Drug Delivery Strategies for Enhancing the Therapeutic Efficacy of Toxin-Derived Anti-Diabetic Peptides

 , ,
, ,
Abstract
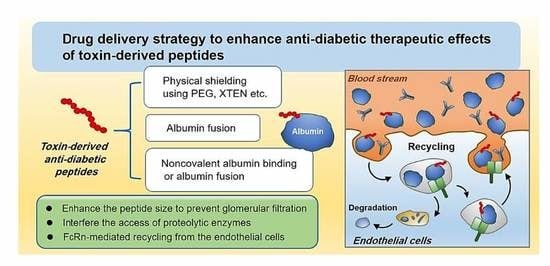
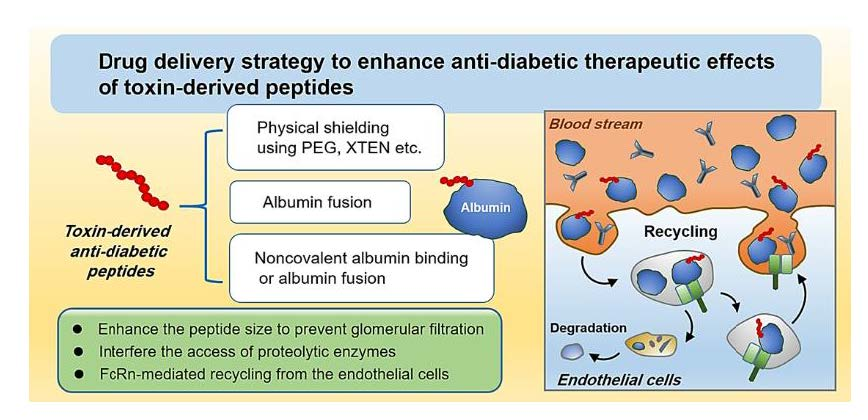
:
1. Introduction
2. Toxin-Derived Anti-Diabetic Peptides from Skin Secretions of Amphibians
2.1. Esculentin-2CHa
2.2. Tigerinin-1R
2.3. Magainin-AM1 and AM2
2.4. Hymenochirin 1B
2.5. Alyteserin-2a
2.6. Brevinin-2-Related Peptide (B2RP)
2.7. Peptide GlycinE-Leucine-Amide (PGLa)-AM1
3. Drug Delivery Challenges for Toxin-Derived Anti-Diabetic Peptides
4. Pharmaceutical Strategies to Improve the Plasma Half-Lives of Anti-Diabetic Peptides
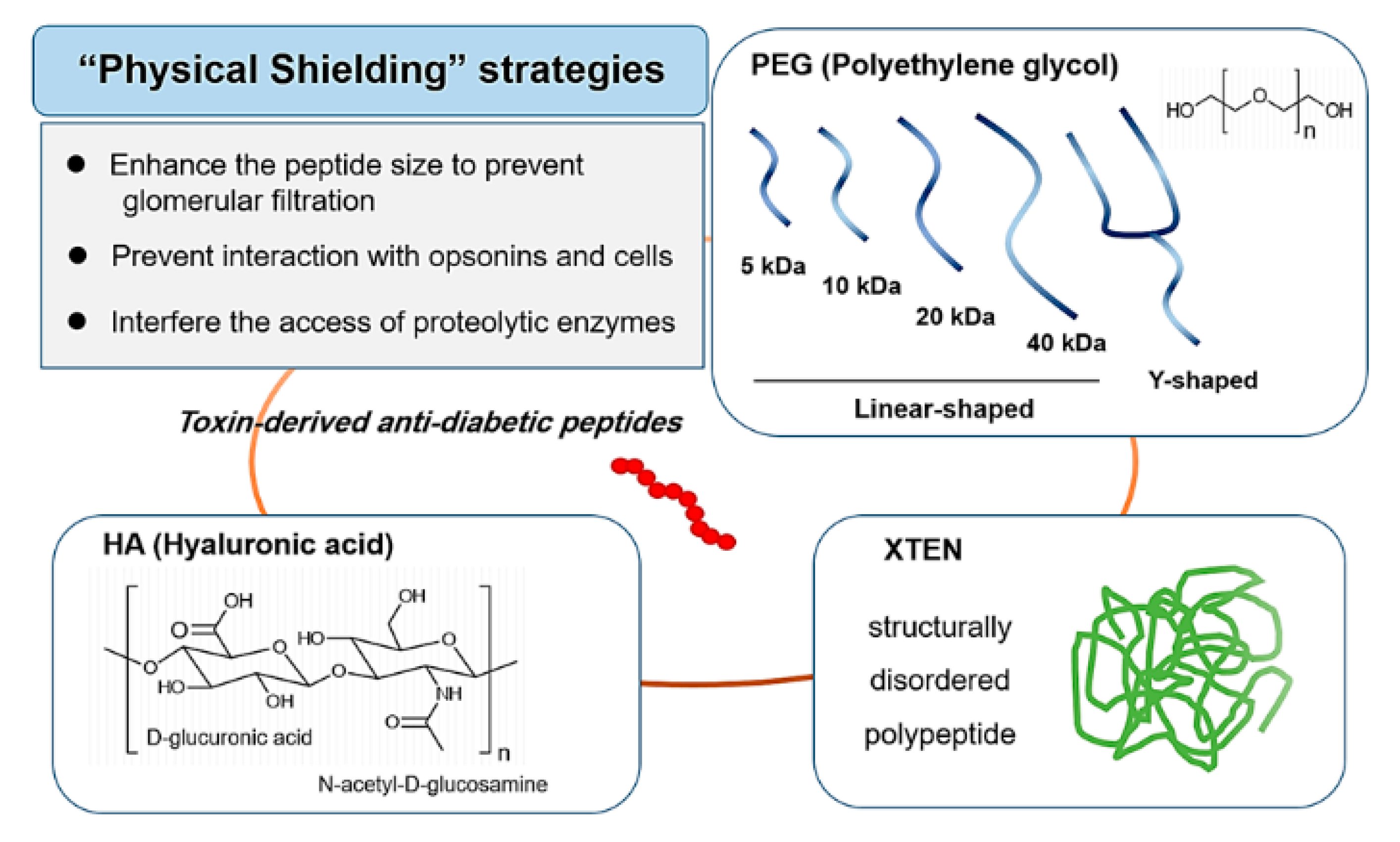
4.1. Physical Shielding
4.1.1. PEGylation
4.1.2. XTEN
4.1.3. HAylation
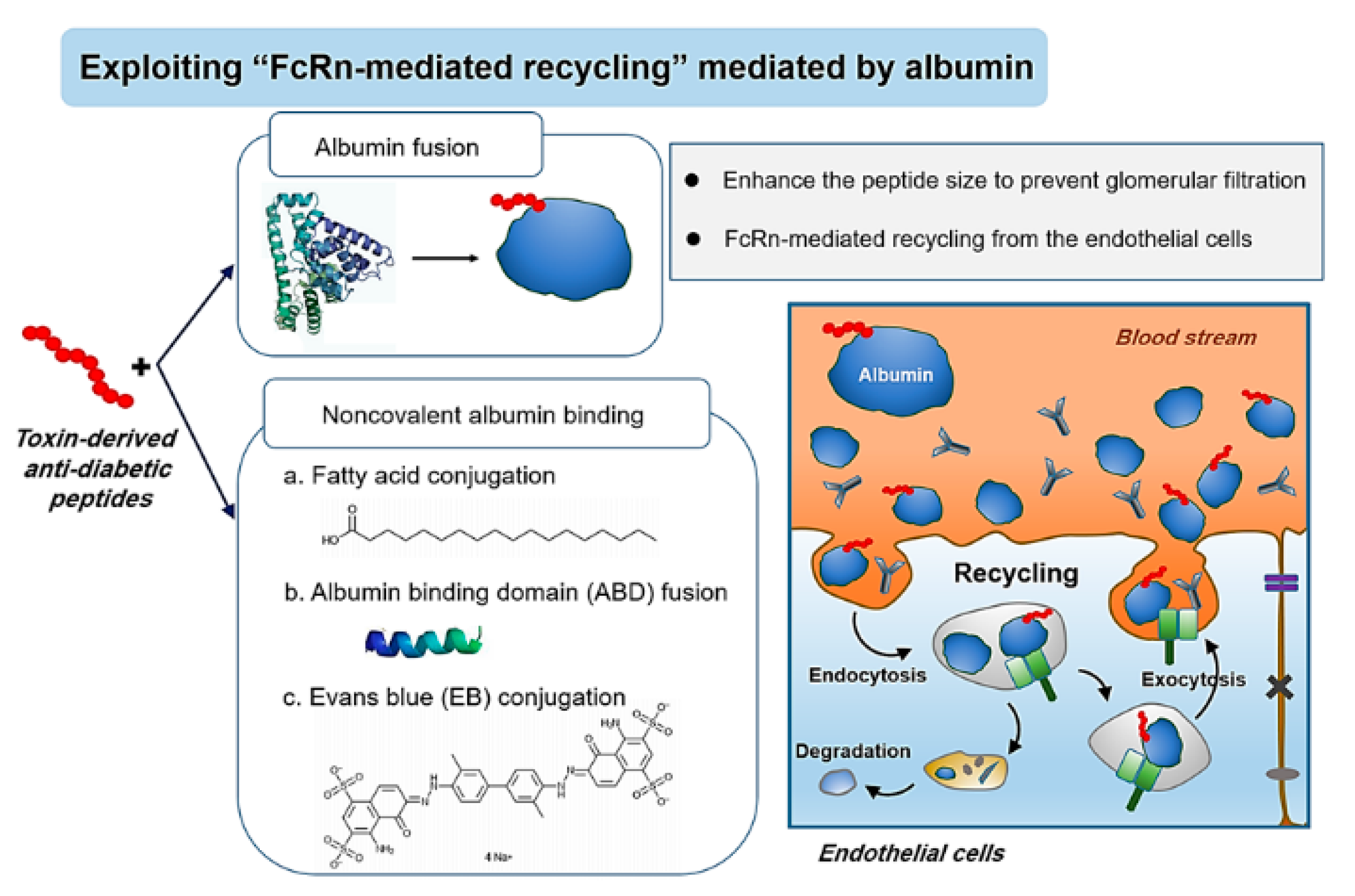
4.2. Exploitation of FcRn-Mediated Recycling
4.2.1. Albumin Fusion
4.2.2. Non-Covalent Albumin Binding
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 7, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bharti, S.K.; Kumar, A. Therapeutic molecules against type 2 diabetes: What we have and what are we expecting? Pharmacol. Rep. 2017, 69, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.; Zraika, S.; Utzschneider, K.; Hull, R. The beta cell lesion in type 2 diabetes: There has to be a primary functional abnormality. Diabetologia 2009, 52, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Jonnalagadda, V.G.; Ram Raju, A.V.; Pittala, S.; Shaik, A.; Selkar, N.A. The prelude on novel receptor and ligand targets involved in the treatment of diabetes mellitus. Adv. Pharm. Bull. 2014, 4, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Min, K.A.; Maharjan, P.; Ham, S.; Shin, M.C. Pro-apoptotic peptides-based cancer therapies: Challenges and strategies to enhance therapeutic efficacy. Arch. Pharm. Res. 2018, 41, 594–616. [Google Scholar] [CrossRef]
- Al-azzawi, S.; Masheta, D. Designing a drug delivery system for improved tumor treatment and targeting by functionalization of a cell-penetrating peptide. J. Pharm. Investig. 2019, 49, 643–654. [Google Scholar] [CrossRef]
- Maharjan, P.; Cho, K.H.; Maharjan, A.; Shin, M.C.; Moon, C.; Min, K.A. Pharmaceutical challenges and perspectives in developing ophthalmic drug formulations. J. Pharm. Investig. 2019, 49, 215–228. [Google Scholar] [CrossRef]
- Breinbauer, R.; Vetter, I.R.; Waldmann, H. From protein domains to drug candidates—natural products as guiding principles in the design and synthesis of compound libraries. Angew. Chem. 2002, 41, 2878–2890. [Google Scholar] [CrossRef]
- Uhlig, T.; Kyprianou, T.; Martinelli, F.G.; Oppici, C.A.; Heiligers, D.; Hills, D.; Calvo, X.R.; Verhaert, P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open Proteomics 2014, 4, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.L. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 2012, 59, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B. Inhibition of dipeptidyl peptidase-4 (DPP-4)-a novel approach to treat type 2 diabetes. Curr. Enz. Inhibit. 2005, 1, 65–73. [Google Scholar] [CrossRef]
