Molecular Mechanism by Which Cobra Venom Cardiotoxins Interact with the Outer Mitochondrial Membrane


Abstract
:1. Introduction
2. Results
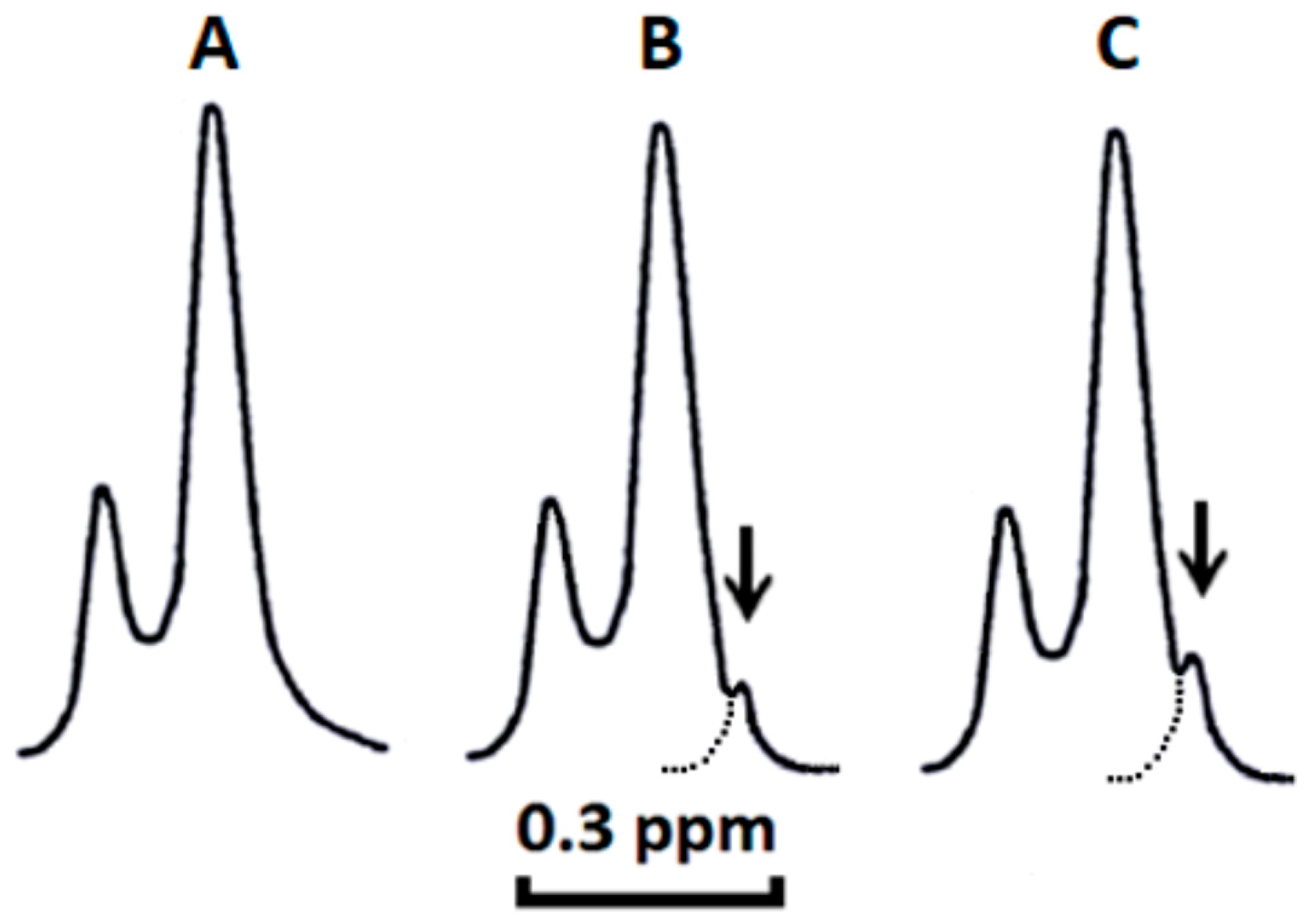
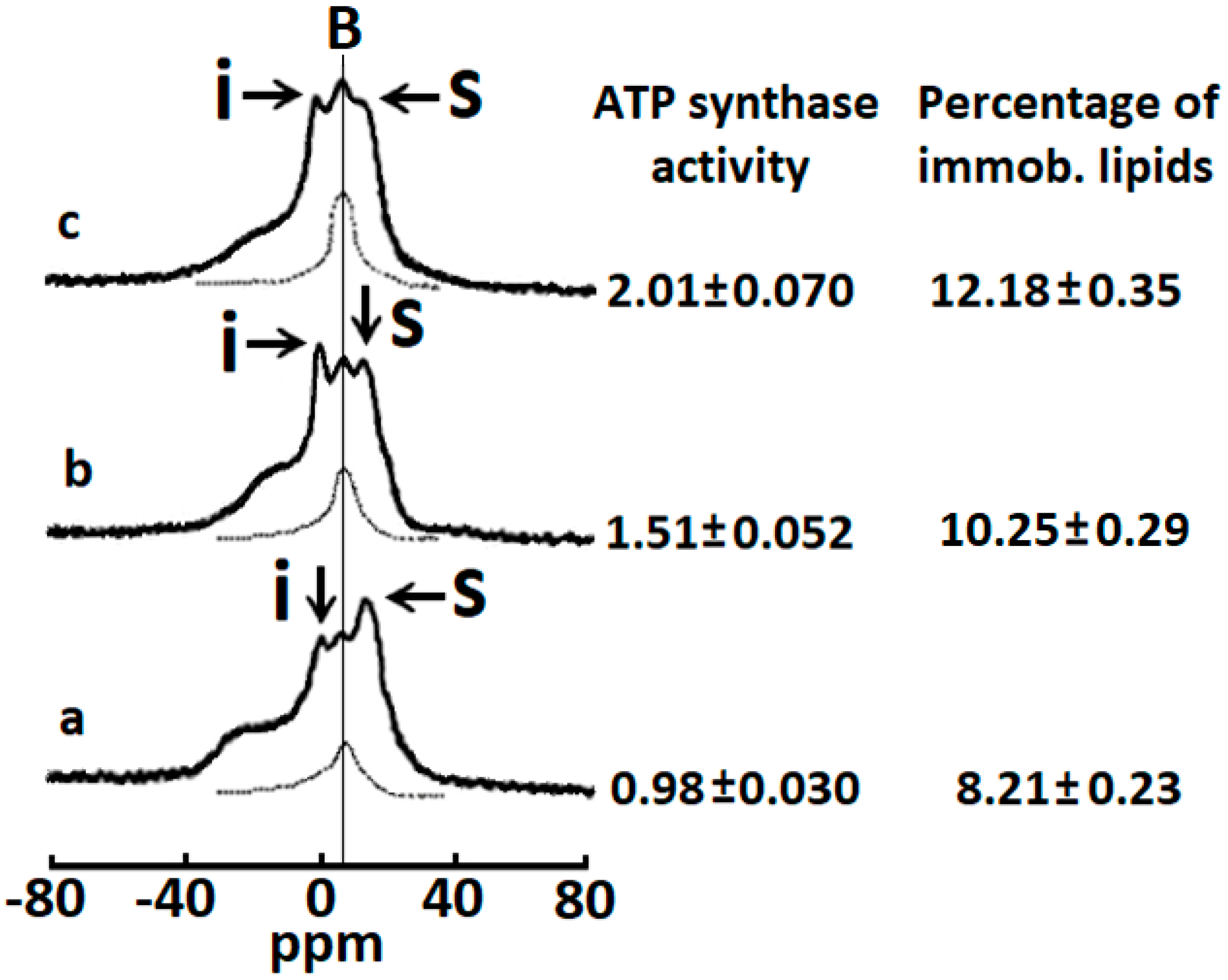
