Temporary Membrane Permeabilization via the Pore-Forming Toxin Lysenin

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
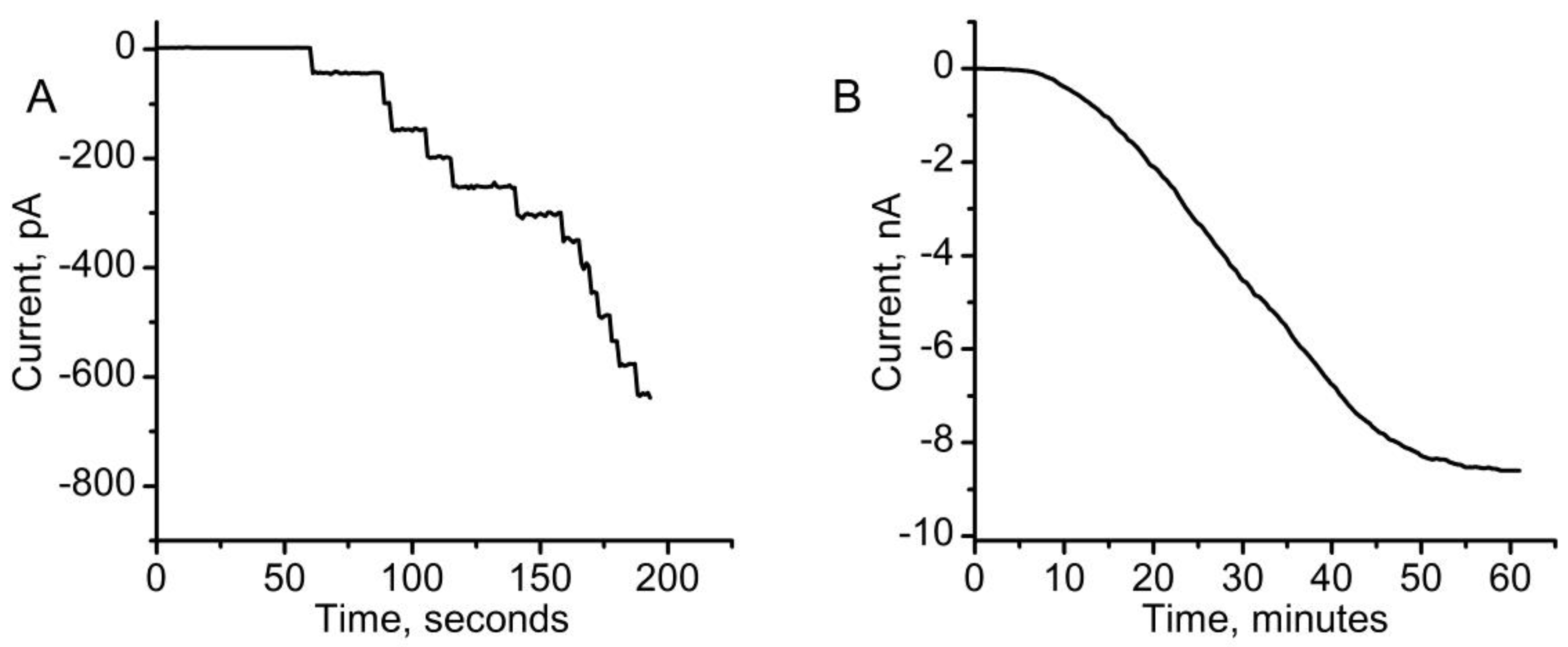
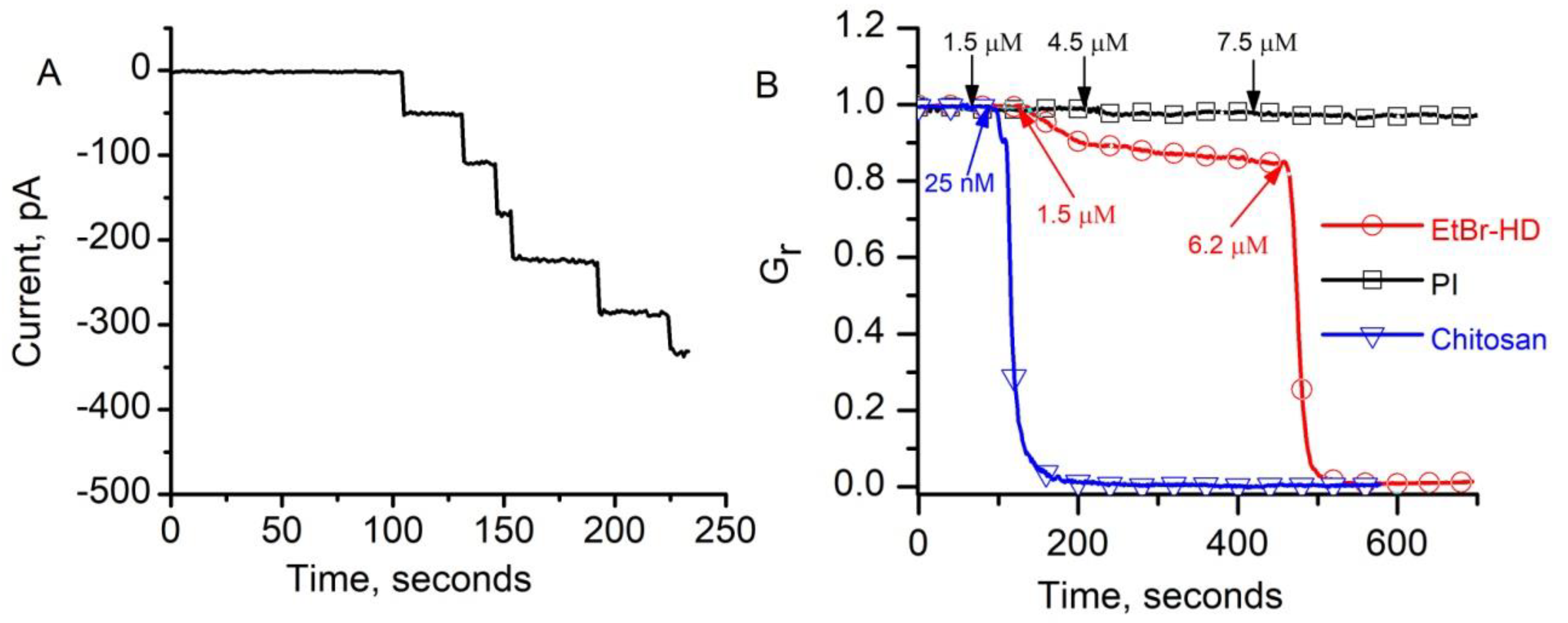
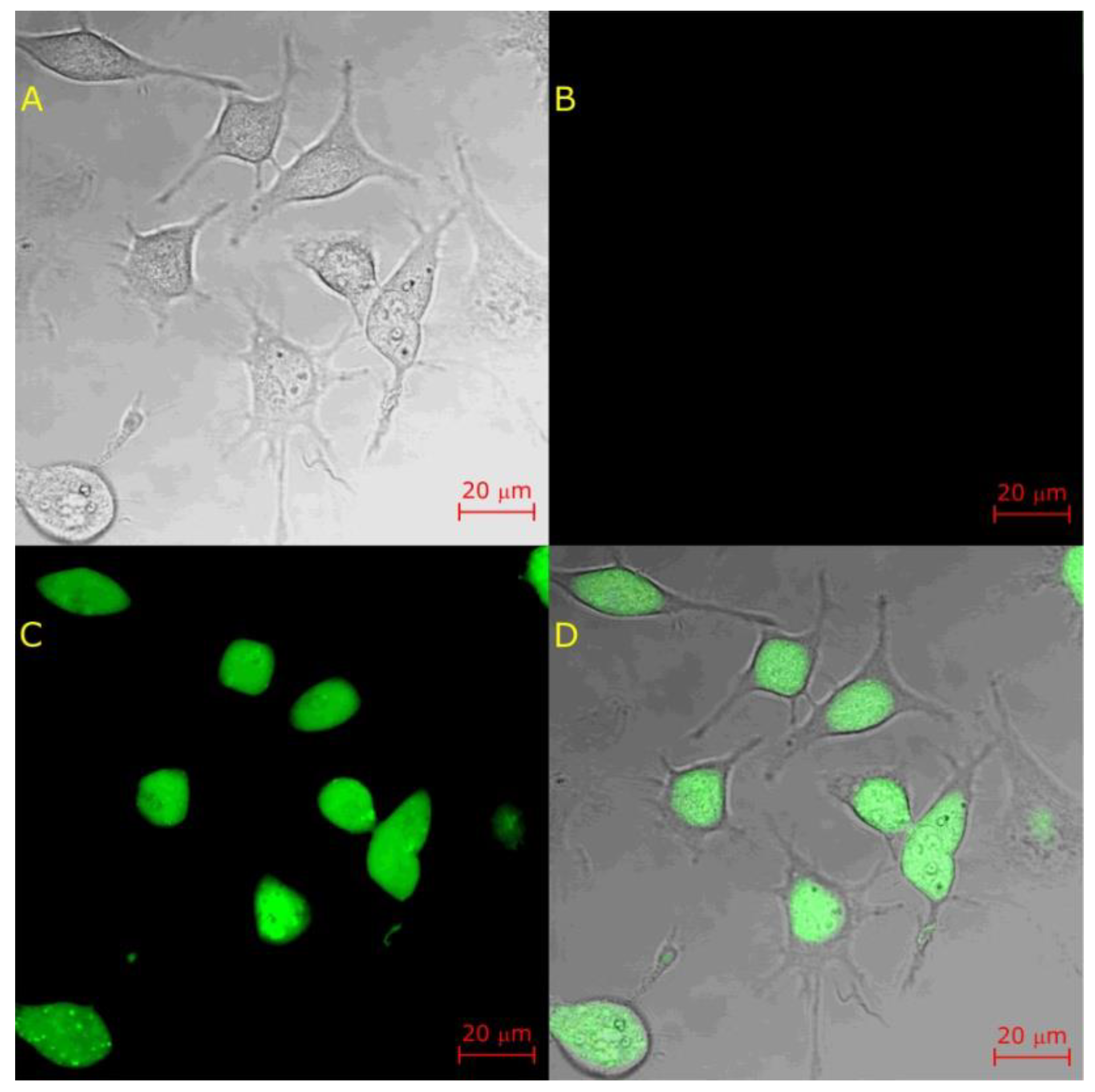{kind=link}
{kind=link}
{kind=link}
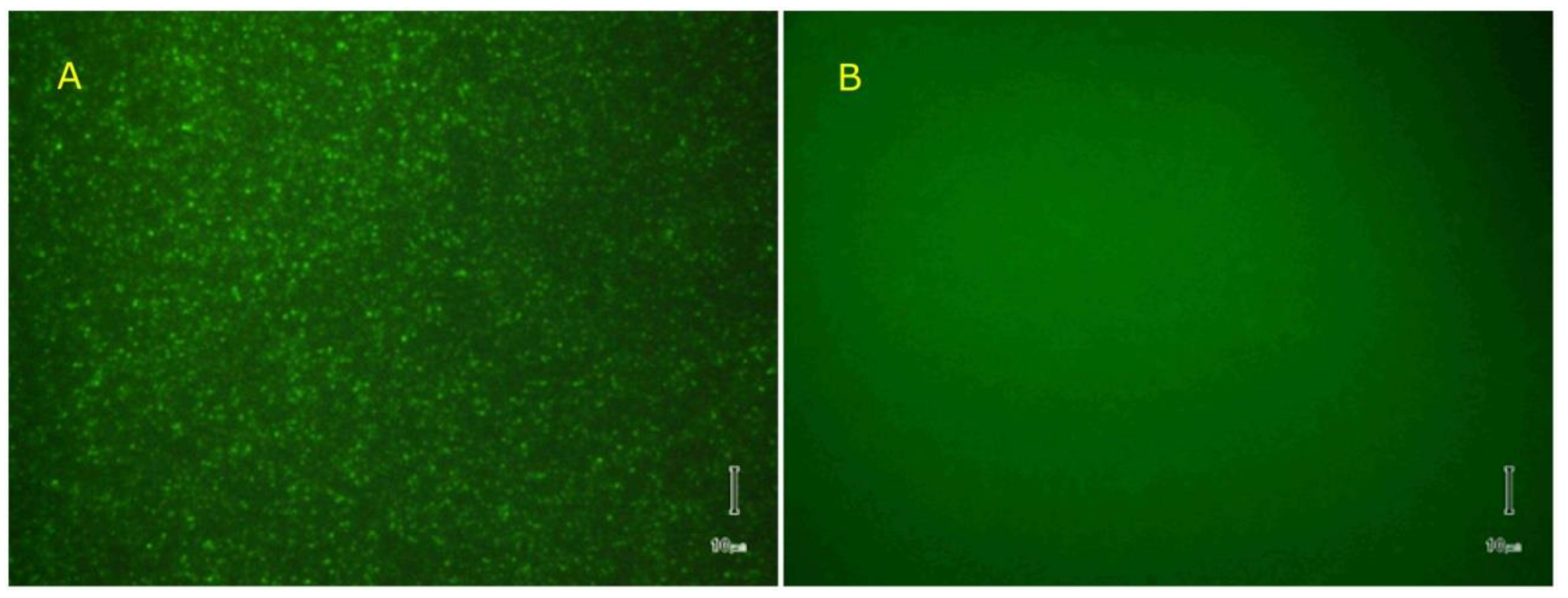{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
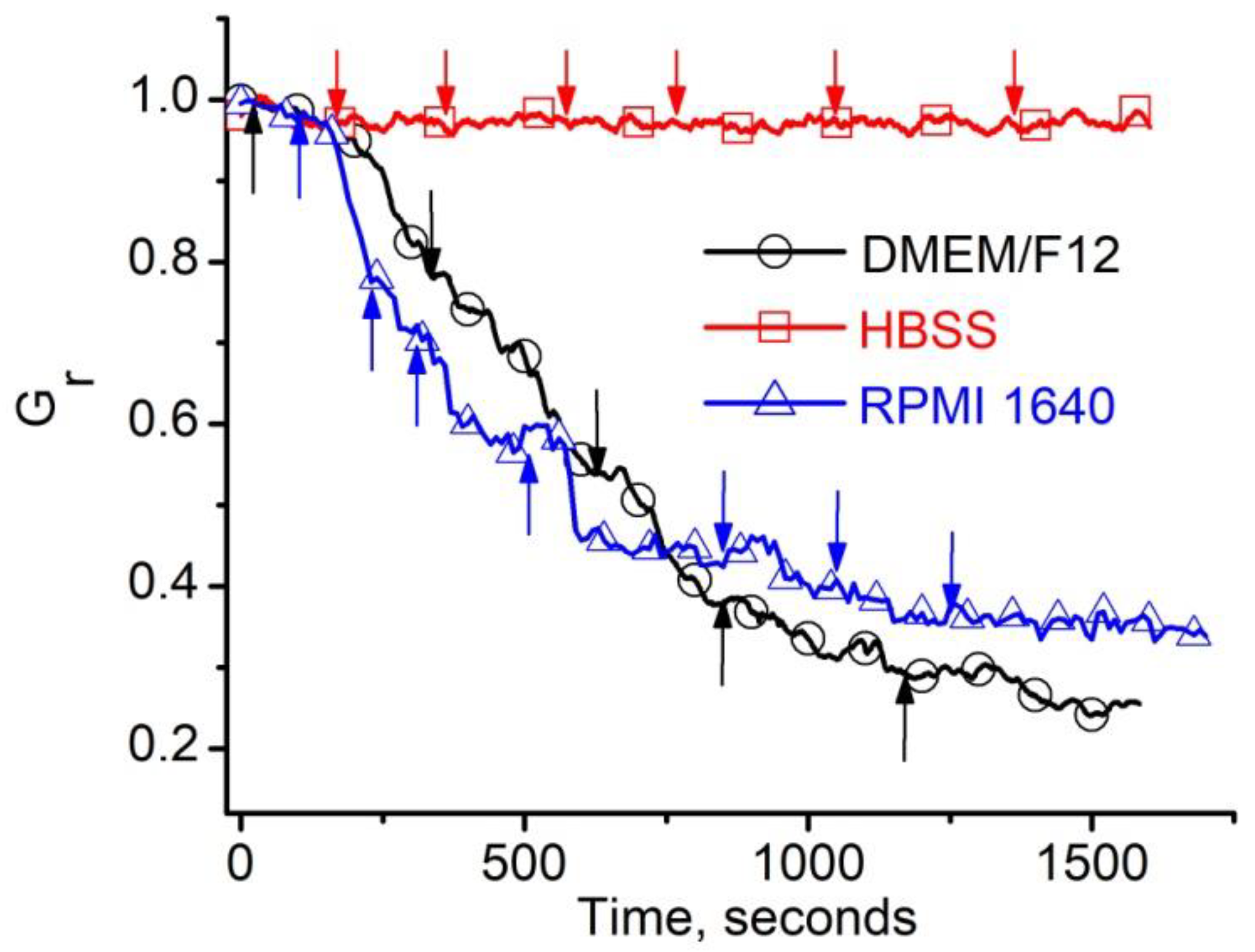
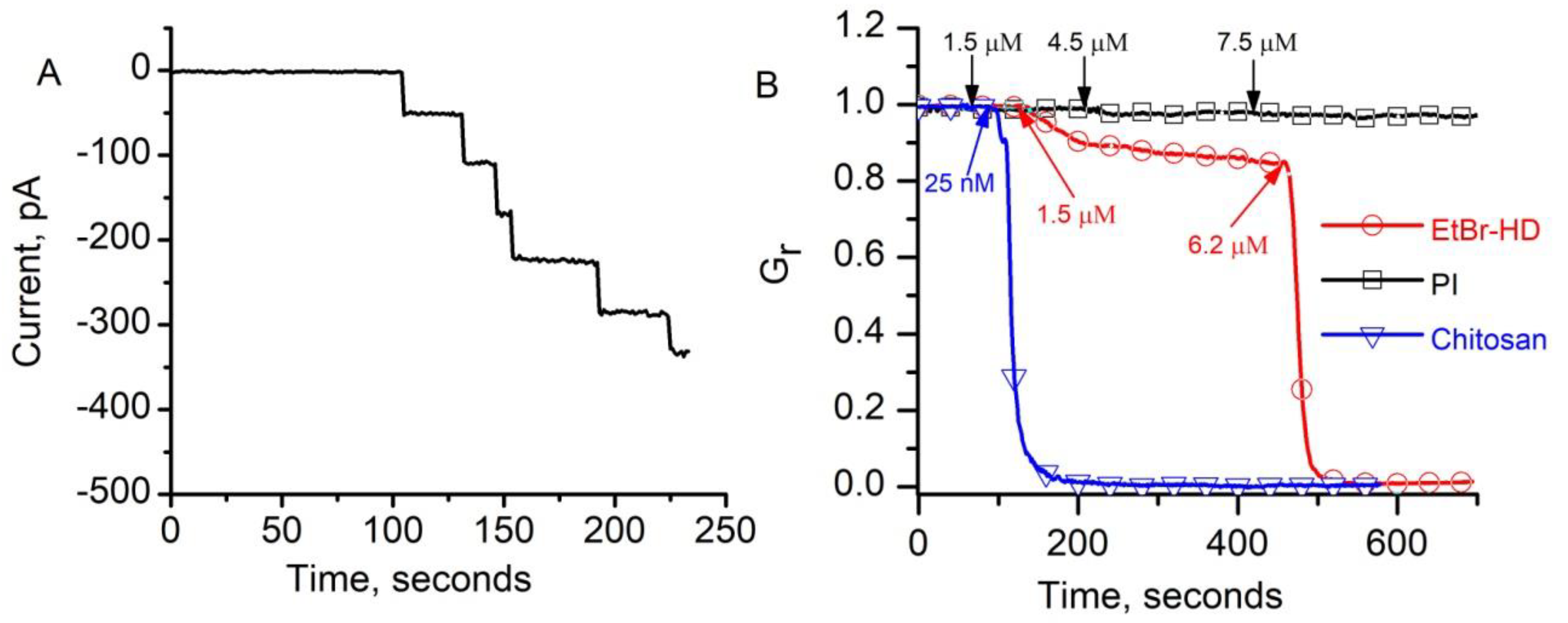
2.1. Investigations of the Effect of Indicators and Culture Media on the Conductance of Lysenin Channels Reconstituted in Artificial Membranes
2.2. Investigations on Jurkat Cells: Viability and Permeabilization Assays
2.2.1. Viability Assessments
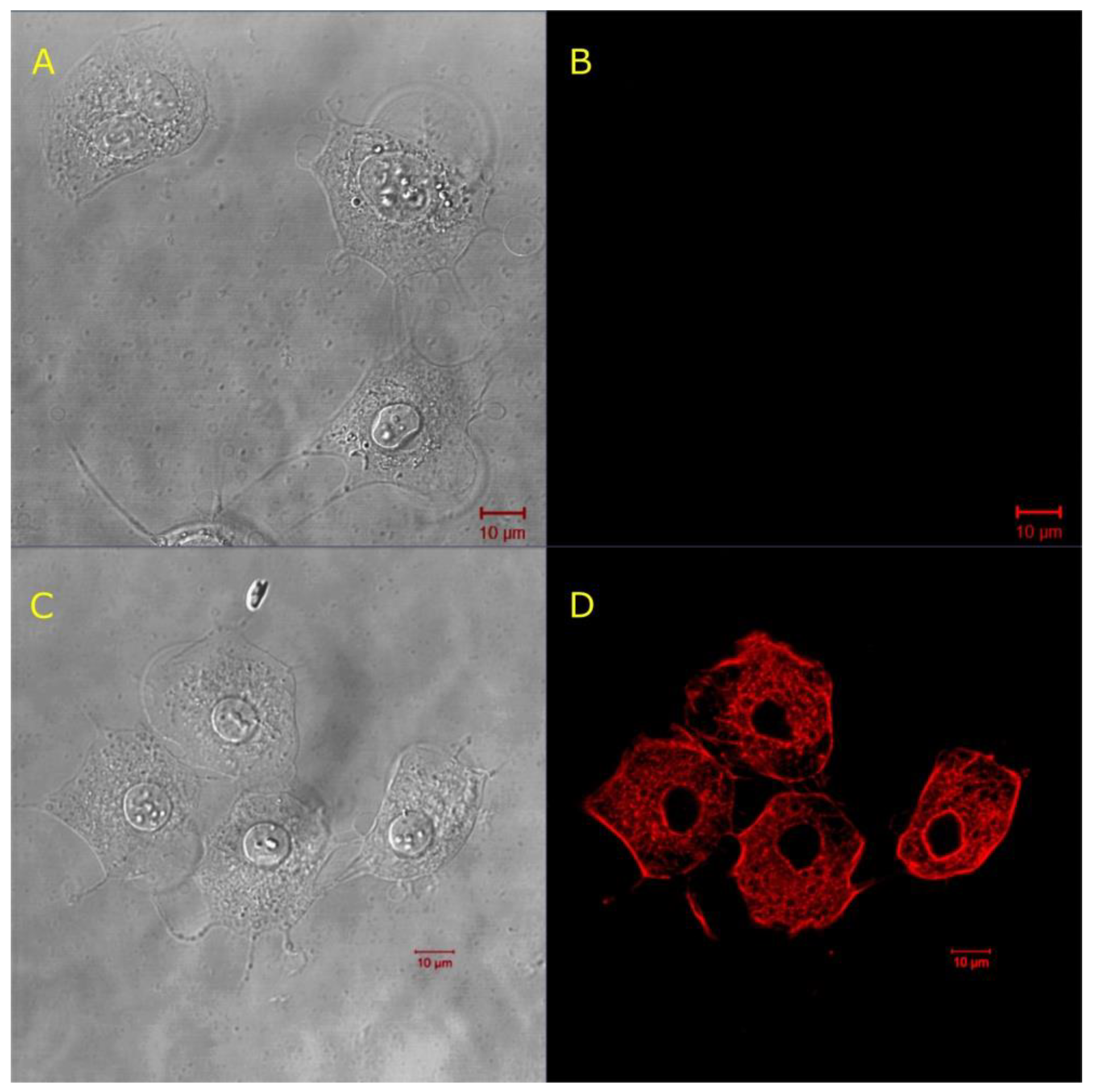
2.2.2. Achievement of Controlled Permeability of Jurkat Cell Membranes
2.3. Investigations on ATDC5 Cells: Permeabilization Assay
Investigations on ATDC5 Cells
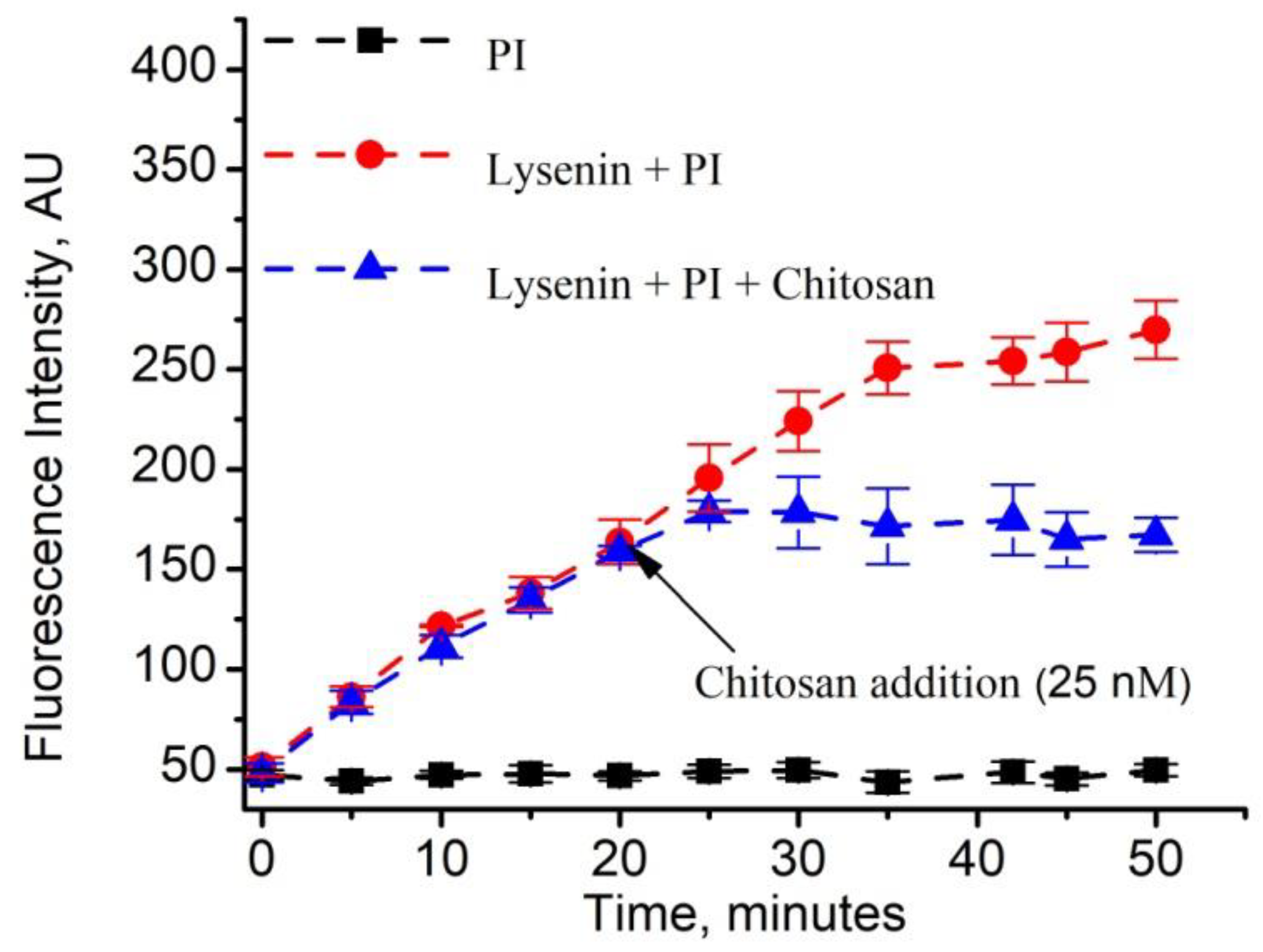
2.4. Reversible Liposome Loading and Unloading via Lysenin Channels
3. Conclusions
4. Materials and Methods
4.1. Bilayer Lipid Membrane Experiments
4.2. Cell Preparation and Analyses
4.2.1. Jurkat Cells Permeabilization and Viability Measurements
4.2.2. ATDC5 Cells Preparation and Experiments
4.3. Liposome Preparation and Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dhakal, K.; Black, B.; Mohanty, S. Introduction of impermeable actin-staining molecules to mammalian cells by optoporation. Sci. Rep. 2014, 4, 6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerner, J.F.; Febvay, S.; Clapham, D.E. Controlled delivery of bioactive molecules into live cells using the bacterial mechanosensitive channel MscL. Nat. Commun. 