- Rieg, T.; Gerasimova, M.; Murray, F.; Masuda, T.; Tang, T.; Rose, M.; Drucker, D.J.; Vallon, V. Natriuretic effect by exendin-4, but not the DPP-4 inhibitor alogliptin, is mediated via the GLP-1 receptor and preserved in obese type 2 diabetic mice. Am. J. Physiol. 2012, 303, F963–F971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Lim, S.M.; Lee, K.C.; Na, D.H. Exendins and exendin analogs for diabetic therapy: A patent review (2012–2015). Expert Opin. Ther. Pat. 2016, 26, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wei, L.; Ma, L.; Huang, X.; Tao, A.; Liu, Z.; Yuan, W. Long-acting preparations of exenatide. Drug Des. Dev. Ther. 2013, 7, 963–970. [Google Scholar] [CrossRef] [Green Version]
- van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-life extension of biopharmaceuticals using chemical methods: Alternatives to PEGylation. J. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef]
- Park, K.A.; Jin, Z.; Lee, J.Y.; An, H.S.; Choi, E.B.; Kim, K.E.; Shin, H.J.; Jeong, E.A.; Min, K.A.; Shin, M.C.; et al. Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice. Pharmaceutics 2020, 12, 159. [Google Scholar] [CrossRef] [Green Version]
- Menting, J.G.; Gajewiak, J.; MacRaild, C.A.; Chou, D.H.-C.; Disotuar, M.M.; Smith, N.A.; Miller, C.; Erchegyi, J.; Rivier, J.E.; Olivera, B.M. A minimized human insulin-receptor-binding motif revealed in a Conus geographus venom insulin. Nat. Struct. Mol. Biol. 2016, 23, 916–920. [Google Scholar] [CrossRef]
- Diaz-Garcia, C.M.; Sanchez-Soto, C.; Hiriart, M. Toxins that modulate ionic channels as tools for exploring insulin secretion. Cell. Mol. Neurobiol. 2010, 30, 1275–1281. [Google Scholar] [CrossRef]
- Conlon, J.M.; Mechkarska, M.; Leprince, J. Peptidomic analysis in the discovery of therapeutically valuable peptides in amphibian skin secretions. Expert Rev. Proteomics 2019, 16, 897–908. [Google Scholar] [CrossRef]
- Marya; Khan, H.; Nabavi, S.M.; Habtemariam, S. Anti-diabetic potential of peptides: Future prospects as therapeutic agents. Life Sci. 2018, 193, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H. Venom peptides as pharmacological tools and therapeutics for diabetes. Neuropharmacology 2017, 127, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; McGahon, M.K.; Moffett, R.C.; Curtis, T.M.; Conlon, J.M.; Abdel-Wahab, Y.H.; Flatt, P.R. Esculentin-2CHa(1-30) and its analogues: Stability and mechanisms of insulinotropic action. J. Endocrinol. 2017, 232, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attoub, S.; Mechkarska, M.; Sonnevend, A.; Radosavljevic, G.; Jovanovic, I.; Lukic, M.L.; Conlon, J.M. Esculentin-2CHa: A host-defense peptide with differential cytotoxicity against bacteria, erythrocytes and tumor cells. Peptides 2013, 39, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Abdel-Wahab, Y.H.; Flatt, P.R.; Mechkarska, M.; Conlon, J.M. Tigerinin-1R: A potent, non-toxic insulin-releasing peptide isolated from the skin of the Asian frog, Hoplobatrachus rugulosus. Diabetes Obes. Metab. 2011, 13, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Frog skin peptides (tigerinin-1R, magainin-AM1, -AM2, CPF-AM1, and PGla-AM1) stimulate secretion of glucagon-like peptide 1 (GLP-1) by GLUTag cells. Biochem. Biophys. Res. Commun. 2013, 431, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M. Host-defense peptides with therapeutic potential from skin secretions of frogs from the family pipidae. Pharmaceuticals 2014, 7, 58–77. [Google Scholar] [CrossRef] [Green Version]
