Simultaneous Determination of Deoxynivalenol, Its Modified Forms, Nivalenol and Fusarenone-X in Feedstuffs by the Liquid Chromatography–Tandem Mass Spectrometry Method


Abstract
:1. Introduction
2. Results and Discussion
2.1. LC-MS/MS Optimisation
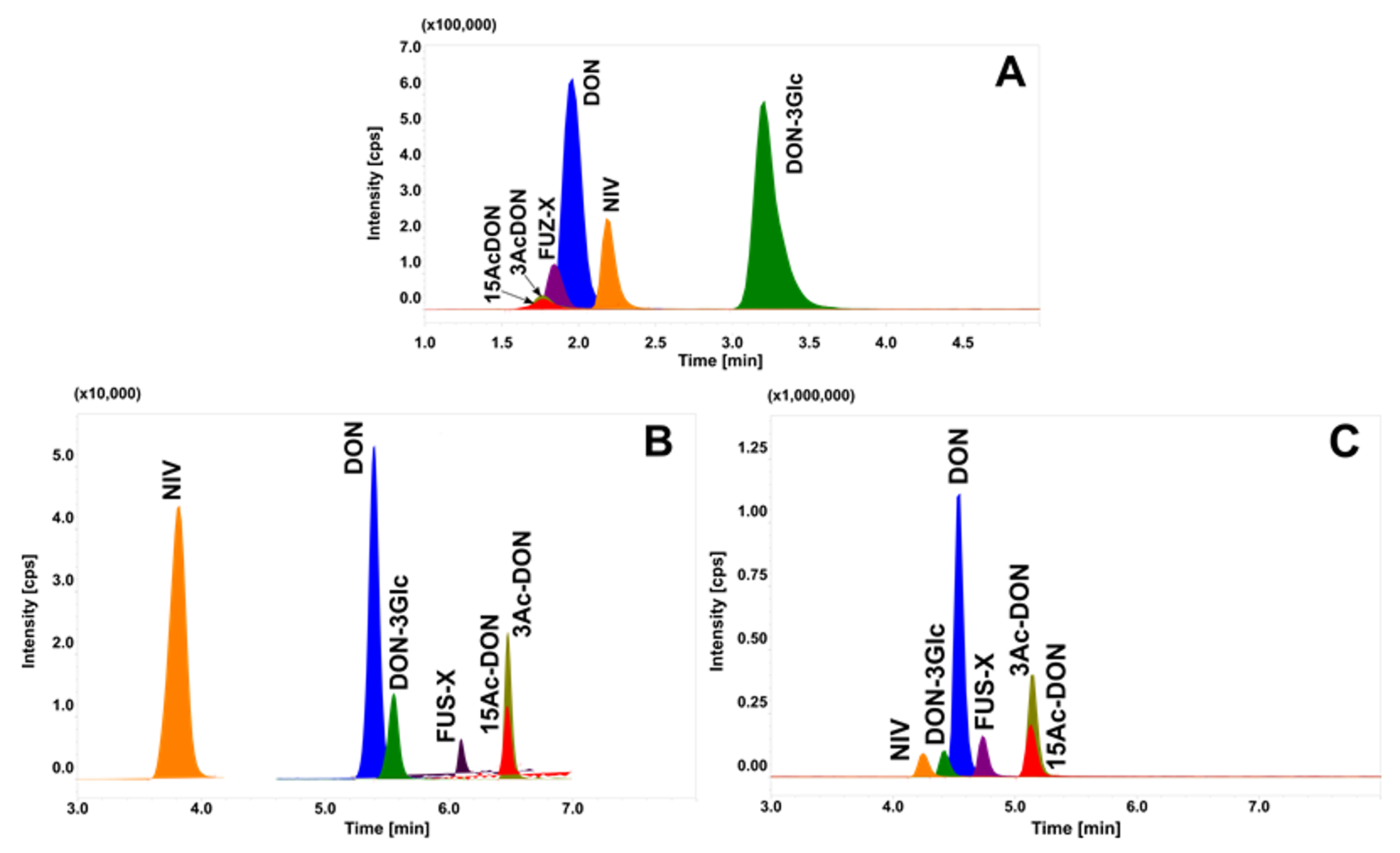
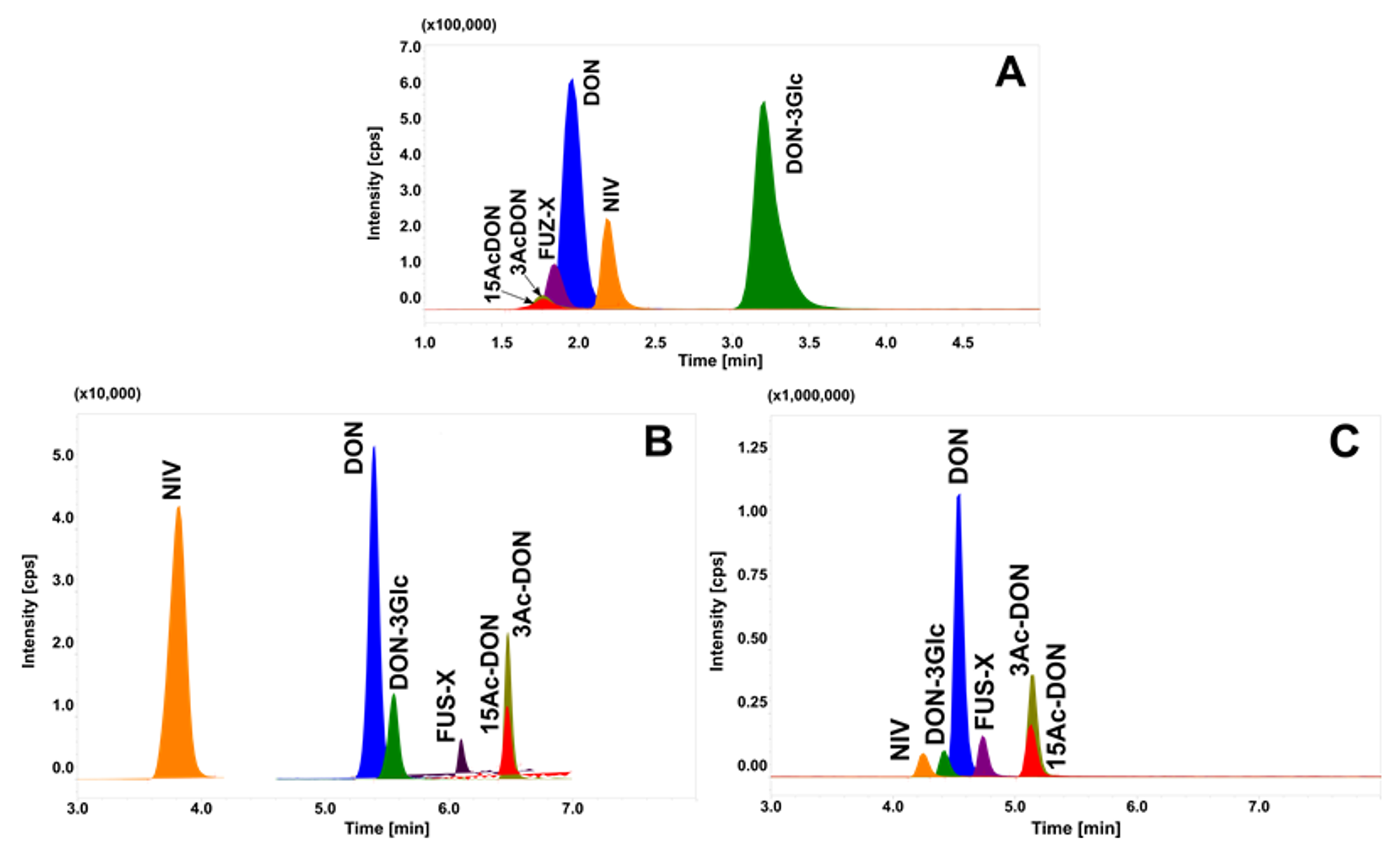
2.2. Chromatographic Separation
2.3. Sample Preparation
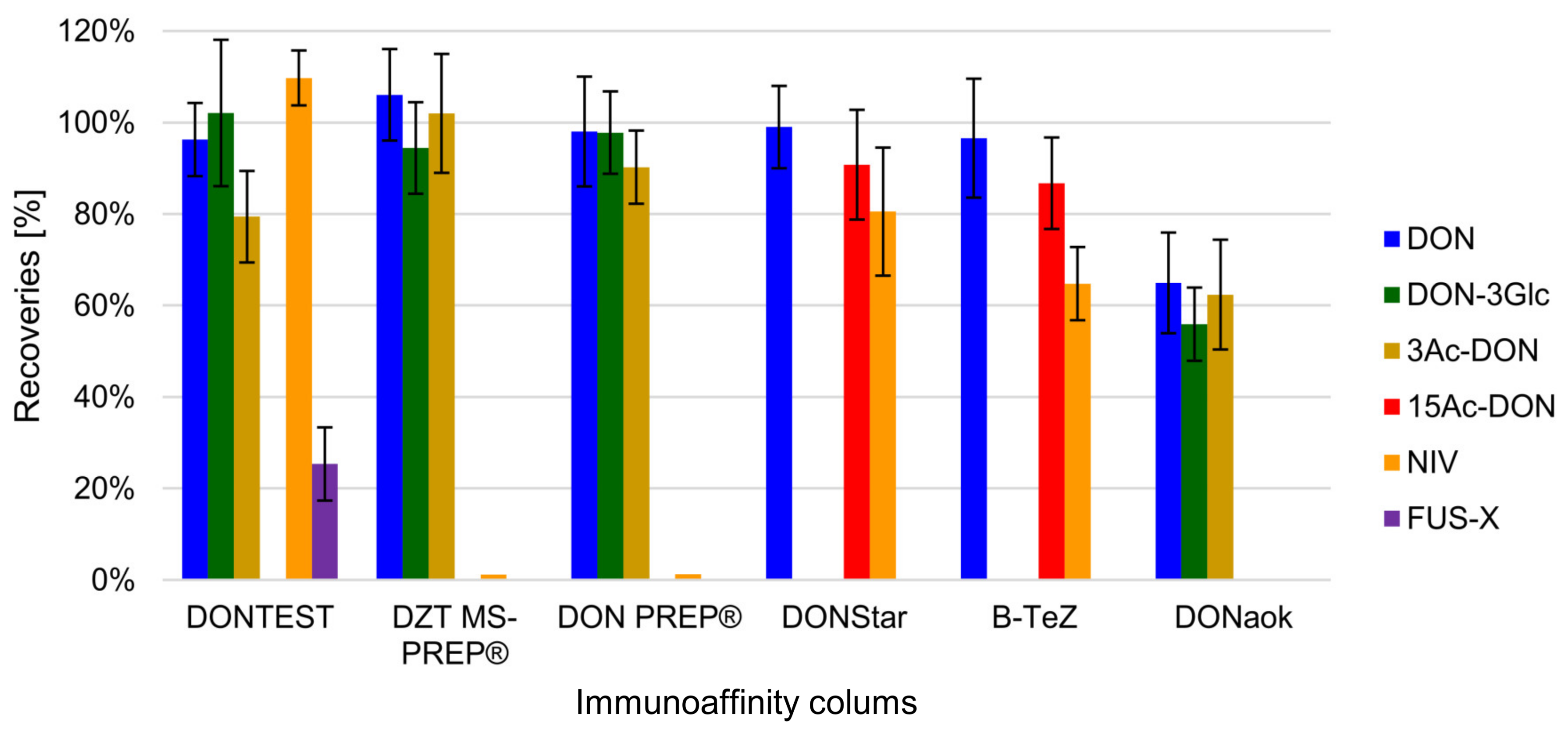
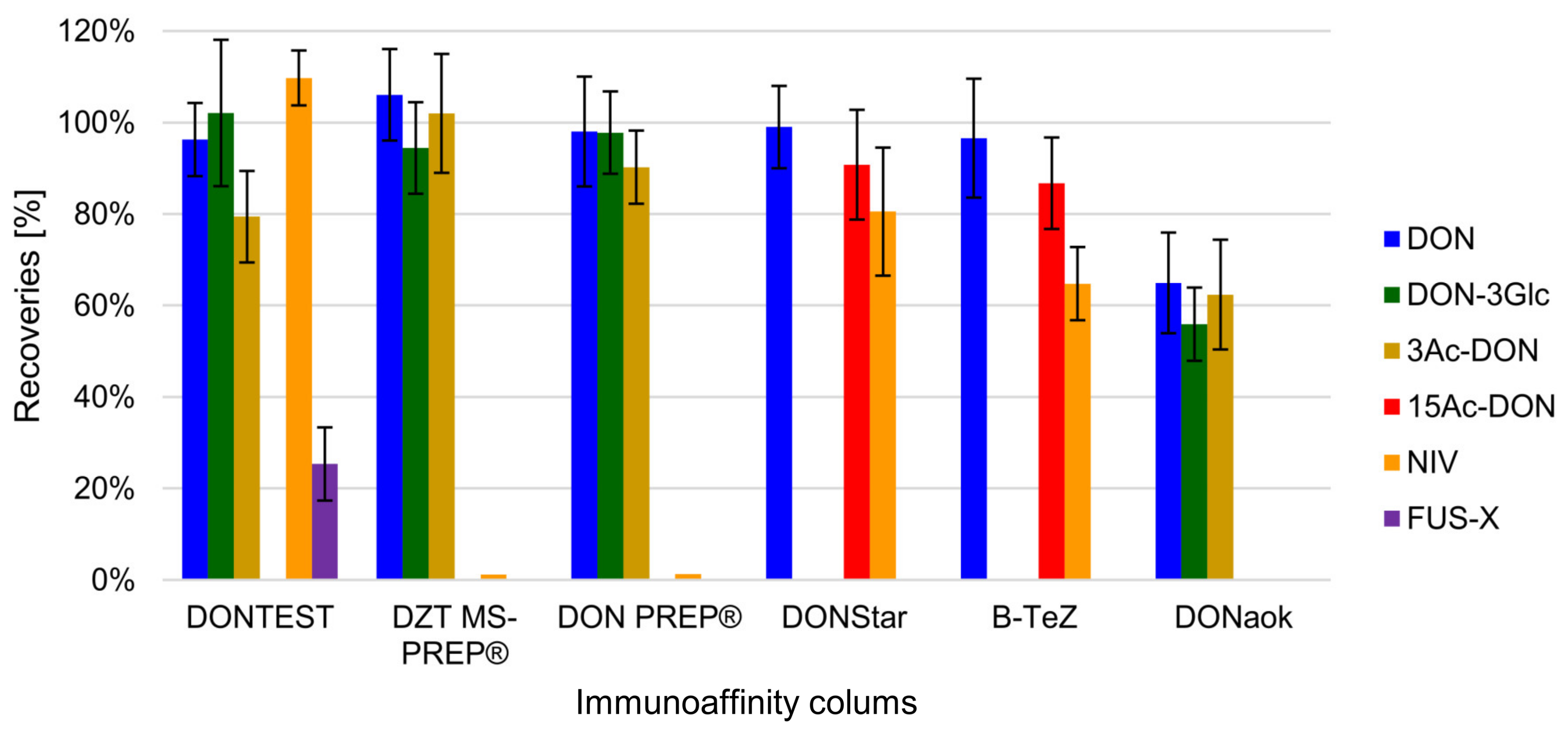
2.3.1. IAC Testing
2.3.2. Comparison of Different Strategies for Sample Preparation and Clean-Up
2.4. Method Validation
2.5. Method Trueness, PT