2012, 3, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoffre, J.M.; Bouakaz, A. Minireview: Biophysical Mechanisms of Cell Membrane Sonopermeabilization. Knowns and Unknowns. Langmuir 2019, 35, 10151–10165. [Google Scholar] [CrossRef] [PubMed]
- Hapala, I. Breaking the Barrier: Methods for Reversible Permeabilization of Cellular Membranes. Crit. Rev. Biotechnol. 1997, 17, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kano, F.; Murata, M. LLO-mediated Cell Resealing System for Analyzing Intracellular Activity of Membrane-impermeable Biopharmaceuticals of Mid-sized Molecular Weight. Sci. Rep. 2018, 8, 1946. [Google Scholar] [CrossRef] [Green Version]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. The EMBO J. 1982, 1, 841–845. [Google Scholar] [CrossRef]
- Russo, M.J.; Bayley, H.; Toner, M. Reversible permeabilization of plasma membranes with an engineered switchable pore. Nat. Biotechnol. 1997, 15, 278–282. [Google Scholar] [CrossRef]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef]
- Teng, K.W.; Ren, P.; Selvin, P.R. Delivery of Fluorescent Probes Using Streptolysin O for Fluorescence Microscopy of Living Cells. Curr. Protoc. Protein Sci. 2018, 93, e60. [Google Scholar] [CrossRef]
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190. [Google Scholar] [CrossRef] [Green Version]
