The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease


1
Laboratory of Nephrology, Department of Immunology and Microbiology, KU Leuven—University of Leuven, B-3000 Leuven, Belgium
2
Department of Nephrology and Renal Transplantation, University Hospitals Leuven, B-3000 Leuven, Belgium
3
Translational Research Center for Gastrointestinal Disorders (TARGID), KU Leuven—University of Leuven, B-3000 Leuven, Belgium
*
Author to whom correspondence should be addressed.
Toxins 2020, 12(5), 285; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050285
Submission received: 30 March 2020
/
Revised: 15 April 2020
/
Accepted: 16 April 2020
/
Published: 29 April 2020
(This article belongs to the Special Issue Comorbidities in Chronic Kidney Disease (CKD))
{kind=link}
Abstract
:Patients with chronic kidney disease (CKD) are at increased risk of bone mineral density loss and vascular calcification. Bone demineralization and vascular mineralization often concur in CKD, similar to what observed in the general population. This contradictory association is commonly referred to as the ‘calcification paradox’ or the bone–vascular axis. Mounting evidence indicates that CKD-associated gut dysbiosis may be involved in the pathogenesis of the bone–vascular axis. A disrupted intestinal barrier function, a metabolic shift from a predominant saccharolytic to a proteolytic fermentation pattern, and a decreased generation of vitamin K may, alone or in concert, drive a vascular and skeletal pathobiology in CKD patients. A better understanding of the role of gut dysbiosis in the bone–vascular axis may open avenues for novel therapeutics, including nutriceuticals.
Key Contribution: Gut dysbiosis is common in patients with CKD and is increasingly recognized to be involved in the pathogenesis of the bone-vascular axis.
1. Introduction
Chronic kidney disease (CKD) is recognized as a major noncommunicable disease of growing epidemic dimensions worldwide. CDK–mineral and bone disorder (CKD–MBD) is one of the many complications associated with CKD. It represents a systemic disorder of mineral and bone metabolism due to CKD, manifested with either one or a combination of the following: (1) abnormalities of calcium, phosphorus (phosphate), parathyroid hormone, or vitamin D metabolism; (2) abnormalities in bone turnover, mineralization, volume, linear growth, or strength; and (3) vascular or other soft-tissue calcification. CKD–MBD explains, at least in part, the high morbidity and mortality of CKD patients [1].
Bone demineralization and vascular mineralization often concur in CKD, as in the general population. This contradictory association is often referred to as the ‘calcification paradox’ or the bone–vascular axis [2]. Mounting evidence indicates that CKD-associated gut dysbiosis may be involved in the pathogenesis of the bone–vascular axis. The present review aims to update the current evidence on the role of gut dysbiosis in the bone–vascular axis.
2. Bone–Vascular Axis
Mounting evidence indicates that CKD is a state of impaired bone quantity [3,4,5,6,7,8,9]. In clinical practice, bone quantity is most commonly assessed by dual-energy X-ray absorptiometry (DXA). A decreased bone quantity [6,10], along with an impaired bone quality [11], contributes to an excessively high fracture risk in CKD patients. Epidemiological evidence demonstrates that the fracture risk increases along with the progression of CKD, with CKD stage-5D patients showing a non-vertebral fracture risk that is up to six times higher than the fracture risk of age- and gender-matched controls [12,13]. Fractures are a major cause of morbidity and, compared to CKD patients without fractures, those with fractures experience a several-fold increased risk of mortality [14,15]. Fractures also impose a large financial burden on healthcare systems.
Vascular calcification is a condition characterized by calcium phosphate crystal deposition in the intima, media, or cardiac valves [16]. Media calcification is most common among patients with CKD, with prevalence and severity paralleling the progression of renal failure [17]. Vascular calcification is observed in more than 60% of patients with CKD stage 5D [16]. Vascular calcification is an active, cell-regulated process. Its pathophysiology varies across vascular beds and remains incompletely understood, despite major progress in the last decade [18,19,20,21]. Vascular calcification is an established independent risk factor for cardiovascular disease (CVD), the leading cause of morbidity and mortality in patients with CKD [22,23].
Many clinical studies have demonstrated an association between low bone mass and vascular calcification in patients with CKD [24,25,26,27,28,29]. The association between osteoporosis and vascular calcification is not specific to CKD. It also is commonly observed in the elderly and in patients with diabetes mellitus or chronic obstructive pulmonary disease [30,31,32,33,34,35]. Importantly, the association remains significant after adjustment for age, which suggests an age-independent relationship [26,27,29,30,31,32,33,36,37]. Vascular calcification and bone mineralization are both actively regulated processes showing many similarities. The co-existence of bone loss with vascular calcification should therefore be considered a paradoxical phenomenon. It is commonly referred to as the ‘calcification paradox’. It most likely reflects direct bone–vascular cross-talk and/or the involvement of common pathogenic factors [2,35].
3. Gut Microbial Ecosystem in Health and CKD
The human gut harbors a complex and dynamic microbial ecosystem that is shaped by diet and host factors [38]. The human microbiome project has shown that the composition of the microbial ecosystem is quite different from one individual to the other. This variability in composition is not continuous and random, but stratified. Nutrient intake patterns are associated with both the degree of diversity and certain clusters of microbial species that are often found to act in concert. The microbial ecosystem thrives on the nutritional leftovers brought to them via the digestive tract. This requires substantial metabolic flexibility, as nutrient availability is dependent on host nutrient intake and digestion. A complex web of overlapping metabolic pathways allows access to nutritional sources inaccessible to mammalian metabolism, thereby supplementing the host metabolism.
The gut microbiota provides the host with a variety of functions including the digestion of complex dietary components, production of vitamins, maturation of the immune system, protection against pathogens, and regulation of host metabolism [39]. A compelling set of bidirectional links between the gut microbiota and the host (patho) physiology has emerged, and metabolites produced by the microbiota are increasingly implicated as crucial executors of the microbial influence on the host. Of note, microbial metabolites do account for about 10% of circulating metabolites [40].
CKD is associated with a disturbed gut microbiota composition and metabolism [41,42,43]. These disturbances reflect the aggregate consequences of CKD, more specifically, the effects of kidney dysfunction combined with the effects of therapeutic interventions and dietary modifications. Kidney dysfunction has a major impact on a number of physiological systems, including the gastrointestinal tract. More specifically, gastrointestinal assimilation and motility, both known to modify the colonic microenvironment, may be disturbed in CKD [44,45]. CKD, furthermore, causes an increased influx of urea, uric acid, and oxalate into the colon. Urea is converted to ammonia and subsequently to ammonium hydroxide, which can raise the colonic pH and result in mucosal damage. Patients with CKD, furthermore, often consume a diet low in dietary fiber to avoid hyperkaliemia. These and other dietary measures may importantly impact on gut microbiota composition and metabolism. Finally, not only antibiotics, but also non-antibiotic drugs are increasingly recognized to extensively affect human gut bacteria [46]. This is especially relevant in the setting of CKD, as pill burden in these patients is huge.
Using bacterial DNA isolated from fecal samples, Vaziri et al. showed highly significant differences in the abundance of over 200 bacterial operational taxonomic units between hemodialysis patients and healthy controls [41]. Additional studies demonstrated that patients with End Stage Kidney Disease (ESKD) had an increased number of bacteria that possess urease, uricase, and p-cresol- and indole-forming enzymes, and a contraction of families or genera possessing butyrate-forming enzymes (e.g., Roseburiae, Lactobacillaceae, and Prevotellaceae) [47,48]. Metabolomics studies showed clear differences in the levels of fecal metabolites (including phenols, indoles, and aldehydes) between patients with CKD and healthy controls. Of interest, the differences in fecal metabolite profiles were greater between patients on hemodialysis and unrelated healthy individuals than between patients on hemodialysis and household members exposed to the same diet [43]. Gryp et al., conversely, failed to observe increasing levels of p-cresyl sulfate, p-cresyl glucuronide, indoxyl sulfate, indole-3-acetic acid levels, and their precursors in stool and urine samples of patients along with the progression of CKD. In addition, anaerobic culture of fecal samples showed no difference in ex vivo p-cresol, indole, and indole-3-acetic acid generation (https://0-doi-org.brum.beds.ac.uk/10.1016/j.kint.2020.01.028). The use of animal models enables the effects of CKD to be separated from those of therapeutic interventions and diet. Studies with uremic rats confirm that renal dysfunction itself induces profound changes in the gut microbiota composition [41] and metabolism [43]. Taken together, current evidence indicates that CKD causes a microbial metabolism shift away from saccharolytic fermentation and towards proteolytic fermentation. Given some contradictory findings, additional prospective studies are required to confirm this shift.