- Owolabi, B.O.; Ojo, O.O.; Srinivasan, D.K.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. In vitro and in vivo insulinotropic properties of the multifunctional frog skin peptide hymenochirin-1B: A structure-activity study. Amino Acids 2016, 48, 535–547. [Google Scholar] [CrossRef]
- Conlon, J.M.; Demandt, A.; Nielsen, P.F.; Leprince, J.; Vaudry, H.; Woodhams, D.C. The alyteserins: Two families of antimicrobial peptides from the skin secretions of the midwife toad Alytes obstetricans (Alytidae). Peptides 2009, 30, 1069–1073. [Google Scholar] [CrossRef]
- Bevier, C.R.; Sonnevend, A.; Kolodziejek, J.; Nowotny, N.; Nielsen, P.F.; Conlon, J.M. Purification and characterization of antimicrobial peptides from the skin secretions of the mink frog (Rana septentrionalis). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2004, 139, 31–38. [Google Scholar] [CrossRef]
- Conlon, J.M.; Sonnevend, A.; Pál, T.; Vila-Farrés, X. Efficacy of six frog skin-derived antimicrobial peptides against colistin-resistant strains of the Acinetobacter baumannii group. Int. J. Antimicrob. Agents 2012, 39, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Simmaco, M.; Mignogna, G.; Barra, D.; Bossa, F. Novel antimicrobial peptides from skin secretion of the European frog Rana esculenta. FEBS Lett. 1993, 324, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Ojo, O.O.; Srinivasan, D.K.; Owolabi, B.O.; Vasu, S.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Esculentin-2CHa-Related Peptides Modulate Islet Cell Function and Improve Glucose Tolerance in Mice with Diet-Induced Obesity and Insulin Resistance. PLoS ONE 2015, 10, e0141549. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; Ojo, O.O.; Moffett, R.C.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H.A. Anti-diabetic actions of esculentin-2CHa(1-30) and its stable analogues in a diet-induced model of obesity-diabetes. Amino Acids 2017, 49, 1705–1717. [Google Scholar] [CrossRef]
- Sato, Y.; Henquin, J.C. The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: In search of the underlying mechanism. Diabetes 1998, 47, 1713–1721. [Google Scholar] [CrossRef]
- Sai, K.P.; Jagannadham, M.V.; Vairamani, M.; Raju, N.P.; Devi, A.S.; Nagaraj, R.; Sitaram, N. Tigerinins: Novel antimicrobial peptides from the Indian frogRana tigerina. J. Biol. Chem. 2001, 276, 2701–2707. [Google Scholar] [CrossRef] [Green Version]
- Ojo, O.O.; Srinivasan, D.K.; Owolabi, B.O.; Flatt, P.R.; Abdel-Wahab, Y.H. Beneficial effects of tigerinin-1R on glucose homeostasis and beta cell function in mice with diet-induced obesity-diabetes. Biochimie 2015, 109, 18–26. [Google Scholar] [CrossRef]
- Srinivasan, D.; Ojo, O.O.; Abdel-Wahab, Y.H.; Flatt, P.R.; Guilhaudis, L.; Conlon, J.M. Insulin-releasing and cytotoxic properties of the frog skin peptide, tigerinin-1R: A structure-activity study. Peptides 2014, 55, 23–31. [Google Scholar] [CrossRef]