- Wong, F.K.; Haffner, C.; Huttner, W.B.; Taverna, E. Microinjection of membrane-impermeable molecules into single neural stem cells in brain tissue. Nat. Protoc. 2014, 9, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; van de Linde, S.; Lummer, M.; Simonis, M.; Huser, T.; Sauer, M. Instant Live-Cell Super-Resolution Imaging of Cellular Structures by Nanoinjection of Fluorescent Probes. Nano Lett. 2014, 15, 1374–1381. [Google Scholar] [CrossRef]
- Leduc, M.; Guay, D.; Leask, R.L.; Coulombe, S. Cell permeabilization using a non-thermal plasma. New J. Phys. 2009, 11, 115021. [Google Scholar] [CrossRef]
- Provoda, C.J.; Lee, K.-D. Bacterial pore-forming hemolysins and their use in the cytosolic delivery of macromolecules. Adv. Drug Deliv. Rev. 2000, 41, 209–221. [Google Scholar] [CrossRef]
- Saito, G.; Amidon, G.L.; Lee, K.-D. Enhanced cytosolic delivery of plasmid DNA by a sulfhydryl-activatable listeriolysin O/protamine conjugate utilizing cellular reducing potential. Gene Ther. 2003, 10, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, K.W.; Ishitsuka, Y.; Ren, P.; Youn, Y.; Deng, X.; Ge, P.; Lee, S.H.; Belmont, A.S.; Selvin, P.R. Labeling proteins inside living cells using external fluorophores for microscopy. eLife 2016, 5, e20378. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001; pp. 2–22. [Google Scholar]
- Andersen, O.S.; Ingolfsson, H.I.; Lundbaek, J.A. Ion Channels. Wiley Encycl. Chem. Biol. 2008, 1–14. [Google Scholar] [CrossRef]
- Bezanilla, F. Ion channels: From conductance to structure. Neuron 2008, 60, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Cabiaux, V. pH-sensitive toxins: Interactions with membrane bilayers and application to drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 987–997. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; van der Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (w)hole story? Cell. Mol. Life Sci. 2008, 65, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Aroian, R.; van der Goot, F.G. Pore forming toxins and cellular non-immune defenses (CNIDs). Curr. Opin. Microbiol. 2007, 10, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Bashford, C.L. Pore-Forming Toxins: Attack and Defence at the Cell Surface. Cell. Mol. Biol. Lett. 2001, 6, 328–333. [Google Scholar]
- Gilbert, R.J.C. Pore-forming toxins. Cell. Mol. Life Sci. 2002, 59, 832–844. [Google Scholar] [CrossRef]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef]
- Mondal, A.K.; Sreekumar, A.; Kundu, N.; Kathuria, R.; Verma, P.; Gandhi, S.; Chattopadhyay, K. Structural Basis and Functional Implications of the Membrane Pore-Formation Mechanisms of Bacterial Pore-Forming Toxins. Adv. Exp. Med. Biol. 2018, 1112, 281–291. [Google Scholar] [CrossRef]
- Mondal, A.K.; Chattopadhyay, K. Taking Toll on Membranes: Curious Cases of Bacterial β-Barrel Pore-Forming Toxins. Biochemistry 2020, 59, 163–170. [Google Scholar] [CrossRef]
- Barry, E.L.; Gesek, F.A.; Friedman, P.A. Introduction of antisense oligonucleotides into cells by permeabilization with streptolysin O. Biotechniques 1993, 15, 1016–1020. [Google Scholar]