CKD-induced changes to the composition and function of the intestinal microbiota also impair the intestinal barrier function, a condition commonly referred to as leaky gut [38]. A leaky gut in CKD is evidenced by the observation of increased concentrations of bacterial components, such as endotoxin or DNA, in the circulation of patients with increasing CKD stage. The levels of bacterial components are the highest in patients with ESKD treated with dialysis [49,50]. Although circulating bacterial components in patients on dialysis might derive from external sources such as dialysate fluids, the intestinal microbiota is by far the most likely source of these components in patients with CKD not on dialysis [50]. One study showed that after a few days of feeding uremic rodents with a non-pathogenic but green fluorescent Escherichia coli strain, green fluorescent bacterial colonies could be cultured from mouse livers, demonstrating that CKD facilitates the translocation across the intestinal barrier not only of bacterial components but also of entire living bacteria [51,52]. Our current understanding of the effects of CKD on the intestinal barrier function is in line with studies from the 1990s that demonstrated that orally ingested high-molecular-mass polyethylene glycols cross the intestinal barrier and enter the circulation and urine of uremic animals and patients [53]. Some but not all studies in animal models of CKD have demonstrated superficial mucosal erosions or disrupted tight junctions between intestinal epithelial cells in several parts of the gastrointestinal tract [52,54,55], in line with autopsy findings of patients on maintenance hemodialysis, which frequently show subtle pathologies indicative of diffuse gastrointestinal wall inflammation. Both an increased exposure to urea-derived ammonia and ammonium hydroxide [56] and a decreased generation of butyrate may contribute to a leaky gut [57]. Butyrate maintains the barrier function by at least two not mutually exclusive mechanisms. Butyrate is the primary energy source for colonic epithelial cells and undergoes fatty-acid oxidation to such an extent that these cells are slightly hypoxic. This leads to hypoxia-inducible factor-1-mediated upregulation of tight junction genes [58]. In addition, butyrate functions as a histone deacetylase (HDAC) inhibitor, and this has been shown to upregulate tight junction genes as well as the major intestinal mucin MUC2 [59,60] gene and to downregulate the expression of pro-inflammatory cytokines [61]. Treatment of uremic rats with the symbiont Bifidobacterium animalis subsp. lactis Bi-07 attenuated epithelial erosion and decreased intestinal inflammation [52].
4. Gut–Bone–Vascular Axis in CKD
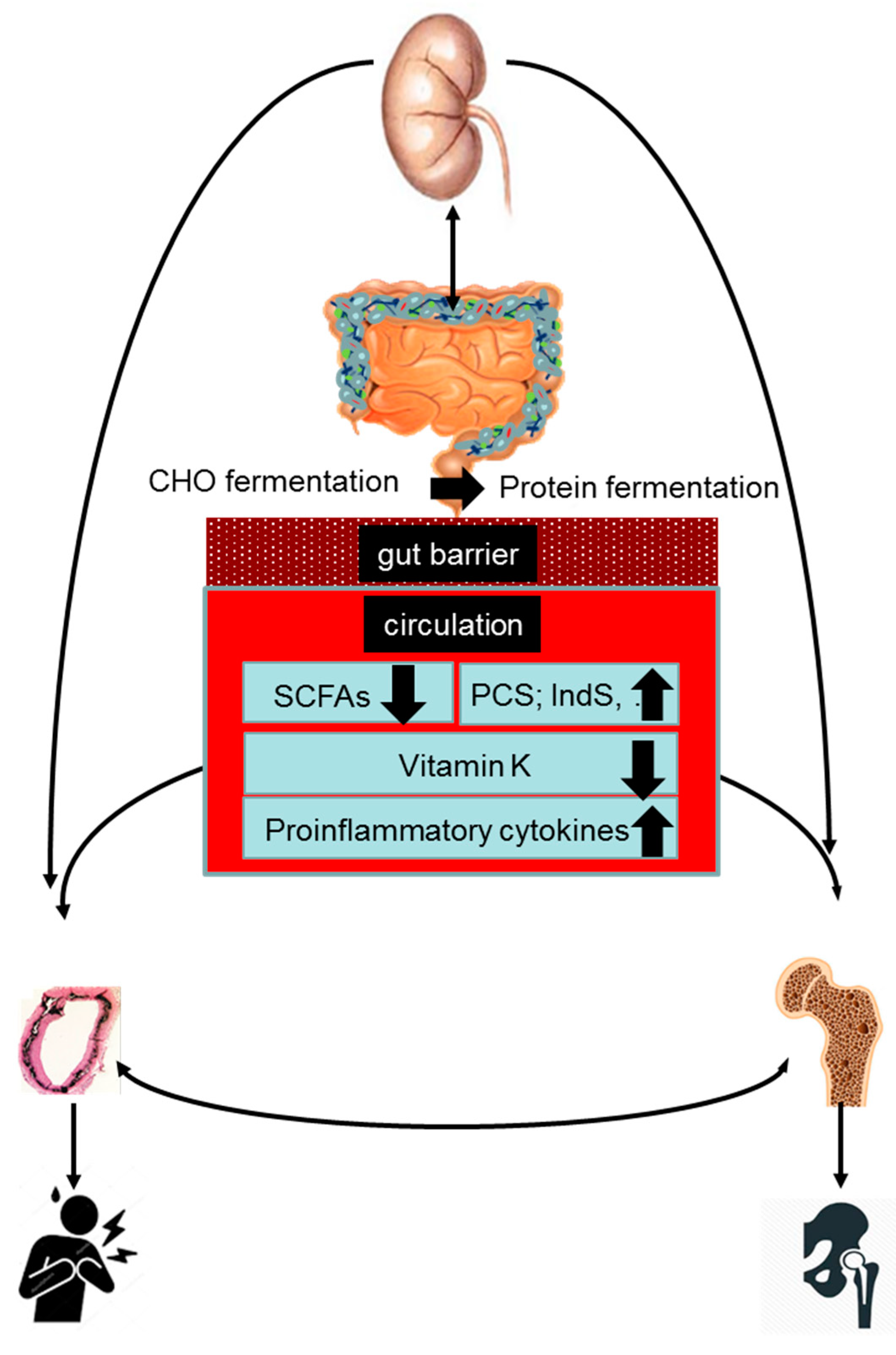
Acknowledging that the gut microbiome is a key regulator of bone [62,63,64] and cardiovascular [65,66,67] health, gut dysbiosis may be hypothesized to be involved in the pathogenesis of the bone–vascular axis. The present review discusses mechanisms by which gut dysbiosis may contribute to vascular calcification and bone demineralization in the setting of CKD. We herein will separately discuss the role of increased protein fermentation, decreased carbohydrate fermentation, vitamin K deficiency, and gut-derived inflammation (Figure 1).
5. Role of Increased Protein Fermentation in the Bone–Vascular Axis
End products of protein fermentation such as phenols and indoles are largely [68] transported across the colonic epithelium via active and passive transport mechanisms [57,69] and subsequently metabolized by phase 1 and 2 reactions (e.g., towards p-cresyl sulfate (PCS) and indoxyl sulfate (IndS)) in the colonic epithelium and liver before entering the systemic circulation 70. Whether CKD affects transport kinetics and metabolism of protein fermentation metabolites remains to be investigated. Protein fermentation metabolites are cleared from the circulation by the kidneys, mainly by tubular secretion, since most are strongly protein-bound [70]. Plasma concentrations of PCS and IndS increase along the progression of CKD to reach levels in patients with ESKD being 10- to 50-fold higher than in healthy controls. These high levels reflect both an increased intestinal production and absorption and a decreased renal clearance [71]. At uremic concentrations, PCS and IndS may disturb several biological processes and confer direct and indirect toxicity in various cells and tissues, at least partly by generating intracellular oxidative stress [72].
