondary Metabolites in Feed and Feed Ingredients. Toxins 2013, 5, 504–523. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.-B.; Liu, N.; Yang, L.; Deng, Y.; Wang, J.; Song, S.; Lin, S.; Wu, A.; Zhou, Z.; Hou, J. Multi-mycotoxin analysis of animal feed and animal-derived food using LC–MS/MS system with timed and highly selective reaction monitoring. Anal. Bioanal. Chem. 2015, 407, 7359–7368. [Google Scholar] [CrossRef]
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gómez, J.V.; Gimeno-Adelantado, J.V.; Jiménez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef]
- Mol, H.G.J.; Plaza-Bolanos, P.; Zomer, P.; De Rijk, T.C.; Stolker, A.A.M.; Mulder, P.P.J. Toward a Generic Extraction Method for Simultaneous Determination of Pesticides, Mycotoxins, Plant Toxins, and Veterinary Drugs in Feed and Food Matrixes. Anal. Chem. 2008, 80, 9450–9459. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Sulyok, M.; Beltran, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography–tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Lacina, O.; Zachariasova, M.; Urbanova, J.; Vaclavikova, M.; Cajka, T.; HajšLová, J. Critical assessment of extraction methods for the simultaneous determination of pesticide residues and mycotoxins in fruits, cereals, spices and oil seeds employing ultra-high performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2012, 1262, 8–18. [Google Scholar] [CrossRef]
- Socas-Rodríguez, B.; González-Sálamo, J.; Herrera-Herrera, A.V.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Recent Advances and Developments in the QuEChERS Method. Compr. Anal. Chem. 2017, 76, 319–374. [Google Scholar]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and Easy Multiresidue Method Employing Acetonitrile Extraction/Partitioning and “Dispersive Solid-Phase Extraction” for the Determination of Pesticide Residues in Produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [Green Version]
- Dzuman, Z.; Stranska-Zachariasova, M.; Veprikova, Z.; Godula, M.; HajšLová, J. Multi-analyte high performance liquid chromatography coupled to high resolution tandem mass spectrometry method for control of pesticide residues, mycotoxins, and pyrrolizidine alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2010, 30, 491–509. [Google Scholar] [CrossRef]
- Li, W.; Herrman, T.J.; Dai, S.Y. Determination of aflatoxins in animal feeds by liquid chromatography/tandem mass spectrometry with isotope dilution. Rapid Commun. Mass Spectrom. 2011, 25, 1222–1230. [Google Scholar] [CrossRef]
- Zhang, K.; Wong, J.W.; Krynitsky, A.J.; Trucksess, M.W. Determining Mycotoxins in Baby Foods and Animal Feeds Using Stable Isotope Dilution and Liquid Chromatography Tandem Mass Spectrometry. J. Agric. Food Chem. 2014, 62, 8935–8943. [Google Scholar] [CrossRef]
- Wu, A.-B.; Rao, Q.; Song, S.; Liu, N.; Han, Z.; Hou, J.; Wu, A. Simultaneous determination of major type B trichothecenes and deoxynivalenol-3-glucoside in animal feed and raw materials using improved DSPE combined with LC-MS/MS. J. Chromatogr. B 2014, 963, 75–82. [Google Scholar] [CrossRef]
- Varga, E.; Glauner, T.; Köppen, R.; Mayer, K.; Sulyok, M.; Schuhmacher, R.; Krska, R.; Berthiller, F. Stable isotope dilution assay for the accurate determination of mycotoxins in maize by UHPLC-MS/MS. Anal. Bioanal. Chem. 2012, 402, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Habler, K.; Gotthardt, M.; Schüler, J.; Rychlik, M. Multi-mycotoxin stable isotope dilution LC–MS/MS method for Fusarium toxins in beer. Food Chem. 2017, 218, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Al-Taher, F.; Banaszewski, K.; Jackson, L.S.; Zweigenbaum, J.; Ryu, D.; Cappozzo, J. Rapid Method for the Determination of Multiple Mycotoxins in Wines and Beers by LC-MS/MS Using a Stable Isotope Dilution Assay. J. Agric. Food Chem. 2013, 61, 2378–2384. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Tessiot, S.; Bessaire, T.; Racault, L.; Fiorese, E.; Urbani, A.; Chan, W.-C.; Cheng, P.; Mottier, P. Combining the quick, easy, cheap, effective, rugged and safe approach and clean-up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2014, 1337, 75–84. [Google Scholar] [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal. Bioanal. Chem. 2007, 389, 1505–1523. [Google Scholar] [CrossRef]
- SANTE, J. European Commission Document No. SANTE/11813/. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. 2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 14 February 2020).
- SANTE, J. European Commission Document no. SANTE/12089/. Guidance Document on Identification of Mycotoxins in Food and Feed. 2016. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_sampling_guid-doc-ident-mycotoxins.pdf (accessed on 14 February 2020).