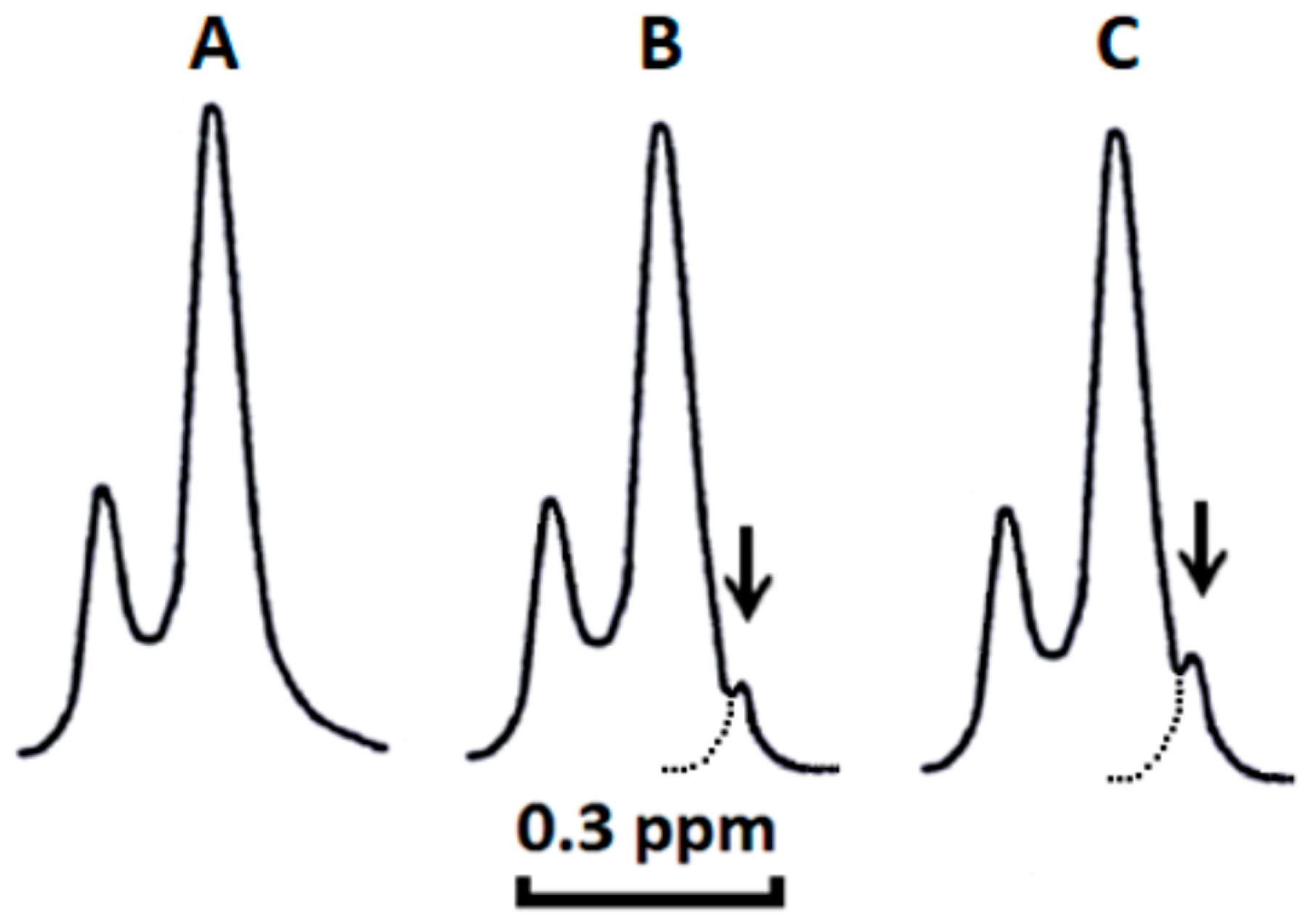
2.1. Cardiotoxins CTI and CTII Induce the Formation of Non-Bilayer