- Conlon, J.M.; Al-Ghaferi, N.; Ahmed, E.; Meetani, M.A.; Leprince, J.; Nielsen, P.F. Orthologs of magainin, PGLa, procaerulein-derived, and proxenopsin-derived peptides from skin secretions of the octoploid frog Xenopus amieti (Pipidae). Peptides 2010, 31, 989–994. [Google Scholar] [CrossRef]
- Ojo, O.O.; Srinivasan, D.K.; Owolabi, B.O.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Magainin-AM2 improves glucose homeostasis and beta cell function in high-fat fed mice. Biochim. Biophys. Acta 2015, 1850, 80–87. [Google Scholar] [CrossRef]
- Mechkarska, M.; Prajeep, M.; Coquet, L.; Leprince, J.; Jouenne, T.; Vaudry, H.; King, J.D.; Conlon, J.M. The hymenochirins: A family of host-defense peptides from the Congo dwarf clawed frog Hymenochirus boettgeri (Pipidae). Peptides 2012, 35, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Owolabi, B.O.; Ojo, O.O.; Srinivasan, D.K.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Glucoregulatory, endocrine and morphological effects of [P5K]hymenochirin-1B in mice with diet-induced glucose intolerance and insulin resistance. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Abdel-Wahab, Y.H.; Flatt, P.R.; Conlon, J.M. Insulinotropic actions of the frog skin host-defense peptide alyteserin-2a: A structure-activity study. Chem. Biol. Drug Des. 2013, 82, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, Y.; Patterson, S.; Flatt, P.; Conlon, J.M. Brevinin-2-related peptide and its [D4K] analogue stimulate insulin release in vitro and improve glucose tolerance in mice fed a high fat diet. Horm. Metab. Res. 2010, 42, 652–656. [Google Scholar] [CrossRef]
- Andreu, D.; Aschauer, H.; Kreil, G.; Merrifield, R.B. Solid-phase synthesis of PYLa and isolation of its natural counterpart, PGLa [PYLa-(4–24)] from skin secretion of Xenopus laevis. Eur. J. Biochem. 1985, 149, 531–535. [Google Scholar] [CrossRef]
- Owolabi, B.O.; Musale, V.; Ojo, O.O.; Moffett, R.C.; McGahon, M.K.; Curtis, T.M.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Actions of PGLa-AM1 and its [A14K] and [A20K] analogues and their therapeutic potential as anti-diabetic agents. Biochimie 2017, 138, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Stepensky, D. Pharmacokinetics of Toxin-Derived Peptide Drugs. Toxins 2018, 10, 483. [Google Scholar] [CrossRef] [Green Version]
- Rekha, M.; Sharma, C.P. Oral delivery of therapeutic protein/peptide for diabetes–future perspectives. Int. J. Pharm. 2013, 440, 48–62. [Google Scholar] [CrossRef]
- Clark, G.C.; Casewell, N.R.; Elliott, C.T.; Harvey, A.L.; Jamieson, A.G.; Strong, P.N.; Turner, A.D. Friends or Foes? Emerging Impacts of Biological Toxins. Trends Biochem. Sci. 2019, 44, 365–379. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Casals, E.; Comenge, J.; Bastus, N.G.; Puntes, V.F. Inorganic engineered nanoparticles and their impact on the immune response. Curr. Drug Metabol. 2009, 10, 895–904. [Google Scholar] [CrossRef]
- Benne, N.; van Duijn, J.; Kuiper, J.; Jiskoot, W.; Slütter, B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J. Control. Release 2016, 234, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Parkes, D.G.; Pittner, R.; Jodka, C.; Smith, P.; Young, A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 2001, 50, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fehmann, H.C.; Goke, R.; Goke, B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr. Rev. 1995, 16, 390–410. [Google Scholar] [CrossRef]