Experimental studies revealed that IndS and PCS may promote vascular calcification through various mechanisms [73,74,75]. These mechanisms include (a) increased shedding of endothelial microparticles [76,77], (b) impaired autophagic flux in endothelial cells [78], (c) downregulation of MiR-29b [79], and (d) suppression of the nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of cellular antioxidant activity [80]. Dahl salt-sensitive hypertensive IndS-administered rats presented aortic calcification and upregulation of osteogenic genes when compared to control rats, indicating a pro-calcifying role of IndS in an in vivo animal model [81]. In a subsequent experiment by the same group, Dahl salt-sensitive hypertensive IndS-administered rats presented markers of senescence in the area of aortic calcification [82]. Recently, Opdebeeck et al. reported that both IndS and PCS independently promote vascular calcification in the adenine-induced CKD rat model. This was demonstrated in the aorta, as well as in peripheral arteries. Uremic toxin-induced vascular calcification was associated with the activation of inflammation and coagulation pathways [83].
In line with these experimental data, the circulating levels of PCS and IndS have been repeatedly associated with cardiovascular morbidity (including arterial stiffness, vascular calcification, ischemic and thrombotic events, and atrial fibrillation) and mortality in patients with CKD across stages of the disease [84,85,86] Also in the general population, clear associations between PCS and IndS concentrations and cardiovascular endpoints have been reported. For example, in a population-based study in Belgium, the prevalence of hypertension increased along with PCS and IndS quartiles [87].
Evidence of the skeletal toxicity of protein fermentation metabolites is much more limited. Protein fermentation metabolites may confer direct toxicity to bone cells and disrupt bone matrix characteristics, thereby compromising bone quality and strength [88,89]. IndS promotes osteoblast apoptosis [90] and inhibits osteoclast differentiation [91]. The latter may occur through aryl hydrocarbon receptor signaling-dependent suppression of receptor activator of nuclear factor kappa-Β ligand (RANKL) production [92]. IndS also causes the deterioration of bone mechanical properties [93,94] and bone architecture. Finally, IndS may induce skeletal resistance to parathyroid hormone (PTH) [95]. Increased protein fermentation may contribute to the high prevalence of a dynamic bone disease in patients with CKD, despite these patients often presenting with PTH levels exceeding the normal upper limit severalfold [96].
Protein fermentation metabolites may also affect bone and vascular health indirectly, e.g., by promoting inflammation (vide infra) and epigenetic silencing of Klotho, an anti-aging protein [97,98,99]. Emerging evidence indicate that Klotho deficiency is involved in the pathogenesis of vascular calcification and bone loss in CKD. Klotho-null mice [100,101] show extensive vascular calcification and a low-turnover osteopenia phenotype. The bone phenotype, most probably, results from systemic disturbances in mineral metabolism associated with disrupted FGF23–Klotho signaling rather than from a functional defect of Klotho in osteocytes [102,103].
6. Role of Decreased Carbohydrate Fermentation in the Bone–Vascular Axis
Fermentation of complex carbohydrates results in the generation of short-chain fatty acids (SCFAs) [104]. The main SCFAs are butyrate, propionate, and acetate, which are found in the intestine in a molar ratio of 60:20:20. SCFAs are efficiently absorbed by the gut mucosa by poorly selective anion-transporting proteins [105]. SCFAs, not used by the colonocytes as a source of energy, enter the portal circulation and subsequently either are metabolized by the liver or enter the systemic circulation. SCFAs entering the systemic circulation have important impacts on host physiology as sources of energy, regulators of gene expression (e.g., via inhibition of HDAC), and signaling molecules that are recognized by specific receptors. Especially butyrate is a pleiotropic molecule, functioning as a ligand for certain G protein-coupled receptor (GPCR, e.g., GPCR41 and 43, also known as free-fatty acid receptor 3 and 2) and as a peroxisome proliferator-activated receptor agonist [57].
Production of both propionate and butyrate is reduced in animal CKD models [106]. Human studies, so far, yielded inconsistent findings with regard to both the overall capacity of microbiota to produce butyrate [107] and the circulating levels of SCFAs [108,109]. Chinese patients with CKD stage 5 showed a reduction in the most abundant butyrate-producing microbial species 48 and almost threefold lower plasma butyrate levels than healthy controls [108]. A comparable study in the Netherlands, however, failed to confirm these findings [107].
An increasing body of evidence implicates SCFAs in the pathogenesis of bone disease [64]. SCFAs may promote a positive bone balance by suppressing osteoclastogenesis and stimulating osteoblastogenesis. Mechanistically, propionate and butyrate induce metabolic reprogramming of osteoclasts, resulting in enhanced glycolysis at the expense of oxidative phosphorylation, thereby downregulating essential osteoclast genes [110]. Butyrate, furthermore, suppresses osteoclast differentiation, most probably by increasing the production of osteoprotegerin (OPG) by human osteoblasts [111,112]. Butyrate is also capable of stimulating bone formation [111,113]. The underlying mechanisms remain poorly defined. Butyrate promotes the differentiation of naïve CD4+ cells into regulatory T cells (Tregs). The expansion of Tregs in the bone marrow leads to increased production of Wnt10b. This Wnt ligand subsequently activates Wnt signaling in osteoblastic cells, leading to osteoblast proliferation, differentiation, and survival [113]. Remarkably, this anabolic effect is only seen in trabecular bone. It is unclear whether, and if so, to what extent, the weak inhibition of HDACs accounts for the bone anabolic effects of butyrate [114].
SCFAs may also protect bone indirectly, e.g., by suppressing inflammation (vide infra) and by increasing insulin-like growth factor 1 (IGF-1), a distinct bone anabolic factor [115]. Finally, the CKD-induced microbial metabolism shift away from saccharolytic fermentation and towards proteolytic fermentation creates a colonic microenvironment (e.g., a higher luminal pH) that may hamper calcium absorption [62]. The contribution of calcium absorption in the colon to the overall calcium influx is probably limited. Nevertheless impaired colonic calcium absorption may contribute to a tight, if not negative, calcium balance, commonly observed in CKD patients free of calcium supplements [116].
Studies exploring the role of SCFAs in vascular (patho)biology are limited. Butyrate activates NRF2 at the transcription level [117,118,119,120]. This effect is mediated by HDAC inhibition. One of the downstream effects of NRF2 activation is the upregulation of the glutathione/glutathione S-transferase (GST) antioxidant system resulting in a beneficial smooth muscle cell (VSMC) redox state [121]. Activation of NRF2 signaling has been shown to alleviate high phosphate-induced calcification of VSMCs [122]. SCFAs also have anti-inflammatory properties and thus may indirectly protect against vascular calcification (vide infra).
7. Role of Vitamin K Deficiency in the Bone–Vascular Axis
Microbiota are capable of producing menaquinones (vitamin K2). To what extent the microbial production of menaquinones (vitamin K2) contributes to the overall vitamin K status of the host remains a matter of ongoing debate [123]. Experimental studies on the effect of oral and colorectal administration of vitamin K on circulating prothrombin concentration in vitamin K-deficient rats demonstrated that the bioavailability of colonic vitamin K is more than 50-fold lower than the bioavailability of oral vitamin K [123]. Conversely, data from germ-free rodents [124] and experimental and clinical studies with broad-spectrum antibiotics indicate that gut microbial metabolism may be important to maintain adequate vitamin K stores in the mammalian host [125,126,127].
Recent data indicate that a large majority of patients with CKD are vitamin K-deficient [128,129,130,131,132,133]. Besides dietary restrictions, therapy with vitamin K antagonists and phosphate chelators, and impaired vitamin K recycling, a decreased microbial production related to gut dysbiosis may account for the high prevalence of functional vitamin K deficiency in CKD [129,130,134,135].
Vitamin K deficiency is a well-recognized risk factor of vascular calcification and arterial stiffness, both in the general population and in CKD patients [136,137]. Accelerated vascular calcification in individuals with functional vitamin K deficiency is explained by incomplete γ-carboxylation and reduced function of matrix Gla protein (MGP) in the vasculature [138]. MGP is a 14 kDa secretory protein synthesized by chondrocytes, VSMCs, endothelial cells (ECs), and fibroblasts. γ-Carboxylated MGP inhibits vascular mineralization both directly, as a part of a complex with fetuin-A (also known as α-2-HS-glycoprotein), and indirectly, by interfering with the binding of bone morphogenetic protein-2 (BMP-2) to its receptor and thereby inhibiting BMP-2-induced osteogenic differentiation.