Immobilized Phospholipids in Mitochondrial Membranes and Increase ATP-Synthase Activity
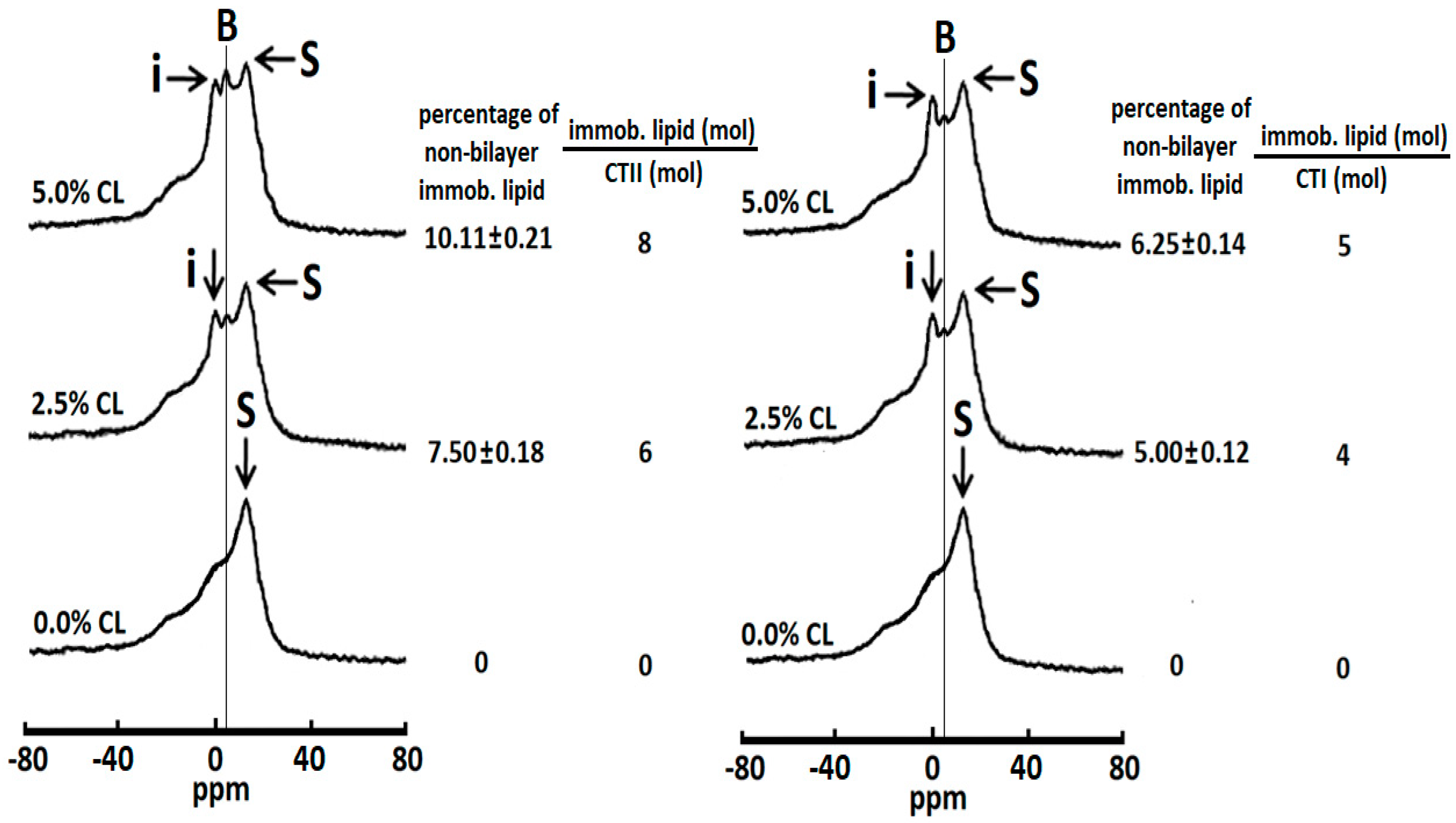
2.2. Cardiotoxins CTII and CTI Induce Formation Non-Bilayer Structures in PC Liposomes