- Madsen, K.; Knudsen, L.B.; Agersoe, H.; Nielsen, P.F.; Thogersen, H.; Wilken, M.; Johansen, N.L. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: Importance of fatty acid length, polarity, and bulkiness. J. Med. Chem. 2007, 50, 6126–6132. [Google Scholar] [CrossRef]
- Plisson, F.; Hill, T.A.; Mitchell, J.M.; Hoang, H.N.; de Araujo, A.D.; Xu, W.; Cotterell, A.; Edmonds, D.J.; Stanton, R.V.; Derksen, D.R. Helixconstraints and amino acid substitution in GLP-1 increase cAMP and insulin secretion but not beta-arrestin 2 signaling. Eur. J. Med. Chem. 2017, 127, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Seo, M.D.; Won, H.S.; Kim, J.H.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial peptides for therapeutic applications: A review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Ma, A.N.; Zhang, L.J.; Luo, X.S.; Zhang, Y.H.; Xu, M.; Wang, Y.X. Site-specific PEGylation of exenatide analogues markedly improved their glucoregulatory activity. Br. J. Pharmacol. 2011, 163, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.-H.; Oh, E.J.; Chae, S.Y.; Lee, K.C.; Hahn, S.K. Long acting hyaluronate–exendin 4 conjugate for the treatment of type 2 diabetes. Biomaterials 2010, 31, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, T.H.; Ma, K.; Lee, E.S.; Kim, D.; Oh, K.T.; Lee, D.H.; Lee, K.C.; Youn, Y.S. Synthesis and evaluation of human serum albumin-modified exendin-4 conjugate via heterobifunctional polyethylene glycol linkage with protracted hypoglycemic efficacy. Bioconjug. Chem. 2010, 21, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Chen, Z.; Chen, Y.Q.; Ma, G.C.; Shan, J.F.; Liu, W.; Zhou, L.F. Preparation and characterization of a novel exendin-4 human serum albumin fusion protein expressed in Pichia pastoris. J. Pept. Sci. 2008, 14, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Meng, Z.; Gan, H.; Gu, R.; Wu, Z.; Gao, L.; Zhu, X.; Sun, W.; Li, J.; et al. A novel exendin-4 human serum albumin fusion protein, E2HSA, with an extended half-life and good glucoregulatory effect in healthy rhesus monkeys. Biochem. Biophys. Res. Commun. 2014, 445, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.Y.; Jin, C.H.; Shin, J.H.; Son, S.; Kim, T.H.; Lee, S.; Youn, Y.S.; Byun, Y.; Lee, M.S.; Lee, K.C. Biochemical, pharmaceutical and therapeutic properties of long-acting lithocholic acid derivatized exendin-4 analogs. J. Control. Release 2010, 142, 206–213. [Google Scholar] [CrossRef]
- Lee, J.G.; Ryu, J.H.; Kim, S.M.; Park, M.Y.; Kim, S.H.; Shin, Y.G.; Sohn, J.W.; Kim, H.H.; Park, Z.Y.; Seong, J.Y.; et al. Replacement of the C-terminal Trp-cage of exendin-4 with a fatty acid improves therapeutic utility. Biochem. Pharmacol. 2018, 151, 59–68. [Google Scholar] [CrossRef]
- Lee, C.; Choi, J.S.; Kim, I.; Byeon, H.J.; Kim, T.H.; Oh, K.T.; Lee, E.S.; Lee, K.C.; Youn, Y.S. Decanoic acid-modified glycol chitosan hydrogels containing tightly adsorbed palmityl-acylated exendin-4 as a long-acting sustained-release anti-diabetic system. Acta Biomater. 2014, 10, 812–820. [Google Scholar] [CrossRef]