- Spiller, D.G.; Tidd, D.M. Nuclear delivery of antisense oligodeoxynucleotides through reversible permeabilization of human leukemia cells with streptolysin O. Antisense Res. Dev. 1995, 5, 13–21. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acker, J.P.; Lu, X.M.; Young, V.; Cheley, S.; Bayley, H.; Fowler, A.; Toner, M. Measurement of trehalose loading of mammalian cells porated with a metal-actuated switchable pore. Biotechnol. Bioeng. 2003, 82, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Martin, T.G.; Naylor, C.E.; Basak, A.K.; Titball, R.W.; Savva, C.G. Cryo-EM structure of lysenin pore elucidates membrane insertion by an aerolysin family protein. Nat. Commun. 2016, 7, 11293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, T.; Aoki, T.; Takeuchi, Y.; Yanagida, T. Lysenin forms a voltage-dependent channel in artificial lipid bilayer membranes. Biochem. Biophys. Res. Commun. 2006, 346, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, K.; Hordejuk, R.; Szymczyk, P.; Kulma, M.; Abdel-Shakor, A.B.; Płucienniczak, A.; Dołowy, K.; Szewczyk, A.; Sobota, A. Lysenin-His, a sphingomyelin-recognizing toxin, requires tryptophan 20 for cation-selective channel assembly but not for membrane binding. Mol. Membr. Biol. 2007, 24, 121–134. [Google Scholar] [CrossRef]
- Podobnik, M.; Savory, P.; Rojko, N.; Kisovec, M.; Wood, N.; Hambley, R.; Pugh, J.; Wallace, E.J.; McNeill, L.; Bruce, M.; et al. Crystal structure of an invertebrate cytolysin pore reveals unique properties and mechanism of assembly. Nat. Commun. 2016, 7, 11598. [Google Scholar] [CrossRef]
- Yilmaz, N.; Kobayashi, T. Visualization of Lipid Membrane Reorganization Induced by a Pore-Forming Toxin Using High-Speed Atomic Force Microscopy. ACS Nano 2015, 9, 7960–7967. [Google Scholar] [CrossRef] [PubMed]