- Kovalsky, P.; Kos, G.; Nährer, K.; Schwab, C.; Jenkins, T.; Schatzmayr, G.; Sulyok, M.; Krska, R. Co-Occurrence of Regulated, Masked and Emerging Mycotoxins and Secondary Metabolites in Finished Feed and Maize—An Extensive Survey. Toxins 2016, 8, 363. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Swine Feed (n = 100) | Poultry Feed (n = 100) | Dairy Feed (n = 100) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Positive Sample | Range (ng/g) | Mean (ng/g) | Positive Sample | Range (ng/g) | Mean (ng/g) | Positive Sample | Range (ng/g) | Mean (ng/g) | |
| AFB1 | 34 | 0.52–14.2 | 1.7 | 77 | 0.27–326.4 | 8.2 | 32 | 0.54–14.9 | 1.6 |
| AFB2 | 13 | 0.51–4.1 | 0.9 | 35 | 0.26–49.9 | 2.4 | 8 | 1.5–2.4 | 0.9 |
| AFG1 | 4 | 0.51–1.6 | 0.5 | 25 | 0.25–0.86 | 0.6 | 12 | 0.5–1.6 | 0.5 |
| AFG2 | 1 | 0.66 | 0.66 | ND | ND | ND | ND | ND | ND |
| T-2 | ND | ND | ND | 4 | 2.1–3.2 | 2.6 | 2 | 5.1–5.5 | 4.5 |
| HT-2 | 7 | 6.0–19.3 | 9.6 | 7 | 2.6–10.0 | 6.6 | 1 | 15.23 | 15.23 |
| OTA | ND | ND | ND | 1 | 3.1 | 3.1 | ND | ND | ND |
| FB1 | 85 | 15.0–464.8 | 102.4 | 96 | 16.0–2645.5 | 451.7 | 62 | 16.2–731.0 | 88.1 |
| FB2 | 77 | 5.0–136.1 | 31.08 | 91 | 6.4–573.3 | 123.2 | 45 | 5.4–252.2 | 26.1 |
| DAS | 2 | 4.2–5.1 | 4.7 | 3 | 2.8–3.6 | 3.1 | 1 | 4.36 | 4.36 |
| 15-AcDON | 16 | 23.0–83.2 | 30.8 | 26 | 12.8–57.1 | 31.7 | 36 | 20.9–68.2 | 35.9 |
| NEO | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| FuSX | ND | ND | ND | 3 | 39.3–61.9 | 48.0 | ND | ND | ND |
| ZEA | 91 | 0.53–169.2 | 17.4 | 72 | 1.3–235.8 | 30.2 | 46 | 0.73–98.4 | 26.1 |
| DON | 43 | 20.1–631.9 | 215 | 31 | 28.7–1430.8 | 304.6 | 37 | 50.2–538.8 | 167.8 |
| NIV | 18 | 21.9–165.4 | 46.0 | 32 | 25.6–626.0 | 103.4 | 6 | 24.0–117.5 | 51.2 |
| 3-AcDON | ND | ND | ND | 1 | 45.6 | 45.6 | 3 | 29.9–46.6 | 36.0 |
| Compounds | Q1 (m/z) | Q3 (m/z) | RT (min) | DP (V) | EP (V) | CE (V) | CXP (V) |
|---|---|---|---|---|---|---|---|
| AFB1 | 313 | 285.00 * | 10.75 | 136.0 | 6.20 | 33.0 | 20.0 |
| 313 | 241 | 136.0 | 6.20 | 51.0 | 20.0 | ||
| [13C17]-AFB1 | 330.1 | 255.1 | 10.75 | 94.5 | 7.65 | 50.3 | 13.5 |
| AFB2 | 315 | 287.10 * | 10.59 | 141.0 | 5.07 | 37.0 | 20.0 |
| 315 | 259 | 141.0 | 5.07 | 41.0 | 24.0 | ||
| [13C17]-AFB2 | 332.1 | 303.2 | 10.59 | 97.1 | 7.59 | 36.8 | 25.0 |
| AFG1 | 329 | 243.00 * | 9.99 | 138.0 | 7.77 | 34.0 | 20.0 |
| 329 | 200 | 138.0 | 7.77 | 51.0 | 18.0 | ||
| [13C17]-AFG1 | 346.2 | 212.1 | 9.99 | 99.9 | 4.90 | 54.2 | 18.1 |
| AFG2 | 331.1 | 313.00 * | 9.29 | 136.0 | 3.87 | 35.0 | 18.0 |
| 331.1 | 189 | 136.0 | 3.87 | 57.0 | 12.0 | ||
| [13C17]-AFG2 | 348.1 | 259.2 | 9.29 | 95.9 | 5.49 | 43.6 | 16.1 |
| T-2 | 484.2 | 215.10 * | 11.08 | 69.9 | 6.05 | 25.0 | 14.8 |
| 484.2 | 185.1 | 69.9 | 6.05 | 28.9 | 16.1 | ||
| [13C24]-T-2 | 508.3 | 229.2 | 11.08 | 77.6 | 7.77 | 26.7 | 16.8 |
| HT-2 | 442.1 | 263.10 * | 11.00 | 66.5 | 4.86 | 18.0 | 24.0 |
| 442.1 | 215 | 66.5 | 4.86 | 18.6 | 24.5 | ||
| [13C22]-HT-2 | 464.3 | 229.1 | 11.00 | 70.0 | 6.10 | 18.1 | 17.5 |