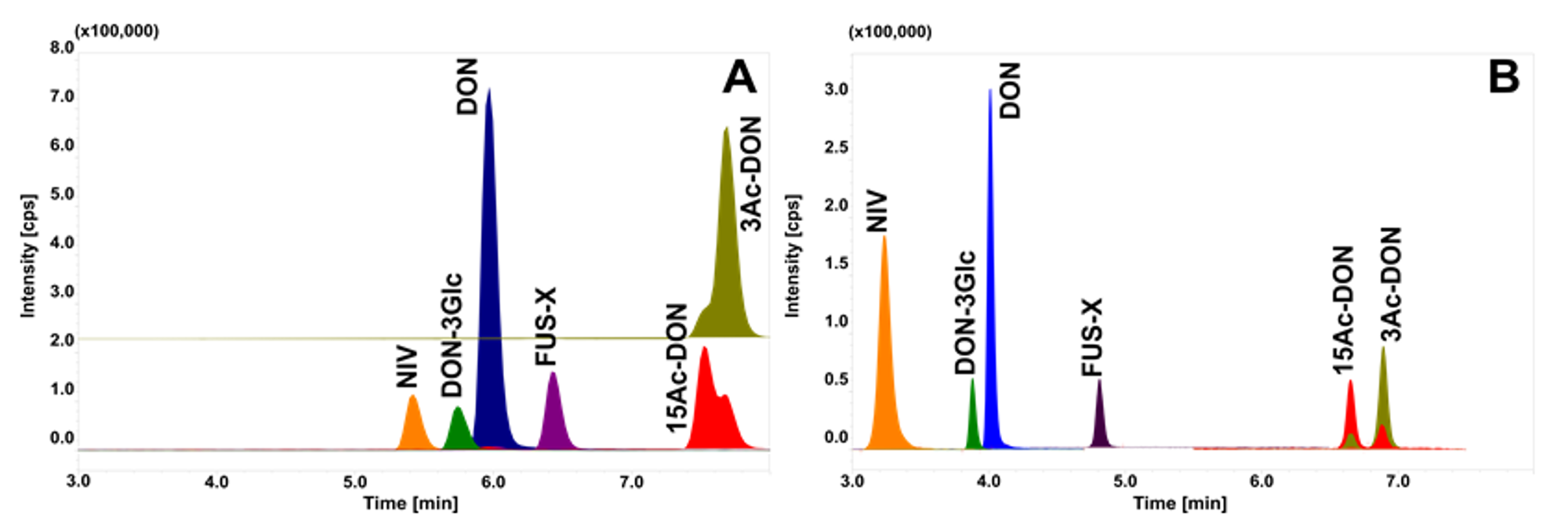
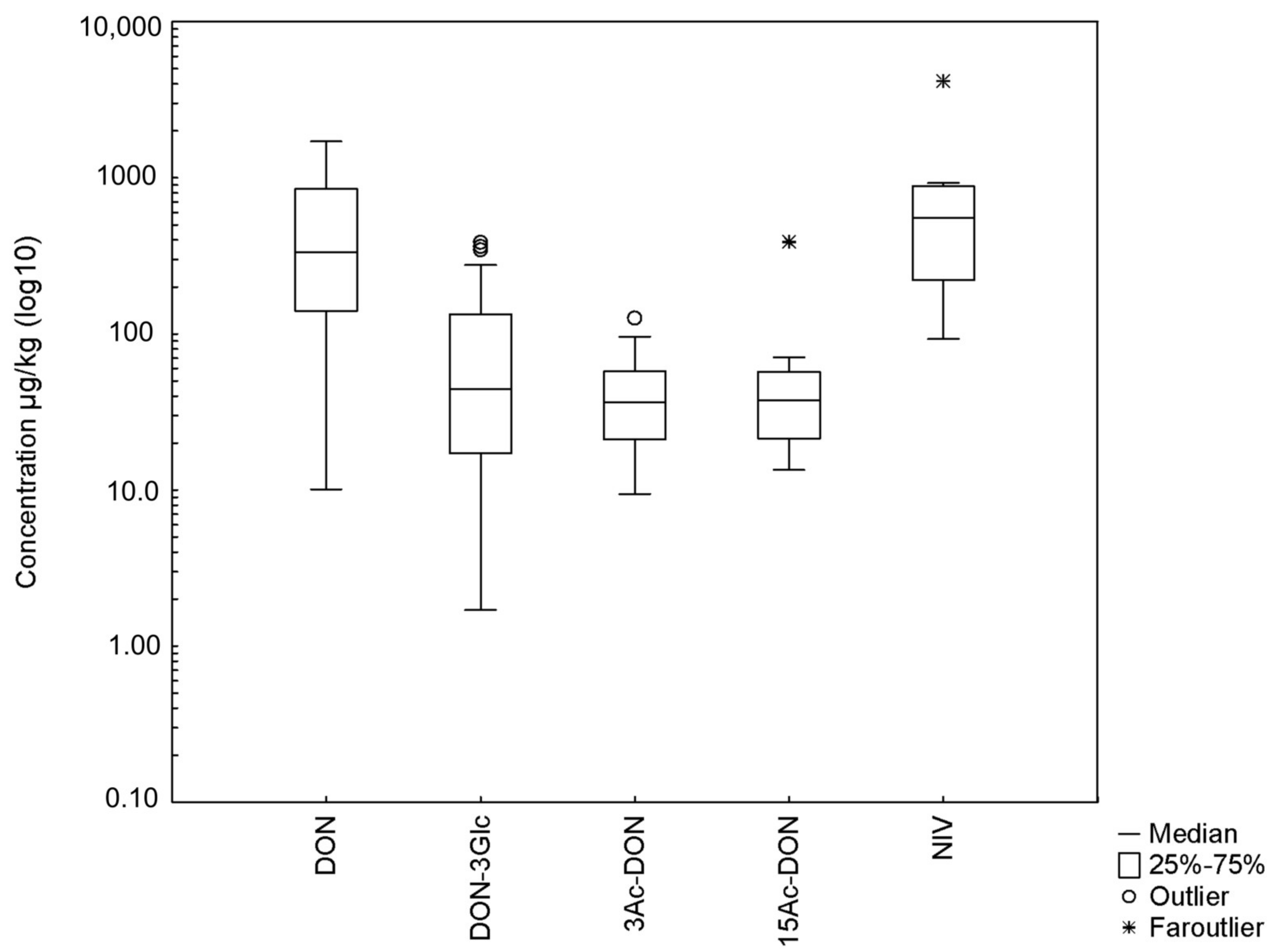
2.6. Application to Real Contaminated Feedstuffs
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Standards
4.2. Mixed Working Solution
4.3. Samples and Reference Materials
4.4. IAC Testing
4.5. Compared Strategies for Sample Preparation and Clean-Up
4.6. LC-MS/MS Analysis
4.7. Method Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Streit, E.; Schatzmayr, G.; Tassis, P.; Tzika, E.; Marin, D.; Taranu, I.; Tabuc, C.; Nicolau, A.; Aprodu, I.; Puel, O.; et al. Current situation of mycotoxin contamination