- Fologea, D.; Krueger, E.; Lee, R.; Naglak, M.; Mazur, Y.; Henry, R.; Salamo, G. Controlled gating of lysenin pores. Biophys. Chem. 2010, 146, 25–29. [Google Scholar] [CrossRef]
- Fologea, D.; Krueger, E.; Mazur, Y.I.; Stith, C.; Okuyama, Y.; Henry, R.; Salamo, G.J. Bi-stability, hysteresis, and memory of voltage-gated lysenin channels. Biochim. et Biophys. Acta. 2011, 1808, 2933–2939. [Google Scholar] [CrossRef] [Green Version]
- Fologea, D.; Al Faori, R.; Krueger, E.; Mazur, Y.I.; Kern, M.; Williams, M.; Mortazavi, A.; Henry, R.; Salamo, G.J. Potential analytical applications of lysenin channels for detection of multivalent ions. Anal. Bioanal. Chem. 2011, 401, 1871–1879. [Google Scholar] [CrossRef]
- Fologea, D.; Krueger, E.; Al Faori, R.; Lee, R.; Mazur, Y.I.; Henry, R.; Arnold, M.; Salamo, G.J. Multivalent ions control the transport through lysenin channels. Biophys. Chem. 2010, 152, 40–45. [Google Scholar] [CrossRef]
- Fologea, D.; Krueger, E.; Rossland, S.; Bryant, S.; Foss, W.; Clark, T. Cationic polymers inhibit the conductance of lysenin channels. Sci. World J. 2013, 2013, 316758. [Google Scholar] [CrossRef] [Green Version]
- Larson, E.M.; Doughman, D.J.; Gregerson, D.S.; Obritsch, W.F. A new, simple, nonradioactive, nontoxic in vitro assay to monitor corneal endothelial cell viability. Invest. Ophthalmol. Vis. Sci. 1997, 38, 1929–1933. [Google Scholar] [PubMed]
- Vetter, J. Toxins of Amanita phalloides. Toxicon 1998, 36, 13–24. [Google Scholar] [CrossRef]