- Zhong, X.; Yang, S.; Liu, T.; Ji, S.; Hu, J.; Li, H. Engineering a novel protease-based Exendin-4 derivative for type 2 antidiabetic therapeutics. Eur. J. Med. Chem. 2018, 150, 841–850. [Google Scholar] [CrossRef]
- Levy, O.E.; Jodka, C.M.; Ren, S.S.; Mamedova, L.; Sharma, A.; Samant, M.; D’Souza, L.J.; Soares, C.J.; Yuskin, D.R.; Jin, L.J.; et al. Novel exenatide analogs with peptidic albumin binding domains: Potent anti-diabetic agents with extended duration of action. PLoS ONE 2014, 9, e87704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wang, G.; Lang, L.; Jacobson, O.; Kiesewetter, D.O.; Liu, Y.; Ma, Y.; Zhang, X.; Wu, H.; Zhu, L. Chemical conjugation of evans blue derivative: A strategy to develop long-acting therapeutics through albumin binding. Theranostics 2016, 6, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Min, K.A.; Jang, D.-J.; Ahn, T.Y.; Min, J.H.; Yu, B.E.; Cho, K.H. Practical approaches on the long-acting injections. J. Pharm. Investig. 2020, 50, 147–157. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Cespi, M.; Bonacucina, G.; Palmieri, G.F. PEGylated polylactide (PLA) and poly (lactic-co-glycolic acid)(PLGA) copolymers for the design of drug delivery systems. J. Pharm. Investig. 2019, 49, 443–458. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K. PEGylation: An approach for drug delivery. A review. Crit. Rev. Ther. Drug Carrier Syst. 2008, 25, 403–447. [Google Scholar] [CrossRef]
- Mezo, A.R.; Low, S.C.; Hoehn, T.; Palmieri, H. PEGylation enhances the therapeutic potential of peptide antagonists of the neonatal Fc receptor, FcRn. Bioorg. Med. Chem. Lett. 2011, 21, 6332–6335. [Google Scholar] [CrossRef]
- Tang, D.; Tian, H.; Wu, J.; Cheng, J.; Luo, C.; Sai, W.; Song, X.; Gao, X.; Yao, W. C-terminal site-specific PEGylated Exendin-4 analog: A long-acting glucagon like Peptide-1 receptor agonist, on glycemic control and beta cell function in diabetic db/db mice. J. Pharmacol. Sci. 2018, 138, 23–30. [Google Scholar] [CrossRef]
- Podust, V.N.; Balan, S.; Sim, B.-C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release 2016, 240, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Mero, A.; Grigoletto, A.; Martinez, G.; Pasut, G. Recent developments in hyaluronic acid-based nanomedicine. Rec. Adv. Biotechnol. 2016, 3, 102–129. [Google Scholar]
- Cho, H.-J. Recent progresses in the development of hyaluronic acid-based nanosystems for tumor-targeted drug delivery and cancer imaging. J. Pharm. Investig. 2020, 50, 115–129. [Google Scholar] [CrossRef]
- Rath, T.; Kuo, T.T.; Baker, K.; Qiao, S.W.; Kobayashi, K.; Yoshida, M.; Roopenian, D.; Fiebiger, E.; Lencer, W.I.; Blumberg, R.S. The immunologic functions of the neonatal Fc receptor for IgG. J. Clin. Immunol. 2013, 33, 9–17. [Google Scholar] [CrossRef]