| OTA | 404 | 239.00 * | 11.12 | 80.0 | 5.78 | 30.9 | 21.0 |
| 404 | 221 | 80.0 | 5.78 | 47.4 | 17.0 | ||
| [13C20]-OTA | 424.2 | 250.1 | 11.12 | 85.1 | 7.95 | 32.3 | 21.4 |
| FB1 | 722.4 | 352.30 * | 10.99 | 165.0 | 7.85 | 49.0 | 26.0 |
| 722.4 | 334.3 | 165.0 | 7.85 | 55.0 | 20.0 | ||
| [13C34]-FB1 | 756.5 | 356.4 | 10.99 | 83.7 | 7.73 | 55.9 | 17.5 |
| FB2 | 706.4 | 336.20 * | 11.12 | 165.0 | 8.00 | 53.0 | 16.0 |
| 706.4 | 318.3 | 165.0 | 8.00 | 53.0 | 18.0 | ||
| [13C34]-FB2 | 740.6 | 358.4 | 11.12 | 87.3 | 11.00 | 50.7 | 23.2 |
| DAS | 384.1 | 307.10 * | 10.78 | 68.8 | 7.67 | 15.60 | 22.70 |
| 384.1 | 247.2 | 68.8 | 7.67 | 18.60 | 17.90 | ||
| [13C19]-DAS | 403.2 | 324.1 | 10.78 | 70.2 | 5.67 | 15.60 | 10.10 |
| 15-AcDON | 339.1 | 261.10 * | 7.61 | 68.8 | 6.16 | 14.20 | 12.90 |
| 339.1 | 137 | 68.8 | 6.16 | 15.80 | 19.90 | ||
| [13C17]-15-AcDON | 373.2 | 338.1 | 7.61 | 60.9 | 8.27 | 17.60 | 23.30 |
| Neosolaniol | 400.2 | 305.10 * | 6.3 | 64.2 | 5.94 | 16.40 | 9.40 |
| 400.2 | 215.1 | 64.2 | 5.94 | 23.80 | 14.90 | ||
| DON | 355.3 | 59.00 * | 4.13 | −58.7 | −4.79 | −20.37 | −17.09 |
| 355.3 | 295 | −58.7 | −4.79 | −13.84 | −8.34 | ||
| [13C15]-DON | 370.4 | 310.1 | 4.13 | −57.7 | −2.72 | −14.37 | −9.14 |
| NIV | 371.3 | 281.00 * | 3.11 | −61.7 | −2.97 | −20.26 | −24.79 |
| 371.3 | 311 | −61.7 | −2.97 | −14.29 | −20.01 | ||
| [13C15]-NIV | 386 | 295.2 | 3.11 | −50.2 | −3.76 | −20.60 | −8.37 |
| 3-AcDON | 397.1 | 337.10 * | 7.38 | −60.1 | −5.91 | −20.22 | −9.04 |
| 397.1 | 307 | −60.1 | −5.91 | −12.47 | −9.94 | ||
| [13C17]-3-AcDON | 414 | 323.2 | 7.38 | −59.3 | −5.60 | −20.04 | −9.25 |
| ZEA | 317 | 175.00 * | 11.14 | −172.9 | −3.57 | −32.59 | −9.20 |
| 317 | 131 | −172.9 | −3.57 | −38.00 | −13.00 | ||
| [13C18]-ZEA | 335 | 140 | 11.14 | −161.9 | −8.80 | −39.16 | −7.28 |
| FusX | 413.1 | 353.10 * | 5.65 | −50.0 | −10.00 | −14.00 | −17.00 |
| 413.1 | 263 | 5.65 | −50.0 | −10.00 | −20.00 | −17.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nualkaw, K.; Poapolathep, S.; Zhang, Z.; Zhang, Q.; Giorgi, M.; Li, P.; Logrieco, A.F.; Poapolathep, A. Simultaneous Determination of Multiple Mycotoxins in Swine, Poultry and Dairy Feeds Using Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry. Toxins 2020, 12, 253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040253
Nualkaw K, Poapolathep S, Zhang Z, Zhang Q, Giorgi M, Li P, Logrieco AF, Poapolathep A. Simultaneous Determination of Multiple Mycotoxins in Swine, Poultry and Dairy Feeds Using Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry. Toxins. 2020; 12(4):253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040253
Chicago/Turabian StyleNualkaw, Kraiwut, Saranya Poapolathep, Zhaowei Zhang, Qi Zhang, Mario Giorgi, Peiwu Li, Antonio Francesco Logrieco, and Amnart Poapolathep. 2020. "Simultaneous Determination of Multiple Mycotoxins in Swine, Poultry and Dairy Feeds Using Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry" Toxins 12, no. 4: 253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040253