- Lengsfeld, A.M.; L#xF6;w, I.; Wieland, T.; Dancker, P.; Hasselbach, W. Interaction of Phalloidin with Actin. Proc. Natl. Acad. Sci. USA 1974, 71, 2803–2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bihan, T.; Gicquaud, C. Stabilization of actin by phalloidin: A differential scanning calorimetric study. Biochem. Biophys. Res. Commun. 1991, 181, 542–547. [Google Scholar] [CrossRef]
- Dancker, P.; Löw, I.; Hasselbach, W.; Wieland, T. Interaction of actin with phalloidin: Polymerization and stabilization of F-actin. Biochim. et Biophys. Acta Protein Struct. 1975, 400, 407–414. [Google Scholar] [CrossRef]
- Harkin, D.G.; Hay, E.D. Effects of electroporation on the tubulin cytoskeleton and directed migration of corneal fibroblasts cultured within collagen matrices. Cell Motil. Cytoskeleton 1996, 35, 345–357. [Google Scholar] [CrossRef]
- Schmit, A.C.; Lambert, A.M. Microinjected fluorescent phalloidin in vivo reveals the F-actin dynamics and assembly in higher plant mitotic cells. Plant Cell 1990, 2, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Tawakoli, P.N.; Al-Ahmad, A.; Hoth-Hannig, W.; Hannig, M.; Hannig, C. Comparison of different live/dead stainings for detection and quantification of adherent microorganisms in the initial oral biofilm. Clin. Oral Investig. 2013, 17, 841–850. [Google Scholar] [CrossRef]
- Brittain, H.G. Submicrogram determination of lanthanides through quenching of calcein blue fluorescence. Anal. Chem. 1987, 59, 1122–1125. [Google Scholar] [CrossRef]