Enriched with 2.5% mol and 5.0% mol CL
2.3. Cardiotoxins CTII and CTI Involve PC Molecules in the Formation of Non-Bilayer Immobilized Phospholipid Structures
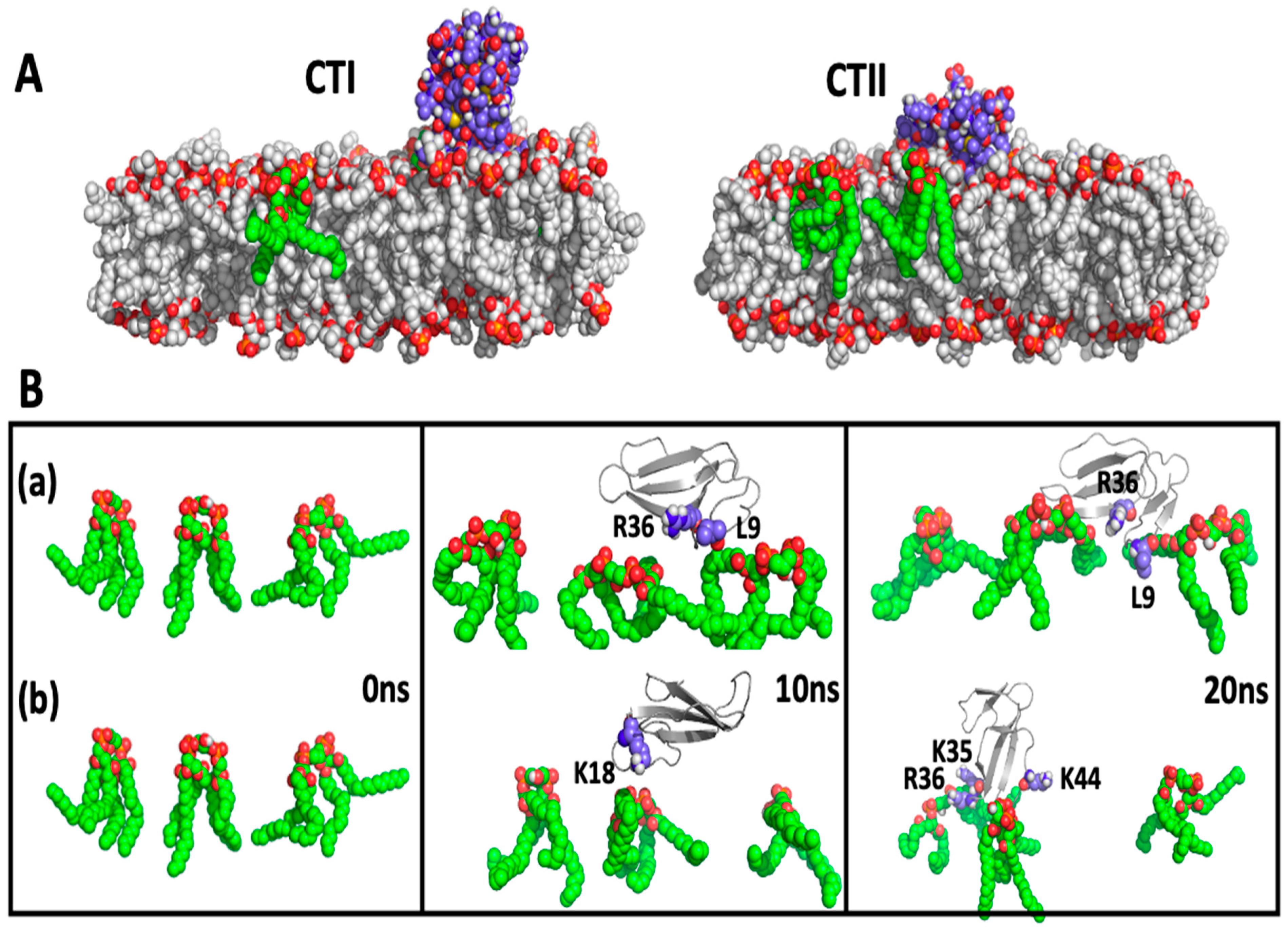