- Sand, K.M.K.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the interaction between FcRn and albumin: Opportunities for design of albumin-based therapeutics. Front. Immunol. 2015, 5, 682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simister, N.E.; Rees, A.R. Isolation and characterization of an Fc receptor from neonatal rat small intestine. Eur. J. Immunol. 1985, 15, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Rath, T.; Baker, K.; Dumont, J.A.; Peters, R.T.; Jiang, H.; Qiao, S.W.; Lencer, W.I.; Pierce, G.F.; Blumberg, R.S. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015, 35, 235–254. [Google Scholar] [CrossRef]
- Nurdiansyah, R.; Rifa’i, M.J. A comparative analysis of serum albumin from different species to determine a natural source of albumin that might be useful for human therapy. J. Taibah Univ. Med. Sci. 2016, 11, 243–249. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Li, Y.; Si, H.; Dong, Y.; Sheng, F.; Yao, X.; Hu, Z. Molecular modeling and spectroscopic studies on the binding of guaiacol to human serum albumin. J. Photochem. Photobiol. A Chem. 2006, 182, 158–167. [Google Scholar] [CrossRef]
- Andersen, J.T.; Dalhus, B.; Viuff, D.; Ravn, B.T.; Gunnarsen, K.S.; Plumridge, A.; Bunting, K.; Antunes, F.; Williamson, R.; Athwal, S.; et al. Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J. Biol. Chem. 2014, 289, 13492–13502. [Google Scholar] [CrossRef] [Green Version]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Seijsing, J.; Lindborg, M.; Hoiden-Guthenberg, I.; Bonisch, H.; Guneriusson, E.; Frejd, F.Y.; Abrahmsen, L.; Ekblad, C.; Lofblom, J.; Uhlen, M.; et al. An engineered affibody molecule with pH-dependent binding to FcRn mediates extended circulatory half-life of a fusion protein. Proc. Natl. Acad. Sci. USA 2014, 111, 17110–17115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Transport of protein-bound hormones into tissues in vivo. Endocr. Rev. 1981, 2, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Borchers, T.; Spener, F.; van der Vusse, G.J. Fatty acids in cell signalling: Modulation by lipid binding proteins. Prostaglandins Leukot. Essent. Fatty Acids 1995, 52, 121–127. [Google Scholar] [CrossRef]
- Glatz, J.F.; Vork, M.M.; Cistola, D.P.; van der Vusse, G.J. Cytoplasmic fatty acid binding protein: Significance for intracellular transport of fatty acids and putative role on signal transduction pathways. Prostaglandins Leukot. Essent. Fatty Acids 1993, 48, 33–41. [Google Scholar] [CrossRef]
- Andersen, J.T.; Pehrson, R.; Tolmachev, V.; Daba, M.B.; Abrahmsén, L.; Ekblad, C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J. Biol. Chem. 2011, 286, 5234–5241. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.A.; Gibbs, A.C.; Conk, M.; Yi, F.; Maguire, D.; Kane, C.; O’Neil, K.T. Fusion to a highly stable consensus albumin binding domain allows for tunable pharmacokinetics. Protein Eng. Des. Sel. 2015, 28, 385–393. [Google Scholar] [CrossRef]
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Xue, X.; Yu, P.; Ni, Y.; Chen, F. Evans Blue Dye: A Revisit of Its Applications in Biomedicine. Contrast Media Mol. Imaging 2018, 2018, 7628037. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wang, G.; Lau, J.; Lang, L.; Jacobson, O.; Ma, Y.; Kiesewetter, D.O.; Zhang, S.; Chen, X. Antidiabetic Effect of Abextide, a Long-Acting Exendin-4 Analogue in Cynomolgus Monkeys. Adv. Healthc. Mater. 2019, 8, e1800686. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Threshold Concentration for Insulin Release from Beta Cells (nM) | Hemolytic Activity (LC50; μM) | Reference |