Low dietary intake of vitamin K, therapy with vitamin K antagonists, and functional vitamin K deficiency, as determined by circulating biomarkers (such as dephosphorylated–uncarboxylated MGP), are associated with low bone mineral density (BMD) and increased risk of fractures, both in the general population [90,91,92] and in patients with CKD [133,139]. Vitamin K-dependent γ-carboxylation of Gla-containing bone proteins such as MGP and osteocalcin (also referred to as bone Gla protein) may positively impact mineralization and bone quality. However, much remains to be learned on the role of MGP and osteocalcin in bone biology [140,141,142]. Vitamin K may affect bone health also directly by targeting the steroid and xenobiotic-sensing nuclear receptor (SXR), expressed in osteoblasts [141,143]. Finally, vitamin K deficiency may trigger micro-inflammation and thus contribute to the calcification paradox (vide infra) [133,144].
8. Role of Inflammation in the Bone–Vascular Axis
CKD is well-recognized as a state of micro-inflammation [25,145]. Several factors contribute to the inflammatory status in CKD. Only in recent years, gut dysbiosis has been recognized as another important culprit [54,146]. The pathways linking gut dysbiosis to inflammation are manifold.
First, gut dysbiosis is associated with a dysfunctional epithelial barrier [69,147]. The disruption of the gut epithelial barrier enables the entry of endotoxin and other microbial components into the systemic circulation, which in turn may elicit an inflammatory response [148]. Several studies in animal models of CKD have documented superficial mucosal erosions, mucin loss, or disrupted tight junctions between intestinal epithelial cells in several parts of the gastrointestinal tract [54,55,149,150], in line with autopsy findings in patients on chronic hemodialysis, which who show subtle pathologies indicative of diffuse gastrointestinal wall inflammation [151]. Besides gut dysbiosis, sympathetic overactivity and intestinal congestion due to hypervolemia as a result of heart failure are hypothesized to contribute to increased intestinal permeability in CKD [38].
Second, both an increased exposure to protein fermentation metabolites and a decreased exposure to SCFAs have been hypothesized to contribute to micro-inflammation in CKD. PCS was shown to activate leucocyte free-radical production [152] and IndS-induced proinflammatory cytokines in human primary macrophages, by a mechanism involving the activation of Delta-like 4 (Dll4)–Notch signaling [153]. Other studies, conversely, failed to confirm the proinflammatory properties of PCS and IndS [154]. Moreover, clinical studies investigating the relationship between serum levels of gut-derived uremic toxins, markers of inflammation, yielded inconsistent findings [155]. The anti-inflammatory immune-regulatory properties of circulating SCFAs are well established and best characterized for butyrate. Butyrate stimulates the production of ketone bodies, including β-hydroxybutyrate, known to suppress the activation of the NACHT leucine-rich repeat and pyd domains-containing 3 (NLRP3) inflammasome [156] and suppresses nuclear factor kappa-Β (NF- kappa-Β) signaling in immune cells [157,158]. Butyrate may also mediate systemic anti-inflammatory effects by inhibition of HDACs [57,159]. However, the clinical relevance of the latter mechanism is questionable, as butyrate circulates only at micromolar levels, which is far below the IC50 for HDAC inhibition.
Finally, vitamin K deficiency is associated with inflammation [133,144]. Both causality of the relationship and its underlying molecular mechanisms remain to be defined.
Inflammation may be a common soil for bone loss and vascular calcification [24,25,160,161,162,163,164,165,166]. The pathophysiological mechanisms linking inflammation to vascular calcification are complex and multifaceted. Inflammatory cytokines and C-reactive protein may (a) promote endothelial-to-mesenchymal transition [161], (b) augment osteo–chondrogenic differentiation of vascular smooth muscle cells through activation of Msx2–Wnt/β-catenin signaling [167] and induction of oxidative stress [168], and (c) repress the production of fetuin-A, an important calcification inhibitor [169]. Vascular calcification, in turn, may elicit an inflammatory response and as such trigger a self-perpetuating vicious circle.
Experimental data indicate that inflammatory cytokines, either circulating or locally produced in the bone, such as TNF-α, IL-6, and IL-1β, may induce increased bone resorption [170,171,172,173]. These effects are mediated, in part, via cytokine-induced increases in RANKL, a key stimulator of bone resorption, expressed by osteoblasts and T cells [174]. TNF-α is also an inhibitor of bone formation [175], further tilting the balance towards bone loss [161]. In disagreement with these data, Barreto et al., reported a positive correlation between TNF-α levels and bone area [176]. These authors speculate that elevated TNF-α expression may represent a homeostatic feedback mechanism to counteract excessive bone mass gain.
9. Therapeutic Options
Targeting gut microbiota composition and metabolism may be appealing to tackle the immense burden of cardiovascular disease and fractures simultaneously. Human trials and experimental murine models have shown that nutrition (e.g., diets high in dietary fiber), probiotics, prebiotics, or supplementation of SCFAs may have beneficial effects on the skeleton [64,177,178] and cardiovascular system [179]. The potential of food as a medicine in the setting of CKD is huge, but many questions remain with regard to the optimal use of nutriceuticals. A ‘one-size-fits-all’ approach is unlikely to be successful. Omics, undoubtedly, will prove useful in personalizing nutritional therapy [180,181].
Author Contributions
Writing—original draft preparation, P.E.; writing—review and editing, P.E., S.D., K.V., B.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Opdebeeck, B.; David, K.; D’Haese, P.C. Bone-Vascular Axis in Chronic Kidney Disease. Adv. Chronic. Kidney Dis. 2019, 26, 472–483. [Google Scholar] [CrossRef]
- Stein, M.S.; Packham, D.K.; Ebeling, P.R.; Wark, J.D.; Becker, G.J. Prevalence and risk factors for osteopenia in dialysis patients. Am. J. Kidney Dis. 1996, 28, 515–522. [Google Scholar] [CrossRef]
- Rix, M.; Andreassen, H.; Eskildsen, P.; Langdahl, B.; Olgaard, K. Bone mineral density and biochemical markers of bone turnover in patients with predialysis chronic renal failure. Kidney Int. 1999, 56, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Urena, P.; Bernard-Poenaru, O.; Ostertag, A.; Baudoin, C.; Cohen-Solal, M.; Cantor, T.; de Vernejoul, M.C. Bone mineral density, biochemical markers and skeletal fractures in haemodialysis patients. Nephrol. Dial. Transplant. 2003, 18, 2325–2331. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Claes, K.; Meijers, B.; Laurent, M.R.; Bammens, B.; Naesens, M.; Sprangers, B.; Pottel, H.; Cavalier, E.; Kuypers, D. Bone mineral density, bone turnover markers, and incident fractures in de novo kidney transplant recipients. Kidney Int. 2019, 95, 1461–1470. [Google Scholar] [CrossRef]