and co-occurrence in animal feed focus on Europe. Toxins 2012, 4, 788–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita-Konishi, Y.; Park, B.J.; Kobayashi-Hattori, K.; Tanaka, T.; Chonan, T.; Yoshikava, K.; Kumagai, S. Effect of Cooking Process on the Deoxynivalenol Content and Its Subsequent Cytotoxicity in Wheat Products. Biosci. Biotechnol. Biochem. 2006, 70, 1764–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, J.J. Deoxynivalenol: Toxicity, mechanisms and animal health risks. Anim. Feed Sci. Technol. 2007, 137, 283–298. [Google Scholar] [CrossRef]
- Rychlik, M.; Humpf, H.U.; Marko, D.; Dänicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; de Saeger, S.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef]
- Freire, L.; Sant’Ana, A.S. Modified mycotoxins: An updated review on their formation, detection, occurrence, and toxic effects. Food Chem. Toxicol. 2018, 111, 189–205. [Google Scholar] [CrossRef]
- Broekaert, N.; Devreese, M.; Demeyere, K.; Berthiller, F.; Michlmayr, H.; Varga, E.; Adam, G.; Meyer, E.; Croubels, S. Comparative in vitro cytotoxicity of modified deoxynivalenol on porcine intestinal epithelial cells. Food Chem. Toxicol. 2016, 95, 103–109. [Google Scholar] [CrossRef]
- European Commission. Commission Recommendation 2006/576/EC of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding; Office of the European Union: Brussels, Belgium, 2006; Volume 229, pp. 7–9. [Google Scholar]
- European Commission. Commision Recommendation 2016/ 1319 of 29 July 2016—Amending Recommendation 2006/ 576/ EC as Regards Deoxynivalenol, Zearalenone and Ochratoxin A in Pet Food; Office of the European Union: Brussels, Belgium, 2016; Volume 73, pp. 58–60. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016H1319&from=EN (accessed on 29 May 2020).