|---|---|---|---|---|
| Esculentin-2CHa | GFSSIFRGVAKFASKGLGKDLAKLGVDLVACKISKQC | 100 | 150 | [23,24] |
| Tigerinin-1R | RVCSAIPLPICH | 0.1 | 500 | [25] |
| Magainin–AM1 | GIKEFAHSLGKFG KAFVGGILNQ | -1 | - | [26] |
| Magainin–AM2 | GVSKILHSAGKFGKAFLGEIMKS | - | >100 | [27] |
| Hymenochirin-1b | IKLSPETKDNLKKVLKGAIKGAIAVAKMV | 1 | 210 | [28] |
| Alyteserin-2a | ILGKLLSTAAGLLSNL | 30 | 135 | [29] |
| Brevinin-2-related peptide (B2RP) | GIWDTIKSMGKVFAGKILQNL | 1000 | - | [30] |
| Brevinin-2-related peptide (B2RP) | GMASKAGSVLGKVAKVALKAAL | 100 | - | [31] |
| Type of Modification | Plasma Half-Life (Tested Animal Species, Administration Route) | Duration of Hypoglycemic Action (Tested Animal Species) | Reference |
|---|---|---|---|
| PEGylation with different sizes (5, 10, 20, and 40 kDa) of linear PEGs | 6.1 to 76.4 h (rat, i.v. (intravenous)) | t1/2: 23 h (for 20 kDa PEG conjugated exendin-4) (mouse) | [60] |
| PEGylation with Y-shaped 40 kDa PEG | 38 h (mouse, s.c. (subcutaneous)) 88 h (monkey, s.c.) | -1 | [61] |
| XTENylation | 12 h (mouse) 32 h (rat) 60 h (monkey, s.c. and i.v.) 128 h (human) | - | [62] |
| Hyaluronic acid (HA) conjugation | - | 3 days (mouse) | [63] |
| Human serum albumin (HSA) chemical conjugate via PEG linker | 24.2 h (mouse, i.p. (intraperitoneal)) | 31 h (mouse) | [64] |
| HSA fusion | 56.7 h (monkey, i.v.) 77.4 h (monkey, s.c.) | - | [65] |
| HSA fusion to tandem dimeric exendin-4 | 54 h (monkey, s.c.) | 42 h (monkey) | [66] |
| Lithocholic acid conjugate | 9.7 h (rat, s.c.) | ≥24 h (mouse) | [67] |
| Capric acid conjugate of exendin-4 analog (Ex-4(1-32) K-Cap) | 2.25 h (mouse, s.c.) | ≥40 h (mouse) | [68] |
| Decanoic acid-modified glycol chitosan hydrogel encapsulation (DAGC-Exendin-4-C16) | - | 8 days (mouse) | [69] |
| Albumin-binding domain (ABD) fusion | 8.1 h (rat) | 8.7 h (mouse) | [70] |
| ABD035 fusion | 16 h (rat, i.v.) | - | [71] |
| Albumin-binding peptide (ABP; SA21) fusion | 11 h (rat, i.v.) | - | [71] |
| Truncated Evans blue (tBE) conjugation | 35 h (mouse, s.c.) | 36 h (mouse) | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amatya, R.; Park, T.; Hwang, S.; Yang, J.; Lee, Y.; Cheong, H.; Moon, C.; Kwak, H.D.; Min, K.A.; Shin, M.C. Drug Delivery Strategies for Enhancing the Therapeutic Efficacy of Toxin-Derived Anti-Diabetic Peptides. Toxins 2020, 12, 313. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050313
Amatya R, Park T, Hwang S, Yang J, Lee Y, Cheong H, Moon C, Kwak HD, Min KA, Shin MC. Drug Delivery Strategies for Enhancing the Therapeutic Efficacy of Toxin-Derived Anti-Diabetic Peptides. Toxins. 2020; 12(5):313. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050313
Chicago/Turabian StyleAmatya, Reeju, Taehoon Park, Seungmi Hwang, JaeWook Yang, Yoonjin Lee, Heesun Cheong, Cheol Moon, Hyun Duck Kwak, Kyoung Ah Min, and Meong Cheol Shin. 2020. "Drug Delivery Strategies for Enhancing the Therapeutic Efficacy of Toxin-Derived Anti-Diabetic Peptides" Toxins 12, no. 5: 313. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050313