- Chen, H.; Lips, P.; Vervloet, M.G.; van Schoor, N.M.; de Jongh, R.T. Association of renal function with bone mineral density and fracture risk in the Longitudinal Aging Study Amsterdam. Osteoporos. Int 2018, 29, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Klawansky, S.; Komaroff, E.; Cavanaugh, P.F., Jr.; Mitchell, D.Y.; Gordon, M.J.; Connelly, J.E.; Ross, S.D. Relationship between age, renal function and bone mineral density in the US population. Osteoporos. Int. 2003, 14, 570–576. [Google Scholar] [CrossRef]
- Ishani, A.; Blackwell, T.; Jamal, S.A.; Cummings, S.R.; Ensrud, K.E. The effect of raloxifene treatment in postmenopausal women with CKD. J. Am. Soc. Nephrol. 2008, 19, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Malluche, H.H.; Porter, D.S.; Monier-Faugere, M.C.; Mawad, H.; Pienkowski, D. Differences in bone quality in low- and high-turnover renal osteodystrophy. J. Am. Soc. Nephrol. 2012, 23, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Jadoul, M.; Albert, J.M.; Akiba, T.; Akizawa, T.; Arab, L.; Bragg-Gresham, J.L.; Mason, N.; Prutz, K.G.; Young, E.W.; Pisoni, R.L. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006, 70, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.M.; Naves, D.M.; Cannata Andia, J.B. Bone metabolism, vascular calcifications and mortality: Associations beyond mere coincidence. J. Nephrol. 2005, 18, 458–463. [Google Scholar]
- Tentori, F.; McCullough, K.; Kilpatrick, R.D.; Bradbury, B.D.; Robinson, B.M.; Kerr, P.G.; Pisoni, R.L. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 2014, 85, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Naves, M.; Diaz-Lopez, J.B.; Gomez, C.; Rodriguez-Rebollar, A.; Rodriguez-Garcia, M.; Cannata-Andia, J.B. The effect of vertebral fracture as a risk factor for osteoporotic fracture and mortality in a Spanish population. Osteoporos. Int. 2003, 14, 520–524. [Google Scholar] [CrossRef]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef]
- Budoff, M.J.; Rader, D.J.; Reilly, M.P.; Mohler III, E.R.; Lash, J.; Yang, W.; Rosen, L.; Glenn, M.; Teal, V.; Feldman, H.I. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2011, 58, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G. Vascular calcification in chronic kidney disease: Not all arteries are created equal. Kidney Int. 2014, 85, 501–503. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, W.C.; Adams, A.L. Breast arterial calcification in chronic kidney disease: Absence of smooth muscle apoptosis and osteogenic transdifferentiation. Kidney Int. 2014, 85, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Okuno, S.; Ishimura, E.; Kitatani, K.; Fujino, Y.; Kohno, K.; Maeno, Y.; Maekawa, K.; Yamakawa, T.; Imanishi, Y.; Inaba, M.; et al. Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2007, 49, 417–425. [Google Scholar] [CrossRef]
- Claes, K.J.; Heye, S.; Bammens, B.; Kuypers, D.R.; Meijers, B.; Naesens, M.; Vanrenterghem, Y.; Evenepoel, P. Aortic calcifications and arterial stiffness as predictors of cardiovascular events in incident renal transplant recipients. Transpl. Int 2013, 26, 973–981. [Google Scholar] [CrossRef]
- Chen, Z.; Qureshi, A.R.; Ripsweden, J.; Wennberg, L.; Heimburger, O.; Lindholm, B.; Barany, P.; Haarhaus, M.; Brismar, T.B.; Stenvinkel, P. Vertebral bone density associates with coronary artery calcification and is an independent predictor of poor outcome in end-stage renal disease patients. Bone 2016, 92, 50–57. [Google Scholar] [CrossRef]
- Viaene, L.; Behets, G.J.; Heye, S.; Claes, K.; Monbaliu, D.; Pirenne, J.; D’Haese, P.C.; Evenepoel, P. Inflammation and the bone-vascular axis in end-stage renal disease. Osteoporos. Int. 2016, 27, 489–497. [Google Scholar] [CrossRef]
- Naves, M.; Rodriguez-Garcia, M.; Diaz-Lopez, J.B.; Gomez-Alonso, C.; Cannata-Andia, J.B. Progression of vascular calcifications is associated with greater bone loss and increased bone fractures. Osteoporos. Int. 2008, 19, 1161–1166. [Google Scholar] [CrossRef]
- Adragao, T.; Herberth, J.; Monier-Faugere, M.C.; Branscum, A.J.; Ferreira, A.; Frazao, J.M.; Dias, C.J.; Malluche, H.H. Low bone volume--a risk factor for coronary calcifications in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Cejka, D.; Weber, M.; Diarra, D.; Reiter, T.; Kainberger, F.; Haas, M. Inverse association between bone microarchitecture assessed by HR-pQCT and coronary artery calcification in patients with end-stage renal disease. Bone 2014, 64, 33–38. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.C.; Carvalho, A.B.; Cuppari, L.; Cendoroglo, M.; Draibe, S.A.; Moyses, R.M.; Neves, K.R.; Jorgetti, V.; Blair, A.; et al. Coronary calcification in hemodialysis patients: The contribution of traditional and uremia-related risk factors. Kidney Int. 2005, 67, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Arfai, K.; Liu, X.; Sayre, J.; Gilsanz, V. Aortic calcification and the risk of osteoporosis and fractures. J. Clin. Endocrinol. Metab. 2004, 89, 4246–4253. [Google Scholar] [CrossRef]
- Tanko, L.B.; Christiansen, C.; Cox, D.A.; Geiger, M.J.; McNabb, M.A.; Cummings, S.R. Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J. Bone Miner. Res. 2005, 20, 1912–1920. [Google Scholar] [CrossRef] [Green Version]
- Hyder, J.A.; Allison, M.A.; Wong, N.; Papa, A.; Lang, T.F.; Sirlin, C.; Gapstur, S.M.; Ouyang, P.; Carr, J.J.; Criqui, M.H. Association of coronary artery and aortic calcium with lumbar bone density: The MESA Abdominal Aortic Calcium Study. Am. J. Epidemiol. 2009, 169, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulos, C.E.; Papaioannou, I.; D’Cruz, D.P. Osteoporosis—A risk factor for cardiovascular disease? Nat. Rev. Rheumatol. 2012, 8, 587–598. [Google Scholar] [CrossRef]
- Flipon, E.; Liabeuf, S.; Fardellone, P.; Mentaverri, R.; Ryckelynck, T.; Grados, F.; Kamel, S.; Massy, Z.A.; Dargent-Molina, P.; Brazier, M. Is vascular calcification associated with bone mineral density and osteoporotic fractures in ambulatory, elderly women? Osteoporos. Int. 2011. [Google Scholar] [CrossRef]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- London, G.M.; Marty, C.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; de Vernejoul, M.C. Arterial Calcifications and Bone Histomorphometry in End-Stage Renal Disease. J. Am. Soc. Nephrol. 2004, 15, 1943–1951. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, M.; Gomez-Alonso, C.; Naves-Diaz, M.; Diaz-Lopez, J.B.; Diaz-Corte, C.; Cannata-Andia, J.B. Vascular calcifications, vertebral fractures and mortality in haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; Desantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.K.I.; Bammens, B.R.M.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Leeuwenhoek 2016, 109, 1389–1396. [Google Scholar] [CrossRef]