- European Food Safety Authority (EFSA). Scientific Opinion on risks for animal and public health related to the presence of nivalenol in food and feed. EFSA J. 2013, 11, 3262. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Risks to human and animal health related to the presence of deoxynivalenol and its acetylated and modified forms in food and feed. EFSA J. 2017, 15. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA J. 2014, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Rao, Q.; Song, S.; Liu, N.; Han, Z.; Hou, J.; Wu, A. Simultaneous determination of major type B trichothecenes and deoxynivalenol-3-glucoside in animal feed and raw materials using improved DSPE combined with LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 963, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cao, X.; Tao, Y.; Wu, Q.; Pan, Y.; Peng, D.; Liu, Z.; Huang, L.; Wang, Y.; Wang, X.; et al. Development of a liquid chromatography—Tandem mass spectrometry with ultrasound-assisted extraction and auto solid-phase clean-up method for the determination of Fusarium toxins in animal derived foods. J. Chromatogr. A 2013, 1311, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Tanaka, T.; Ishikuro, E.; Horie, M.; Nagayama, T.; Nakajima, M.; Naito, S.; Ohnishi, T.; Sugita-Konishi, Y. Inter-laboratory Study of an LC-MS/MS Method for Simultaneous Determination of Deoxynivalenol and Its Acetylated Derivatives, 3-Acetyl-deoxynivalenol and 15-Acetyl-deoxynivalenol in Wheat. Shokuhin Eiseigaku Zasshi 2013, 54, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Vendl, O.; Berthiller, F.; Crews, C.; Krska, R. Simultaneous determination of deoxynivalenol, zearalenone, and their major masked metabolites in cereal-based food by LC-MS-MS. Anal. Bioanal. Chem. 2009, 395, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Stroka, J. Cross-reactivity features of deoxynivalenol (DON)-targeted immunoaffinity columns aiming to achieve simultaneous analysis of DON and major conjugates in cereal samples. Food Addit. Contam. Part A 2016, 33, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Pascale, M.; Panzarini, G.; Powers, S.; Visconti, A. Determination of Deoxynivalenol and Nivalenol in Wheat by Ultra-Performance Liquid Chromatography/Photodiode-Array Detector and Immunoaffinity Column Cleanup. Food Anal. Methods 2014, 7, 555–562. [Google Scholar] [CrossRef]
- Bryła, M.; Ksieniewicz-Woźniak, E.; Waśkiewicz, A.; Szymczyk, K.; Jędrzejczak, R. Natural Occurrence of Nivalenol, Deoxynivalenol, and Deoxynivalenol-3-Glucoside in Polish Winter Wheat. Toxins 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Trombete, F.; Barros, A.; Vieira, M.; Saldanha, T.; Venâncio, A.; Fraga, M. Simultaneous Determination of Deoxynivalenol, Deoxynivalenol-3-Glucoside and Nivalenol in Wheat Grains by HPLC-PDA with Immunoaffinity Column Cleanup. Food Anal. Methods 2016, 9, 2579–2586. [Google Scholar] [CrossRef] [Green Version]
- De Boevre, M.; van Poucke, C.; Ediage, E.N.; Vanderputten, D.; van Landschoot, A.; de Saeger, S. Ultra-High-Performance Supercritical Fluid Chromatography as a Separation Tool for Fusarium Mycotoxins and Their Modified Forms. J. AOAC Int. 2018, 101, 627–632. [Google Scholar] [CrossRef]
- Righetti, L.; Paglia, G.; Galaverna, G.; Dall’Asta, C. Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research. Toxins 2016, 8, 361. [Google Scholar] [CrossRef]
- De Boevre, M.; di Mavungu, J.D.; Maene, P.; Audenaert, K.; Deforce, D.; Haesaert, G.; Eeckhout, M.; Callebaut, A.; Berthiller, F.; van Peteghem, C.; et al. Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolites in different cereals and cereal-derived food. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; de Saeger, S.; Shi, W.; Li, C.; Zhang, S.; Cao, X.; Shen, J. Determination of deoxynivalenol in cereals by immunoaffinity clean-up and ultra-high performance liquid chromatography tandem mass spectrometry. Methods 2012, 56, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Suman, M.; Bergamini, E.; Catellani, D.; Manzitti, A. Development and validation of a liquid chromatography/linear ion trap mass spectrometry method for the quantitative determination of deoxynivalenol-3-glucoside in processed cereal-derived products. Food Chem. 2013, 136, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Veršilovskis, A.; Geys, J.; Huybrechts, B.; Goossens, E.; de Saeger, S.; Callebaut, A. Simultaneous determination of masked forms of deoxynivalenol and zearalenone after oral dosing in rats by LC-MS/MS. World Mycotoxin J. 2012, 5, 303–318. [Google Scholar] [CrossRef]