- Sukumwang, N.; Umawaza, K. Earthworm-Derived Pore-Forming Toxin Lysenin and Screening of Its Inhibitors. Toxins 2013, 5, 1392–1402. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Bryant, S.L.; Thomas, C.; Richtsmeier, D.; Pu, X.; Tinker, J.; Fologea, D. Stochastic sensing of Angiotensin II with lysenin channels. Scient. Rep. 2017, 7, 2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, P.J.; Adolph, S.K.; Subramanian, A.K.; Arouri, A.; Andresen, T.L.; Mouritsen, O.G.; Madsen, R.; Madsen, M.W.; Peters, G.N.H.; Clausen, M.H. Liposomal Formulation of Retinoids Designed for Enzyme Triggered Release. J. Med. Chem. 2010, 53, 3782–3792. [Google Scholar] [CrossRef] [PubMed]



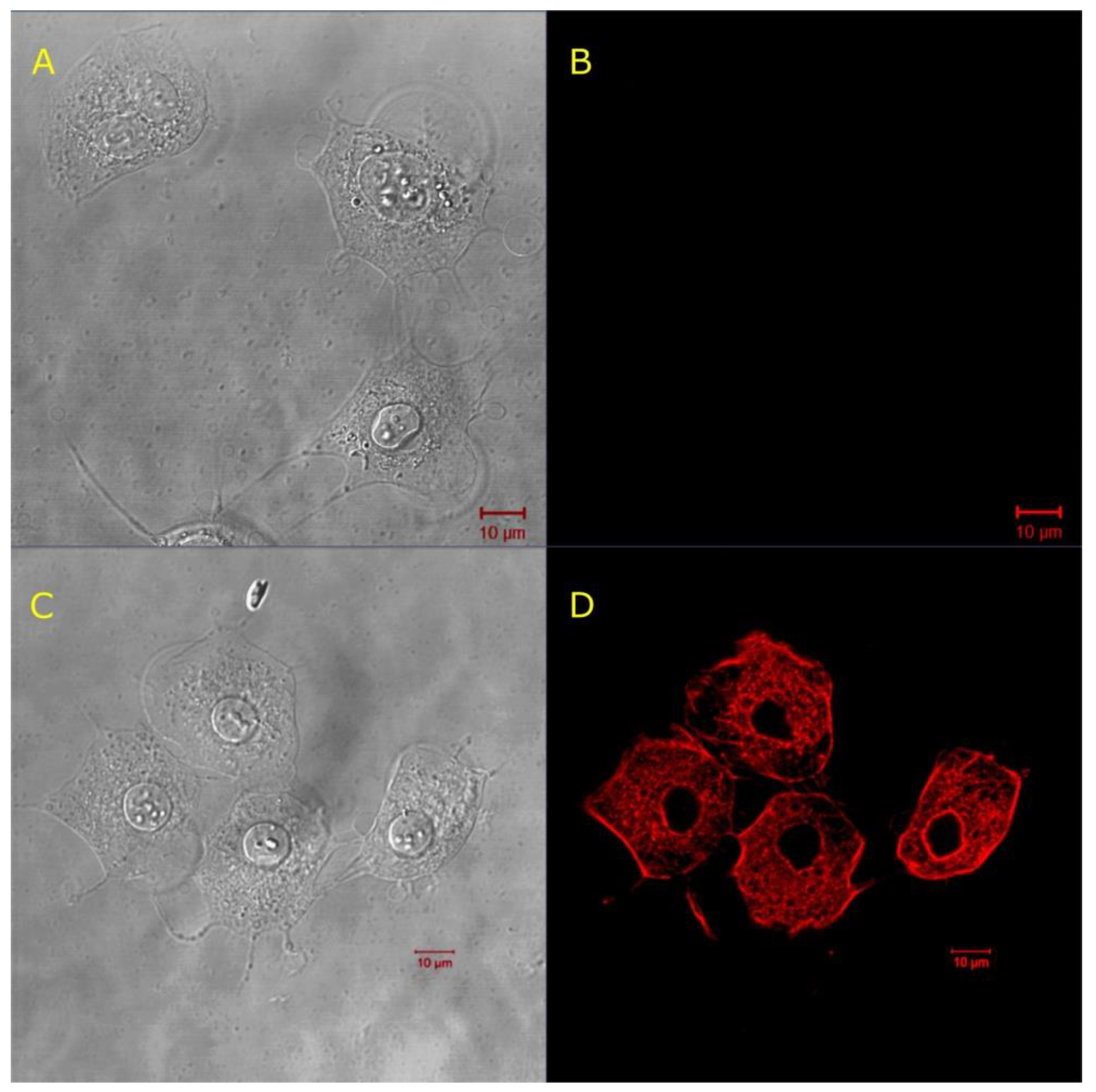
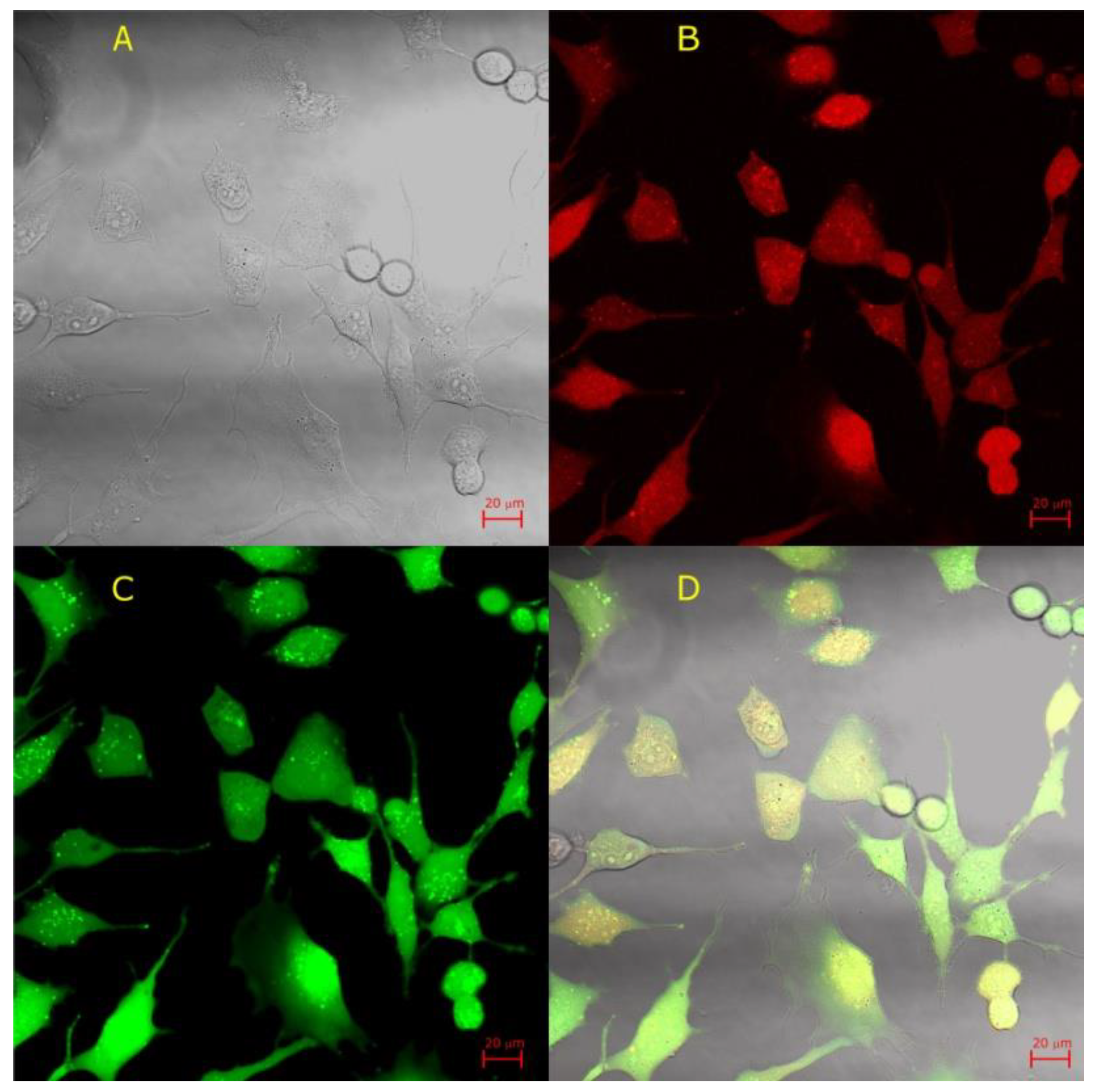
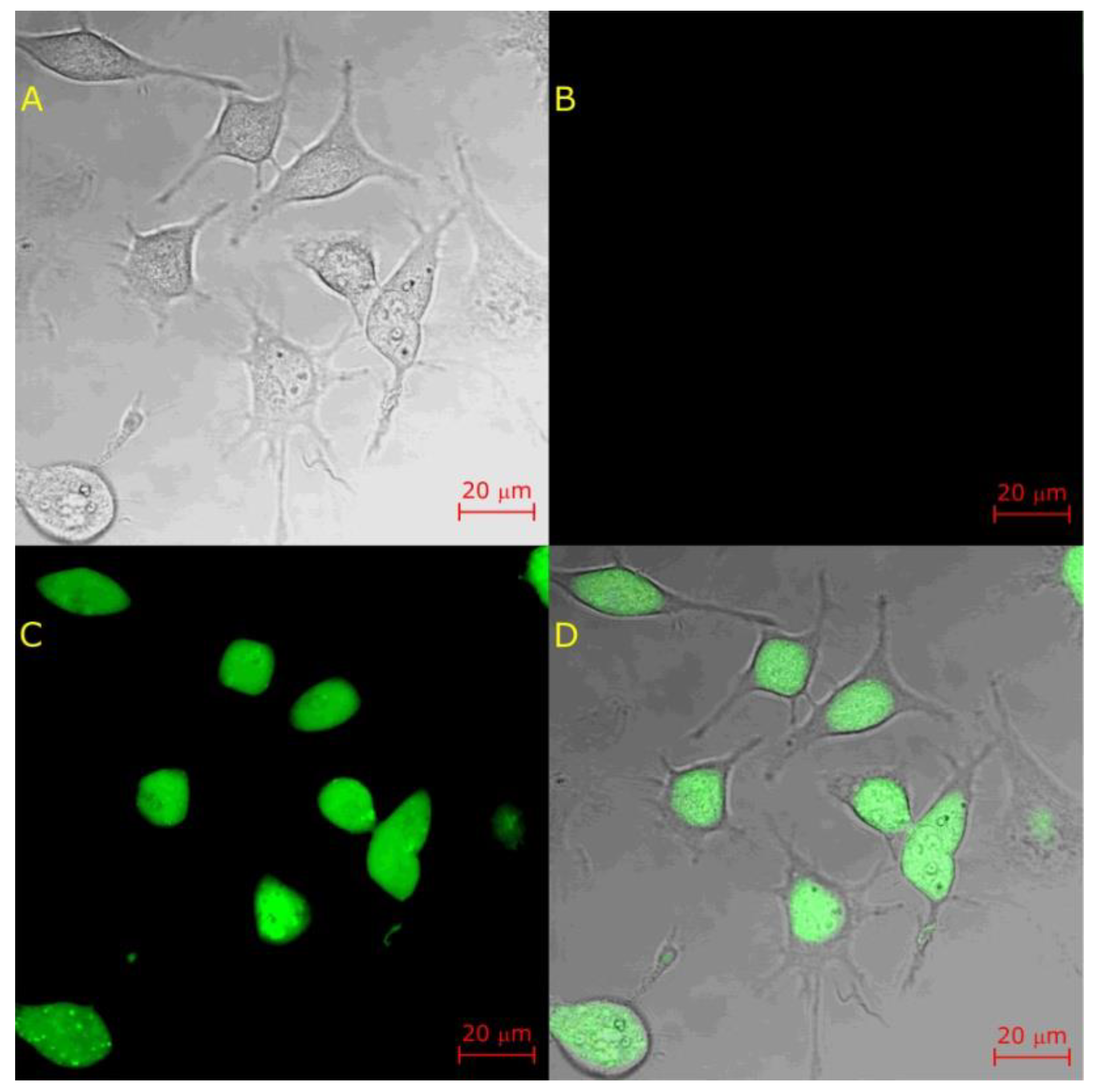
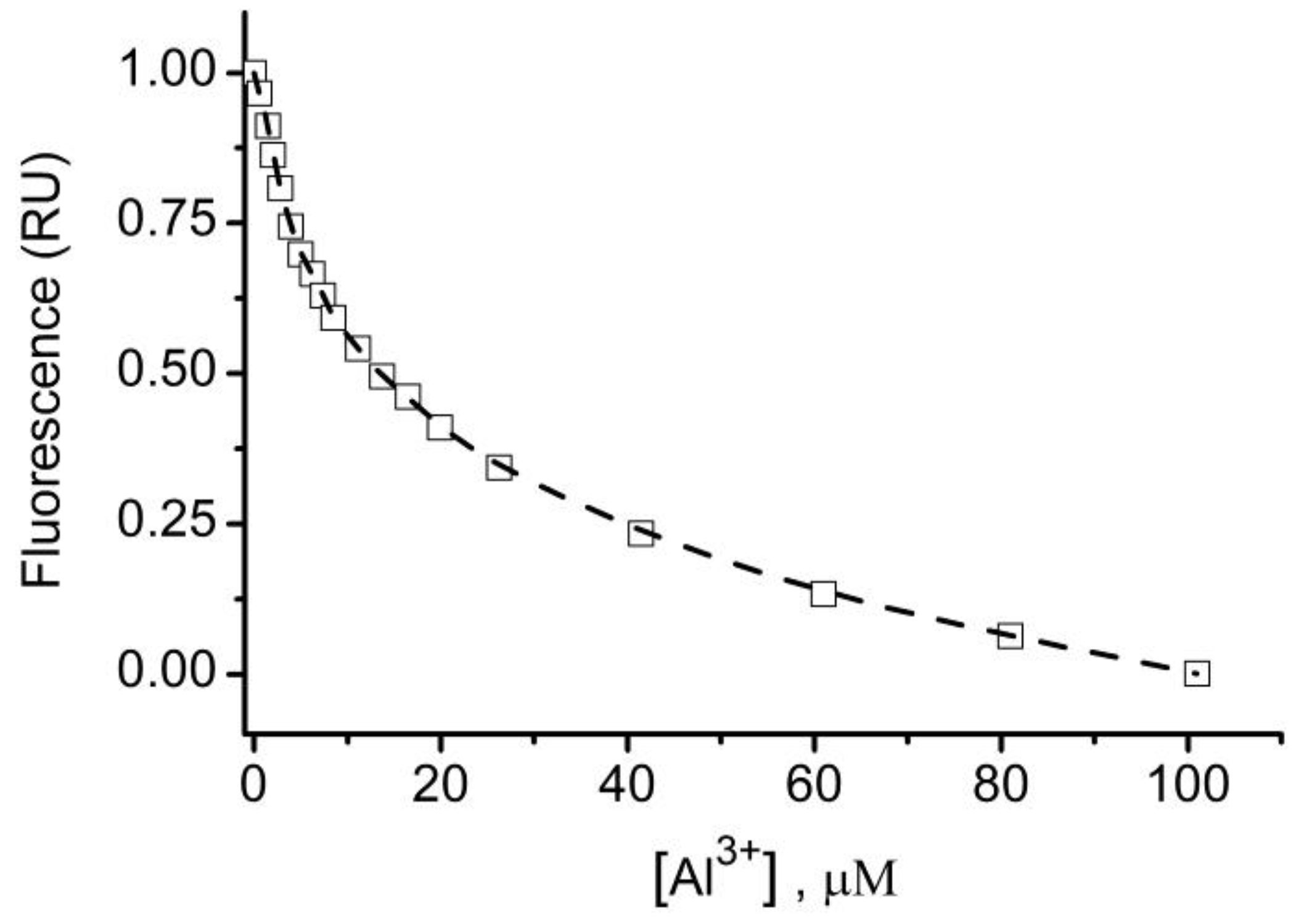

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, N.; Thomas, C.A.; Richtsmeier, D.; Bogard, A.; Hermann, R.; Walker, M.; Abatchev, G.; Brown, R.J.; Fologea, D. Temporary Membrane Permeabilization via the Pore-Forming Toxin Lysenin. Toxins 2020, 12, 343. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050343
Shrestha N, Thomas CA, Richtsmeier D, Bogard A, Hermann R, Walker M, Abatchev G, Brown RJ, Fologea D. Temporary Membrane Permeabilization via the Pore-Forming Toxin Lysenin. Toxins. 2020; 12(5):343. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050343
Chicago/Turabian StyleShrestha, Nisha, Christopher A. Thomas, Devon Richtsmeier, Andrew Bogard, Rebecca Hermann, Malyk Walker, Gamid Abatchev, Raquel J. Brown, and Daniel Fologea. 2020. "Temporary Membrane Permeabilization via the Pore-Forming Toxin Lysenin" Toxins 12, no. 5: 343. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050343