- Poesen, R.; Ramezani, A.; Claes, K.; Augustijns, P.; Kuypers, D.; Barrows, I.R.; Muralidharan, J.; Evenepoel, P.; Meijers, B.; Raj, D.S. Associations of Soluble CD14 and Endotoxin with Mortality, Cardiovascular Disease, and Progression of Kidney Disease among Patients with CKD. Clin. J. Am. Soc. Nephrol. 2015, 10, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar, V.S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Wang, Z.; Liu, H.; Jiang, H.; Wang, M.; Liang, S.; Shi, K.; Feng, J. Probiotic Bifidobacterium animalis subsp. lactis Bi-07 alleviates bacterial translocation and ameliorates microinflammation in experimental uraemia. Nephrology (Carlton.) 2014, 19, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Magnusson, K.E.; Sundqvist, T.; Denneberg, T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut 1991, 32, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.; Liu, S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach Knudsen, K.E.; Laerke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host. Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Hatayama, H.; Iwashita, J.; Kuwajima, A.; Abe, T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem. Biophys. Res. Commun. 2007, 356, 599–603. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; Seppen, J.; Muncan, V.; van den Brink, G.R.; Lambers, T.T.; van Tol, E.A.; de Jonge, W.J. The SCFA butyrate stimulates the epithelial production of retinoic acid via inhibition of epithelial HDAC. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1138–G1146. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.M. Diet, gut microbiome, and bone health. Curr. Osteoporos. Rep. 2015, 13, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.J.; Guss, J.D.; Luna, M.; Goldring, S.R. Links Between the Microbiome and Bone. J. Bone Miner. Res. 2016, 31, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Jones, R.M.; Schett, G.; Pacifici, R. The gut-bone axis: How bacterial metabolites bridge the distance. J. Clin. Investig. 2019, 129, 3018–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and Cardiovascular Disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Claus, D.; Geypens, B.; Hiele, M.; Geboes, K.; Rutgeerts, P.; Ghoos, Y. Amount and fate of egg protein escaping assimilation in the small intestine of humans. AJP-Gastrointest. Liver Physiol. 1999, 277, G935–G943. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Farre, R.; Dejongh, S.; Vicario, M.; Evenepoel, P. Intestinal Barrier Function in Chronic Kidney Disease. Toxins (Basel) 2018, 10, 298. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Viaene, L.; Verbeke, K.; Claes, K.; Bammens, B.; Sprangers, B.; Naesens, M.; Vanrenterghem, Y.; Kuypers, D.; Evenepoel, P.; et al. Renal clearance and intestinal generation of p-cresyl sulfate and indoxyl sulfate in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins (Basel) 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.K.; Van, K.S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Buendia, P.; Montes de Oca, A.; Madueno, J.A.; Merino, A.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Rodriguez, M.; Carracedo, J. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015, 29, 173–181. [Google Scholar] [CrossRef]
- Rodrigues, S.D.; Santos, S.S.; Meireles, T.; Romero, N.; Glorieux, G.; Pecoits-Filho, R.; Zhang, D.D.; Nakao, L.S. Uremic toxins promote accumulation of oxidized protein and increased sensitivity to hydrogen peroxide in endothelial cells by impairing the autophagic flux. Biochem. Biophys. Res. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-kappaB/Nrf2 expression. Int Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De, M.A.; De, L.W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and Cardiovascular Risk in Mild-to-Moderate Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Viaene, L.; Thijs, L.; Jin, Y.; Liu, Y.; Gu, Y.; Meijers, B.; Claes, K.; Staessen, J.; Evenepoel, P. Heritability and Clinical Determinants of Serum Indoxyl Sulfate and p-Cresyl Sulfate, Candidate Biomarkers of the Human Microbiome Enterotype. PLoS ONE 2014, 9, e79682. [Google Scholar] [CrossRef] [Green Version]
- Kazama, J.J.; Iwasaki, Y.; Fukagawa, M. Uremic osteoporosis. Kidney Int. Suppl. (2011) 2013, 3, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Iwasaki, Y.; Yamato, H.; Mori, Y.; Komaba, H.; Watanabe, H.; Maruyama, T.; Fukagawa, M. p-Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone 2013, 56, 347–354. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kwak, K.A.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC. Pharmacol. Toxicol. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, D.; Perna, A.F.; Oliva, A.; Vanholder, R.; Pletinck, A.; Guastafierro, S.; Di, N.A.; Vigorito, C.; Capasso, G.; Jankowski, V.; et al. Impact of the uremic milieu on the osteogenic potential of mesenchymal stem cells. PLoS ONE 2015, 10, e0116468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Fukagawa, M. Changes in chemical composition of cortical bone associated with bone fragility in rat model with chronic kidney disease. Bone 2011, 48, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Shimoda, H.; Fukagawa, M. Accumulated uremic toxins attenuate bone mechanical properties in rats with chronic kidney disease. Bone 2013, 57, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Bover, J.; Urena, T.P. Parathyroid hormone metabolism and signaling in health and chronic kidney disease. Kidney Int. 2016, 90, 1184–1190. [Google Scholar] [CrossRef]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int, J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Mencke, R.; Hillebrands, J.L. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res. Rev. 2017, 35, 124–146. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Manabe, N.; Miyaura, C.; Chikuda, H.; Nakamura, K.; Kuro-o, M. Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J. Clin. Investig. 1999, 104, 229–237. [Google Scholar] [CrossRef]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.; Bivi, N.; Farrow, E.; Lezcano, V.; Plotkin, L.I.; White, K.E.; Bellido, T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011, 49, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaba, H.; Kaludjerovic, J.; Hu, D.Z.; Nagano, K.; Amano, K.; Ide, N.; Sato, T.; Densmore, M.J.; Hanai, J.I.; Olauson, H.; et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 2017, 92, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Stumpff, F. A look at the smelly side of physiology: Transport of short chain fatty acids. Pflugers Arch. 2018, 470, 571–598. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Terpstra, M.L.; Sinnige, M.J.; Hugenholtz, F.; Peters-Sengers, H.; Remmerswaal, E.B.; Geerlings, S.E.; Bemelman, F.J. Butyrate production in patients with end-stage renal disease. Int, J. Nephrol. Renovasc. Dis. 2019, 12, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lv, D.; Jiang, S.; Jiang, J.; Liang, M.; Hou, F.; Chen, Y. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. (Lond.) 2019, 133, 1857–1870. [Google Scholar] [CrossRef]
- Jadoon, A.; Mathew, A.V.; Byun, J.; Gadegbeku, C.A.; Gipson, D.S.; Afshinnia, F.; Pennathur, S. Gut Microbial Product Predicts Cardiovascular Risk in Chronic Kidney Disease Patients. Am. J. Nephrol. 2018, 48, 269–277. [Google Scholar] [CrossRef]
- Lucas, S.; Omata, Y.; Hofmann, J.; Bottcher, M.; Iljazovic, A.; Sarter, K.; Albrecht, O.; Schulz, O.; Krishnacoumar, B.; Kronke, G.; et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 2018, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Katono, T.; Kawato, T.; Tanabe, N.; Suzuki, N.; Iida, T.; Morozumi, A.; Ochiai, K.; Maeno, M. Sodium butyrate stimulates mineralized nodule formation and osteoprotegerin expression by human osteoblasts. Arch. Oral Biol. 2008, 53, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Montalvany-Antonucci, C.C.; Duffles, L.F.; de Arruda, J.A.A.; Zicker, M.C.; de Oliveira, S.; Macari, S.; Garlet, G.P.; Madeira, M.F.M.; Fukada, S.Y.; Andrade, I.; et al. Short-chain fatty acids and FFAR2 as suppressors of bone resorption. Bone 2019, 125, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The Microbial Metabolite Butyrate Stimulates Bone Formation via T Regulatory Cell-Mediated Regulation of WNT10B Expression. Immunity 2018, 49, 1116–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005, 20, 2254–2263. [Google Scholar] [CrossRef]