- Andrade, P.D.; Dantas, R.R.; de Moura-Alves, T.L.D.; Caldas, E.D. Determination of multi-mycotoxins in cereals and of total fumonisins in maize products using isotope labeled internal standard and liquid chromatography/tandem mass spectrometry with positive ionization. J. Chromatogr. A 2017, 1490, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Bai, B.; Jin, P.; Fan, K.; Guo, W.; Zhao, Z.; Han, Z. Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products. Molecules 2016, 21, 747. [Google Scholar] [CrossRef] [PubMed]
- Radová, Z.; Holadová, K.; Hajšlová, J. Comparison of two clean-up principles for determination of trichothecenes in grain extract. J. Chromatogr. A 1998, 829, 259–267. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; Visconti, A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in cereals and derived products by liquid chromatography-tandem mass spectrometry after multi-toxin immunoaffinity clean up. J. Chromatogr. A 2014, 1354, 139–143. [Google Scholar] [CrossRef]
- Nathanail, A.V.; Sarikaya, E.; Jestoi, M.; Godula, M.; Peltonen, K. Determination of deoxynivalenol and deoxynivalenol-3-glucoside in wheat and barley using liquid chromatography coupled to mass spectrometry: On-line clean-up versus conventional sample preparation techniques. J. Chromatogr. A 2014, 1374, 31–39. [Google Scholar] [CrossRef]
- Berthiller, F.; Dalla’sta, C.; Corradini, R.; Marchelli, R.; Sulyok, M.; Krska, R.; Adam, A.; Schuhmacher, R. Occurrence of deoxynivalenol and its 3-ß-D-glucoside in wheat and maize. Food Addit. Contam. 2009, 26, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekaert, N.; Devreese, M.; de Mil, T.; Fraeyman, S.; de Baere, S.; de Saeger, S.; de Backer, P.; Croubels, S. Development and validation of an LC – MS / MS method for the toxicokinetic study of deoxynivalenol and its acetylated derivatives in chicken and pig plasma. J. Chromatogr. B 2014, 971, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Ohnishi, T.; Kadota, T.; Sugita-Konishi, Y. Development of a Purification Method for Simultaneous Determination of Deoxynivalenol and Its Acetylated and Glycosylated Derivatives in Corn Grits and Corn Flour by Liquid Chromatography–Tandem Mass Spectrometry. J. Food Prot. 2012, 75, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Neuhof, T.; Ganzauer, N.; Koch, M.; Nehls, I. A Comparison of Chromatographic Methods for the Determination of Deoxynivalenol in Wheat. Chromatographia 2009, 69, 1457–1462. [Google Scholar] [CrossRef]
- Slobodchikova, I.; Vuckovic, D. Liquid chromatography—High resolution mass spectrometry method for monitoring of 17 mycotoxins in human plasma for exposure studies. J. Chromatogr. A 2018, 1548, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Goryacheva, I.Y.; de Saeger, S. Immunochemical detection of masked mycotoxins: A short review. World Mycotoxin J. 2012, 5, 281–287. [Google Scholar] [CrossRef]
- Veršilovskis, A.; Huybrecht, B.; Tangni, E.K.; Pussemier, L.; de Saeger, S.; Callebaut, A. Cross-reactivity of some commercially available deoxynivalenol (DON) and zearalenone (ZEN) immunoaffinity columns to DON- and ZEN-conjugated forms and metabolites. Food Addit. Contam. Part A Chem. Anal. Control. Exp. Risk Assess. 2011, 28, 1687–1693. [Google Scholar] [CrossRef]
- Uhlig, S.; Stanic, A.; Hussain, F.; Miles, C.O. Selectivity of commercial immunoaffinity columns for modified forms of the mycotoxin 4-deoxynivalenol (DON). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1061, 322–326. [Google Scholar] [CrossRef]
- Panasiuk, L.; Jedziniak, P.; Pietruszka, K.; Piatkowska, M.; Bocian, L. Frequency and levels of regulated and emerging mycotoxins in silage in Poland. Mycotoxin Res. 2019, 35, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Jedziniak, P.; Panasiuk, Ł.; Pietruszka, K.; Posyniak, A. Multiple mycotoxins analysis in animal feed with LC-MS/MS: Comparison of extract dilution and immunoaffinity clean-up. J. Sep. Sci. 2019, 42, 1240–1247. [Google Scholar] [CrossRef]
- Malachova, A.; Sulyok, M.; Schuhmacher, R.; Berthiller, F.; Hajslova, J.; Veprikova, Z.; Zachariasova, M.; Lattanzio, V.M.T.; de Saeger, S.; di Mavungu, J.D.; et al. Collaborative investigation of matrix effects in mycotoxin determination by high performance liquid chromatography coupled to mass spectrometry. Qual. Assur. Saf. Crops Foods 2013, 5, 91–103. [Google Scholar] [CrossRef]
- Geng, Z.; Yang, D.; Zhou, M.; Zhang, P.; Wang, D.; Liu, F.; Zhu, Y.; Zhang, M. Determination of Deoxynivalenol-3-Glucoside in Cereals by Hydrophilic Interaction Chromatography with Ultraviolet Detection. Food Anal. Methods 2013, 7, 1139–1146. [Google Scholar] [CrossRef]