- Yan, J.; Herzog, J.W.; Tsang, K.; Brennan, C.A.; Bower, M.A.; Garrett, W.S.; Sartor, B.R.; Aliprantis, A.O.; Charles, J.F. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7554–E7563. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Viaene, L.; Meijers, B. Calcium balance in chronic kidney disease: Walking the tightrope. Kidney Int. 2012, 81, 1057–1059. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Jia, Y.; Yang, S.; Zhao, N.; Hu, Y.; Hong, J.; Gao, S.; Zhao, R. Sodium butyrate protects against high-fat diet-induced oxidative stress in rat liver by promoting expression of nuclear factor E2-related factor 2. Br. J. Nutr. 2019, 122, 400–410. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, Z.; Zhang, H.; Liang, W.; Huang, W.; Zhang, H.; Li, Y.; Wang, Z.; Wang, J.; Jia, Y.; et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Biol. Med. 2018, 124, 454–465. [Google Scholar] [CrossRef]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with Nrf2 and p53. Mol. Cell Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef]
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Ranganna, K.; Mathew, O.P.; Yatsu, F.M.; Yousefipour, Z.; Hayes, B.E.; Milton, S.G. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007, 274, 5962–5978. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenen-van Dooren, M.M.; Ronden, J.E.; Soute, B.A.; Vermeer, C. Bioavailability of phylloquinone and menaquinones after oral and colorectal administration in vitamin K-deficient rats. Biochem. Pharmacol. 1995, 50, 797–801. [Google Scholar] [CrossRef]
- Komai, M.; Shirakawa, H.; Kimura, S. Newly developed model for vitamin K deficiency in germfree mice. Int, J. Vitam. Nutr. Res. 1988, 58, 55–59. [Google Scholar]
- Allison, P.M.; Mummah-Schendel, L.L.; Kindberg, C.G.; Harms, C.S.; Bang, N.U.; Suttie, J.W. Effects of a vitamin K-deficient diet and antibiotics in normal human volunteers. J. Lab. Clin. Med. 1987, 110, 180–188. [Google Scholar] [PubMed]
- Frick, P.G.; Riedler, G.; Brogli, H. Dose response and minimal daily requirement for vitamin K in man. J. Appl. Physiol 1967, 23, 387–389. [Google Scholar] [CrossRef]
- Guss, J.D.; Taylor, E.; Rouse, Z.; Roubert, S.; Higgins, C.H.; Thomas, C.J.; Baker, S.P.; Vashishth, D.; Donnelly, E.; Shea, M.K.; et al. The microbial metagenome and bone tissue composition in mice with microbiome-induced reductions in bone strength. Bone 2019, 127, 146–154. [Google Scholar] [CrossRef]
- Krueger, T.; Westenfeld, R.; Ketteler, M.; Schurgers, L.J.; Floege, J. Vitamin K deficiency in CKD patients: A modifiable risk factor for vascular calcification? Kidney Int. 2009, 76, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Holden, R.M.; Morton, A.R.; Garland, J.S.; Pavlov, A.; Day, A.G.; Booth, S.L. Vitamins K and D status in stages 3-5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 590–597. [Google Scholar] [CrossRef]
- Cranenburg, E.C.; Schurgers, L.J.; Uiterwijk, H.H.; Beulens, J.W.; Dalmeijer, G.W.; Westerhuis, R.; Magdeleyns, E.J.; Herfs, M.; Vermeer, C.; Laverman, G.D. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012, 82, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G.; Westenfeld, R.; Kruger, T.; Cranenburg, E.C.; Magdeleyns, E.J.; Brandenburg, V.M.; Djuric, Z.; Damjanovic, T.; Ketteler, M.; Vermeer, C.; et al. Circulating nonphosphorylated carboxylated matrix gla protein predicts survival in ESRD. J. Am. Soc. Nephrol. 2011, 22, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxma, P.Y.; van den Berg, E.; Geleijnse, J.M.; Laverman, G.D.; Schurgers, L.J.; Vermeer, C.; Kema, I.P.; Muskiet, F.A.; Navis, G.; Bakker, S.J.; et al. Vitamin k intake and plasma desphospho-uncarboxylated matrix Gla-protein levels in kidney transplant recipients. PLoS ONE 2012, 7, e47991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evenepoel, P.; Claes, K.; Meijers, B.; Laurent, M.; Bammens, B.; Naesens, M.; Sprangers, B.; Pottel, H.; Cavalier, E.; Kuypers, D. Poor Vitamin K Status Is Associated With Low Bone Mineral Density and Increased Fracture Risk in End-Stage Renal Disease. J. Bone Miner. Res. 2019, 34, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansz, T.T.; Neradova, A.; van Ballegooijen, A.J.; Verhaar, M.C.; Vervloet, M.G.; Schurgers, L.J.; van Jaarsveld, B.C. The role of kidney transplantation and phosphate binder use in vitamin K status. PLoS ONE 2018, 13, e0203157. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Magdeleyns, E.; Herfs, M.; Schettgen, T.; Brandenburg, V.; Fliser, D.; Vermeer, C.; Floege, J.; Schlieper, G.; Kruger, T. Impaired vitamin K recycling in uremia is rescued by vitamin K supplementation. Kidney Int. 2014, 86, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Delanaye, P.; Krzesinski, J.M.; Warling, X.; Moonen, M.; Smelten, N.; Medart, L.; Pottel, H.; Cavalier, E. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC. Nephrol. 2014, 15, 145. [Google Scholar] [CrossRef] [Green Version]
- Fain, M.E.; Kapuku, G.K.; Paulson, W.D.; Williams, C.F.; Raed, A.; Dong, Y.; Knapen, M.H.J.; Vermeer, C.; Pollock, N.K. Inactive Matrix Gla Protein, Arterial Stiffness, and Endothelial Function in African American Hemodialysis Patients. Am. J. Hypertens. 2018, 31, 735–741. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Fusaro, M.; Noale, M.; Viola, V.; Galli, F.; Tripepi, G.; Vajente, N.; Plebani, M.; Zaninotto, M.; Guglielmi, G.; Miotto, D.; et al. Vitamin K, vertebral fractures, vascular calcifications, and mortality: VItamin K Italian (VIKI) dialysis study. J. Bone Miner. Res. 2012, 27, 2271–2278. [Google Scholar] [CrossRef]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Shiba, S.; Hasegawa, T.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Ouchi, Y.; Amizuka, N.; Inoue, S. Osteoblast-Specific gamma-Glutamyl Carboxylase-Deficient Mice Display Enhanced Bone Formation with Aberrant Mineralization. J. Bone Miner. Res. 2015, 30, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Maruyama-Nagao, A.; Sakuraba, K.; Kawai, S. Level of serum undercarboxylated osteocalcin correlates with bone quality assessed by calcaneal quantitative ultrasound sonometry in young Japanese females. Exp. Ther. Med. 2017, 13, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grun, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, M.K.; Booth, S.L.; Massaro, J.M.; Jacques, P.F.; D’Agostino, R.B., Sr.; Dawson-Hughes, B.; Ordovas, J.M.; O’Donnell, C.J.; Kathiresan, S.; Keaney, J.F.; et al. Vitamin K and vitamin D status: Associations with inflammatory markers in the Framingham Offspring Study. Am. J. Epidemiol. 2008, 167, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Wanner, C.; Metzger, T.; Heimburger, O.; Mallamaci, F.; Tripepi, G.; Malatino, L.; Zoccali, C. Inflammation and outcome in end-stage renal failure: Does female gender constitute a survival advantage? Kidney Int. 2002, 62, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Kotanko, P.; Carter, M.; Levin, N.W. Intestinal bacterial microflora--a potential source of chronic inflammation in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2006, 21, 2057–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef]
- Yang, J.; Lim, S.Y.; Ko, Y.S.; Lee, H.Y.; Oh, S.W.; Kim, M.G.; Cho, W.Y.; Jo, S.K. Intestinal barrier disruption and dysregulated mucosal immunity contribute to kidney fibrosis in chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 419–428. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dure-Smith, B.; Miller, R.; Mirahmadi, M.K. Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am. J. Gastroenterol. 1985, 80, 608–611. [Google Scholar] [PubMed]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Viaene, L.; Evenepoel, P.; Meijers, B.; Vanderschueren, D.; Overbergh, L.; Mathieu, C. Uremia Suppresses Immune Signal-Induced CYP27B1 Expression in Human Monocytes. Am. J. Nephrol. 2012, 36, 497–508. [Google Scholar] [CrossRef]
- Hsu, H.J.; Yen, C.H.; Wu, I.W.; Hsu, K.H.; Chen, C.K.; Sun, C.Y.; Chou, C.C.; Chen, C.Y.; Tsai, C.J.; Wu, M.S.; et al. The association of uremic toxins and inflammation in hemodialysis patients. PLoS ONE 2014, 9, e102691. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World, J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Meijer, K.; de Vos, P.; Priebe, M.G. Butyrate and other short-chain fatty acids as modulators of immunity: What relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 715–721. [Google Scholar] [CrossRef]
- Koh, A.; De, V.F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart, J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef]
- Khosla, S. The bone and beyond: A shift in calcium. Nat. Med. 2011, 17, 430–431. [Google Scholar] [CrossRef] [PubMed]
- New, S.E.; Aikawa, E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panuccio, V.; Enia, G.; Tripepi, R.; Aliotta, R.; Mallamaci, F.; Tripepi, G.; Zoccali, C. Pro-inflammatory cytokines and bone fractures in CKD patients. An exploratory single centre study. BMC. Nephrol. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Wunsch, R.; Turzer, M.; Bahner, M.; Raggi, P.; Querfeld, U.; Mehls, O.; Schaefer, F. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 2002, 106, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Guerin, A.P.; London, G.M.; Marchais, S.J.; Metivier, F. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol. Dial. Transplant. 2000, 15, 1014–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauley, J.A.; Barbour, K.E.; Harrison, S.L.; Cloonan, Y.K.; Danielson, M.E.; Ensrud, K.E.; Fink, H.A.; Orwoll, E.S.; Boudreau, R. Inflammatory Markers and the Risk of Hip and Vertebral Fractures in Men: The Osteoporotic Fractures in Men (MrOS). J. Bone Miner. Res. 2016, 31, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt Calcification Cascade Is Regulated by TNF-+¦GÇôDependent Signals in Diabetic LdlrGêÆ/GêÆ Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany. NY) 2019, 11, 5445–5462. [Google Scholar] [CrossRef]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Böhm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Feyen, J.H.; Elford, P.; Di Padova, F.E.; Trechsel, U. Interleukin-6 is produced by bone and modulated by parathyroid hormone. J. Bone Miner. Res. 1989, 4, 633–638. [Google Scholar] [CrossRef]
- Pfeilschifter, J.; Chenu, C.; Bird, A.; Mundy, G.R.; Roodman, G.D. Interleukin-1 and tumor necrosis factor stimulate the formation of human osteoclastlike cells in vitro. J. Bone Miner. Res. 1989, 4, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Saraiva, M.; Behets, G.; Macedo, A.; Galvao, M.; D’Haese, P.; Drueke, T.B. Evaluation of bone remodeling in hemodialysis patients: Serum biochemistry, circulating cytokines and bone histomorphometry. J. Nephrol. 2009, 22, 783–793. [Google Scholar] [PubMed]
- Cafiero, C.; Gigante, M.; Brunetti, G.; Simone, S.; Chaoul, N.; Oranger, A.; Ranieri, E.; Colucci, S.; Pertosa, G.B.; Grano, M.; et al. Inflammation induces osteoclast differentiation from peripheral mononuclear cells in chronic kidney disease patients: Crosstalk between the immune and bone systems. Nephrol. Dial. Transplant. 2018, 33, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Lacey, D.L.; Dunstan, C.R.; Spelsberg, T.C.; Riggs, B.L.; Khosla, S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 1999, 25, 255–259. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Moyses, R.M.; Neves, C.L.; Jorgetti, V.; Draibe, S.A.; Canziani, M.E.; Carvalho, A.B. Osteoporosis in hemodialysis patients revisited by bone histomorphometry: A new insight into an old problem. Kidney Int. 2006, 69, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Tousen, Y.; Matsumoto, Y.; Nagahata, Y.; Kobayashi, I.; Inoue, M.; Ishimi, Y. Resistant Starch Attenuates Bone Loss in Ovariectomised Mice by Regulating the Intestinal Microbiota and Bone-Marrow Inflammation. Nutrients 2019, 11, 297. [Google Scholar] [CrossRef] [Green Version]
- McCabe, L.; Britton, R.A.; Parameswaran, N. Prebiotic and Probiotic Regulation of Bone Health: Role of the Intestine and its Microbiome. Curr. Osteoporos. Rep. 2015, 13, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Krautkramer, K.A.; Org, E.; Romano, K.A.; Kerby, R.L.; Vivas, E.I.; Mehrabian, M.; Denu, J.M.; Backhed, F.; Lusis, A.J.; et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol. 2018, 3, 1461–1471. [Google Scholar] [CrossRef]
- Lampe, J.W.; Navarro, S.L.; Hullar, M.A.; Shojaie, A. Inter-individual differences in response to dietary intervention: Integrating omics platforms towards personalised dietary recommendations. Proc. Nutr. Soc. 2013, 72, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Veiga, P. Rethinking Diet to Aid Human-Microbe Symbiosis. Trends Microbiol. 2017, 25, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The kidney–gut–bone–vascular axis. Chronic kidney disease is associated with gut dysbiosis, characterized by a metabolic shift towards a predominantly proteolytic fermentation pattern and a leaky gut. Gut dysbiosis may induce bone loss and vascular calcification and as such may play a pathogenic role in the bone–vascular axis in CKD. Underlying pathophysiological mechanisms include increased exposure to protein fermentation metabolites (such as p-cresyl sulfate (PCS) and indoxyl sulfate (IndS)), a leaky gut contributing to inflammation, and deficiency of vitamin K and short-chain fatty acids (SCFAs).
Figure 1.
The kidney–gut–bone–vascular axis. Chronic kidney disease is associated with gut dysbiosis, characterized by a metabolic shift towards a predominantly proteolytic fermentation pattern and a leaky gut. Gut dysbiosis may induce bone loss and vascular calcification and as such may play a pathogenic role in the bone–vascular axis in CKD. Underlying pathophysiological mechanisms include increased exposure to protein fermentation metabolites (such as p-cresyl sulfate (PCS) and indoxyl sulfate (IndS)), a leaky gut contributing to inflammation, and deficiency of vitamin K and short-chain fatty acids (SCFAs).
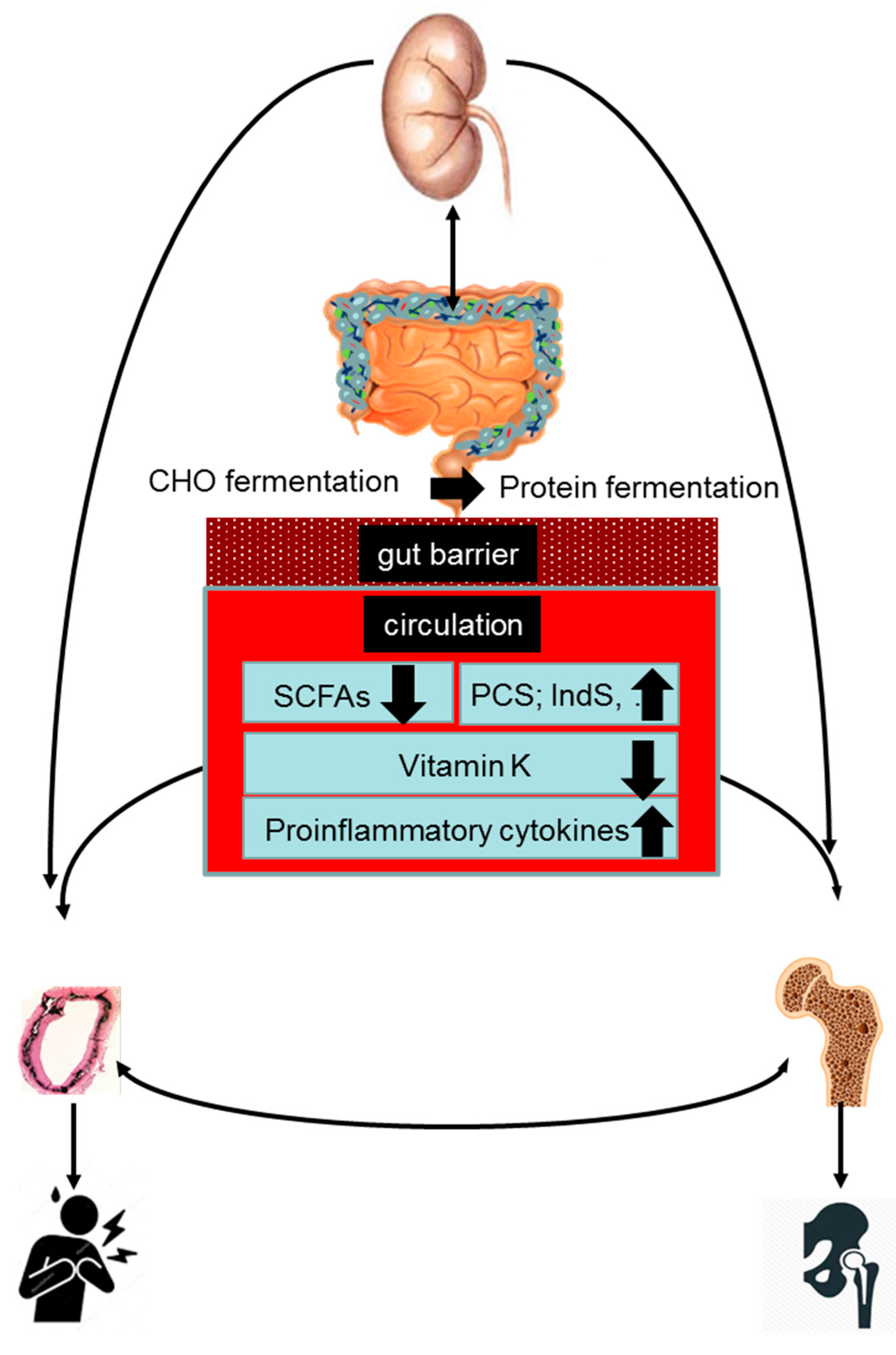
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Evenepoel, P.; Dejongh, S.; Verbeke, K.; Meijers, B. The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins 2020, 12, 285. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050285
AMA Style
Evenepoel P, Dejongh S, Verbeke K, Meijers B. The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins. 2020; 12(5):285. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050285
Chicago/Turabian StyleEvenepoel, Pieter, Sander Dejongh, Kristin Verbeke, and Bjorn Meijers. 2020. "The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease" Toxins 12, no. 5: 285. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050285
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.