- Xu, J.J.; Zhou, J.; Huang, B.F.; Cai, Z.X.; Xu, X.M.; Ren, Y.P. Simultaneous and rapid determination of deoxynivalenol and its acetylate derivatives in wheat flour and rice by ultra high performance liquid chromatography with photo diode array detection. J. Sep. Sci. 2016, 39, 2028–2035. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EC) No 401/2006 of 23 February 2006 Laying down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs; Office of the European Union: Brussels, Belgium, 2006; Volume 2006, pp. 12–34. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission regulation (EC) No 152/2009 of 27 January 2009 Laying down the Methods of Sampling and Analysis for the Official Control of Feed; Office of the European Union: Brussels, Belgium, 2009; Volume 36, pp. 1–130. [Google Scholar]
- Haedrich, J.; Wenzl, T.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; Publications Office of the European Union: Luxembourg, 2016. [Google Scholar] [CrossRef]
- European Commission (EC). Guidance Document on Identification of Mycotoxins in Food and Feed; Office of the European Union: Brussels, Belgium, 2017; pp. 1–4. [Google Scholar]
- Magnusson, B.; Näykki, T.; Hovind, H.; Krysell, M.; Sahlin, E. Nordtest, Handbook for Calculation of Measurement Uncertainty in Environmental Laboratories, 4th ed.; NT TR 537; Nordtest: Taastrup, Denmark, 2017; Volume 4. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| REC (%) | ME (%) | Calibration Range(µg/kg) | R2 | Precision (CV, %) | LOD (µg/kg) | LOQ (µg/kg) | U (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RSD | RSDr | ||||||||||||||||
| 0.5 × VL | 1.0 × VL | 1.5 × VL | 0.5 × VL | 1.0 × VL | 1.5 × VL | 0.5 × VL | 1.0 × VL | 1.5 × VL | 0.5 × VL | 1.0 × VL | 1.5 × VL | ||||||
| DON | 104 ± 4 | 93 ± 6 | 99 ± 6 | 82 ± 12 | 101 ± 17% | 87 ± 8 | 90–1800 | 0.999 | 9 | 9 | 11 | 4 | 7 | 6 | 10.1 | 33.3 | 26.0 |
| DON-3Glc | 93 ± 10 | 92 ± 12 | 96 ± 10 | 78 ± 12 | 61 ± 15% | 74 ± 14 | 10–200 | 0.999 | 20 | 8 | 13 | 10 | 15 | 10 | 1.78 | 5.87 | 12.0 |
| 3Ac-DON | 105 ± 12 | 94 ± 6 | 104 ± 10 | 109 ± 7 | 119 ± 20% | 113 ± 7 | 10–200 | 0.999 | 9 | 9 | 9 | 12 | 7 | 10 | 2.43 | 8.02 | 26.0 |
| 15Ac-DON | 97 ± 12 | 92 ± 16 | 90 ± 18 | 115 ± 15 | 120 ± 16% | 111 ± 12 | 10–200 | 0.999 | 4 | 9 | 13 | 22 | 19 | 21 | 5.65 | 18.6 | 25.0 |
| NIV | 106 ± 14 | 97 ± 15 | 94 ± 15 | 96 ± 6 | 85 ± 7% | 89 ± 6 | 90–1800 | 0.999 | 16 | 13 | 24 | 13 | 19 | 17 | 3.30 | 10.9 | 25.0 |
| FUS-X | 101 ± 12 | 93 ± 12 | 96 ± 11 | 93 ± 6 | 104 ± 15% | 88 ± 8 | 50–200 | 0.998 | 6 | 9 | 10 | 8 | 16 | 14 | 15.0 | 49.5 | 35.0 |
| Reference Sample | Matrix | Analyte | Reference Concentration (µg/kg) | Concentration Uncertainty (µg/kg) | Measured Concentration (µg/kg) a | Calculated z-Score |
|---|---|---|---|---|---|---|
| Proficiency Test EURLPT-MP01 | wheat | DON | 570 | ±14.8 | 502 | −0.48 |
| DON-3Glc | 215 | ±22.2 | 217 | −0.04 | ||
| 3Ac-DON | 35.2 | ±2.40 | 32.0 | −0.37 | ||
| maize | DON | 751 | ±20.4 | 729 | −0.12 | |
| DON-3Glc | 35.0 | ±1.48 | 47.5 | 1.43 | ||
| 3Ac-DON | 92.8 | ±3.71 | 90.2 | −0.11 | ||
| 15Ac-DON | 152 | ±11.5 | 159 | 0.18 | ||
| RM M15362D (Chiron) | maize | DON | 1077 | ±73.0 | 993 | --- |
| DON-3Glc | --- | --- | 431 | --- | ||
| 3Ac-DON | --- | --- | 19.0 | --- | ||
| 15Ac-DON | --- | --- | 202 | --- | ||
| NIV | --- | --- | 203 | --- | ||
| RM TET007RM (Fapas) | wheat | DON | 1810 | ±311 | 1956 | --- |
| DON-3Glc | --- | --- | 125 | --- | ||
| RM TET030RM (Fapas) | maize | DON | 1208 | ±117 | 1077 | --- |
| DON-3Glc | --- | --- | 190 | --- | ||
| 15Ac-DON | --- | --- | 121 | --- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panasiuk, Ł.; Jedziniak, P.; Pietruszka, K.; Posyniak, A. Simultaneous Determination of Deoxynivalenol, Its Modified Forms, Nivalenol and Fusarenone-X in Feedstuffs by the Liquid Chromatography–Tandem Mass Spectrometry Method. Toxins 2020, 12, 362. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060362
Panasiuk Ł, Jedziniak P, Pietruszka K, Posyniak A. Simultaneous Determination of Deoxynivalenol, Its Modified Forms, Nivalenol and Fusarenone-X in Feedstuffs by the Liquid Chromatography–Tandem Mass Spectrometry Method. Toxins. 2020; 12(6):362. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060362
Chicago/Turabian StylePanasiuk, Łukasz, Piotr Jedziniak, Katarzyna Pietruszka, and Andrzej Posyniak. 2020. "Simultaneous Determination of Deoxynivalenol, Its Modified Forms, Nivalenol and Fusarenone-X in Feedstuffs by the Liquid Chromatography–Tandem Mass Spectrometry Method" Toxins 12, no. 6: 362. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060362