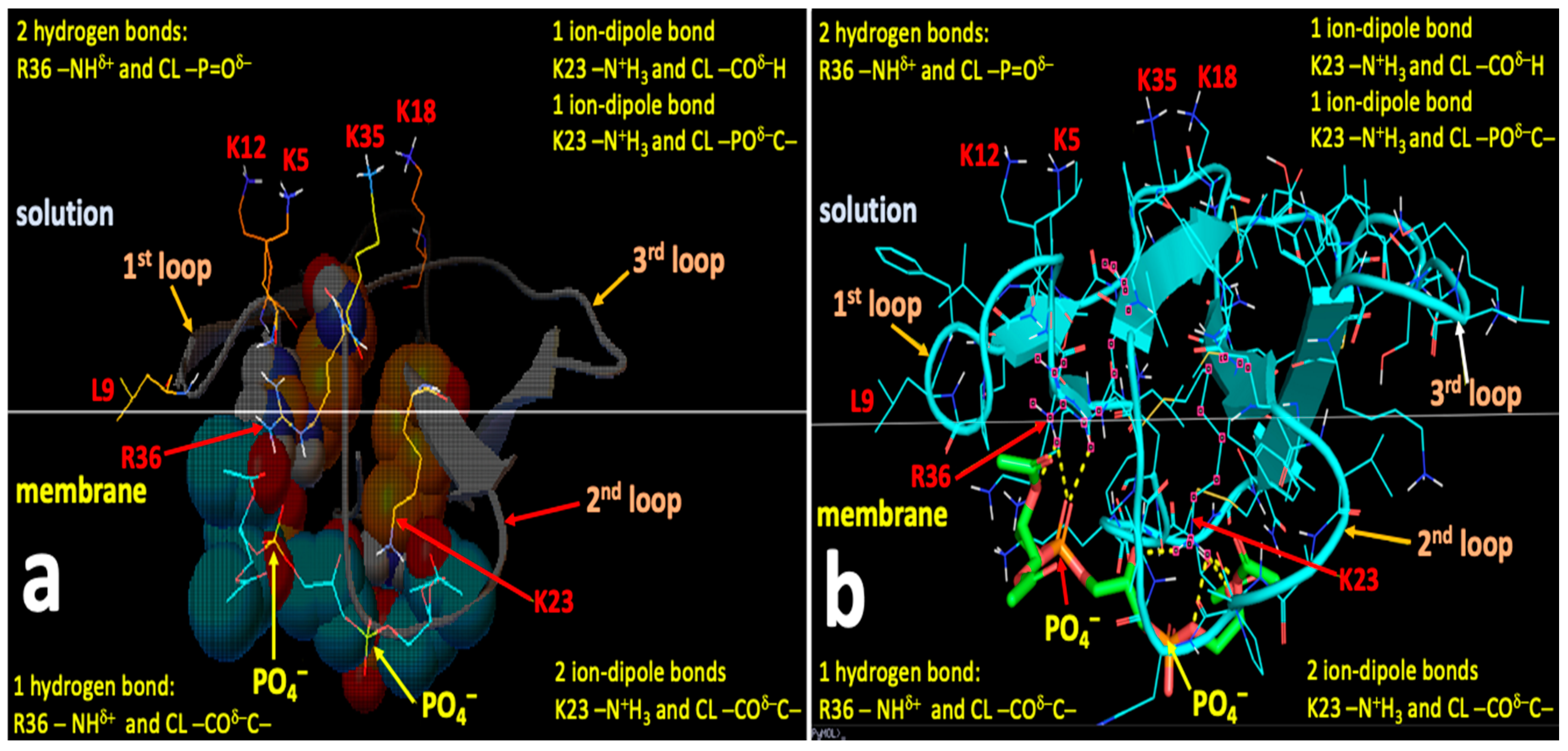
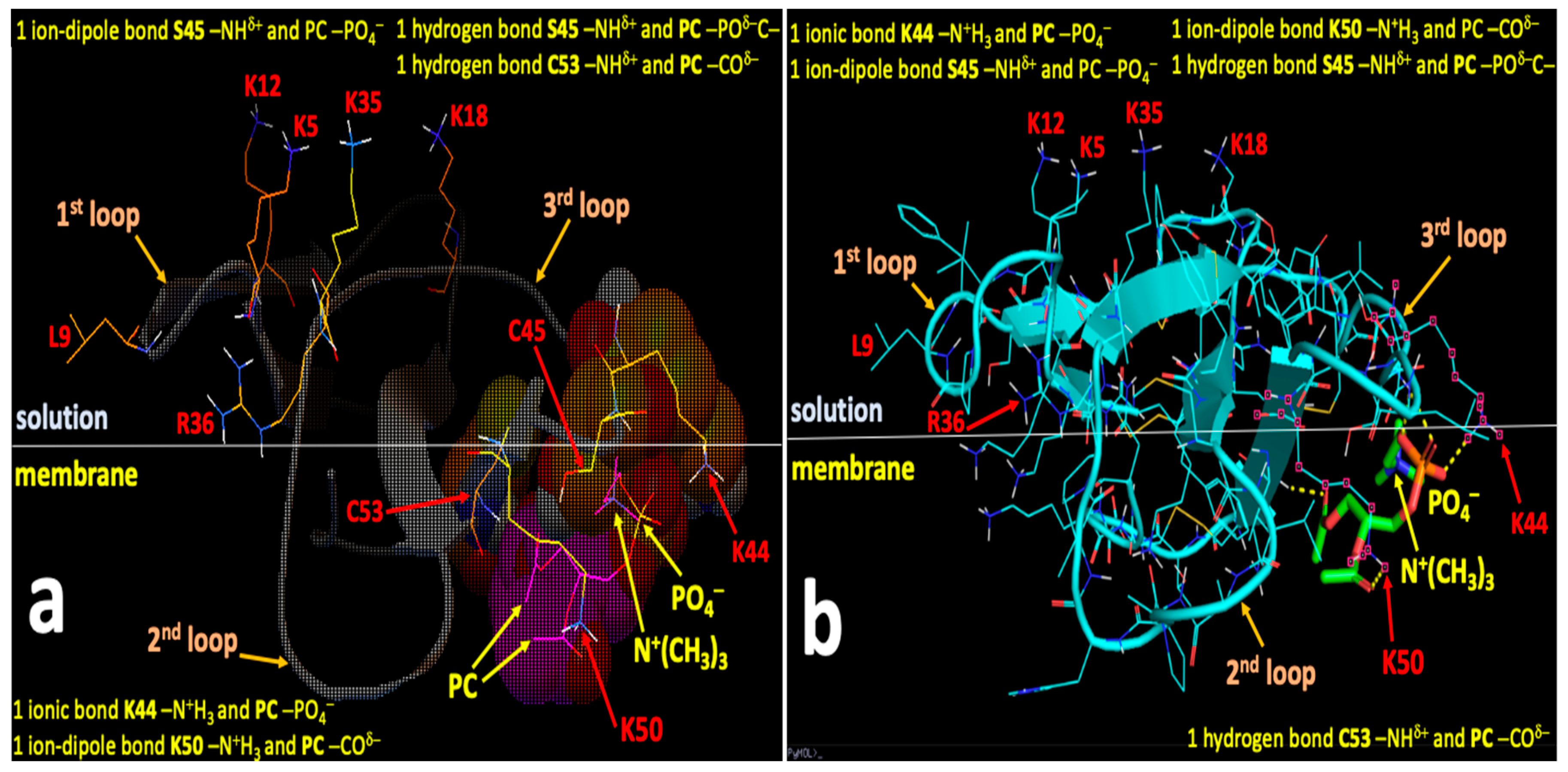
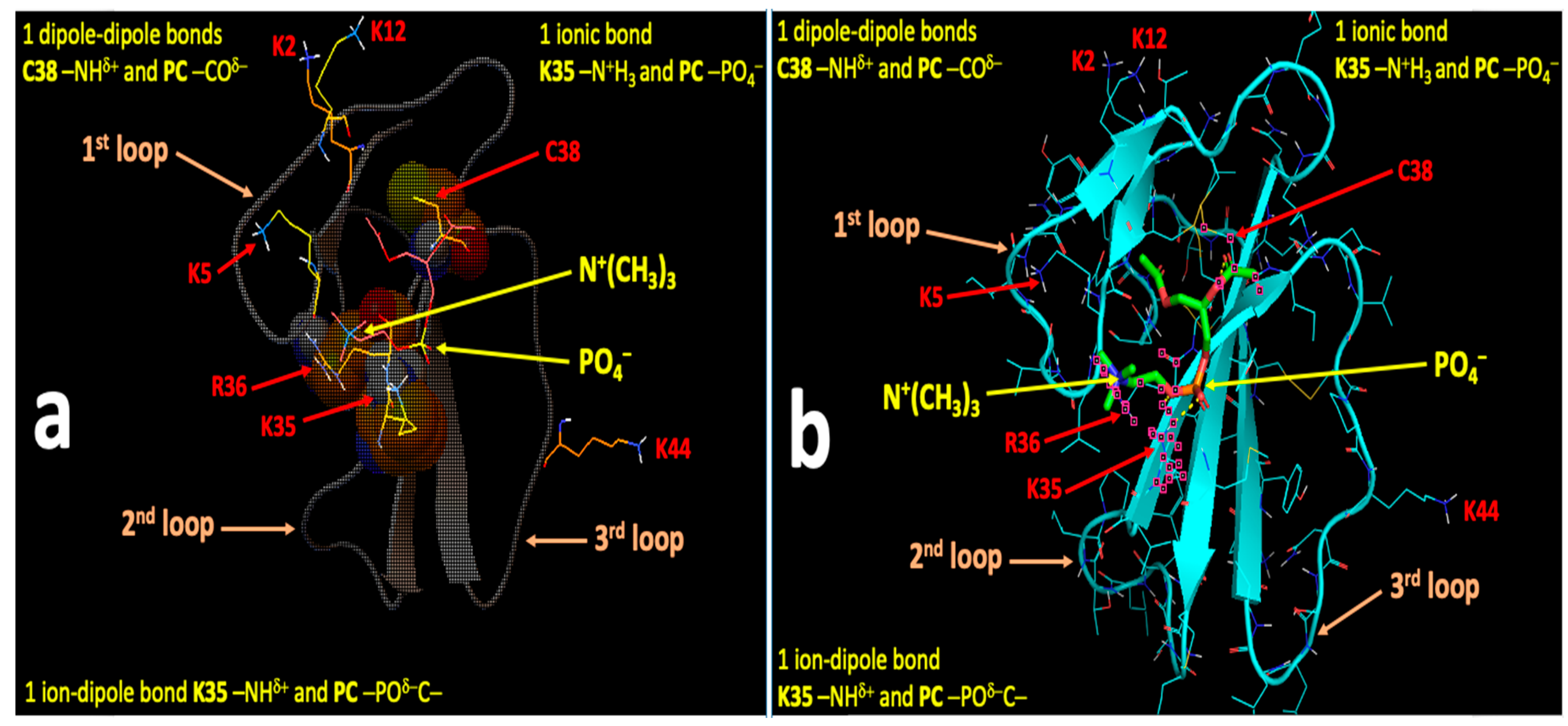
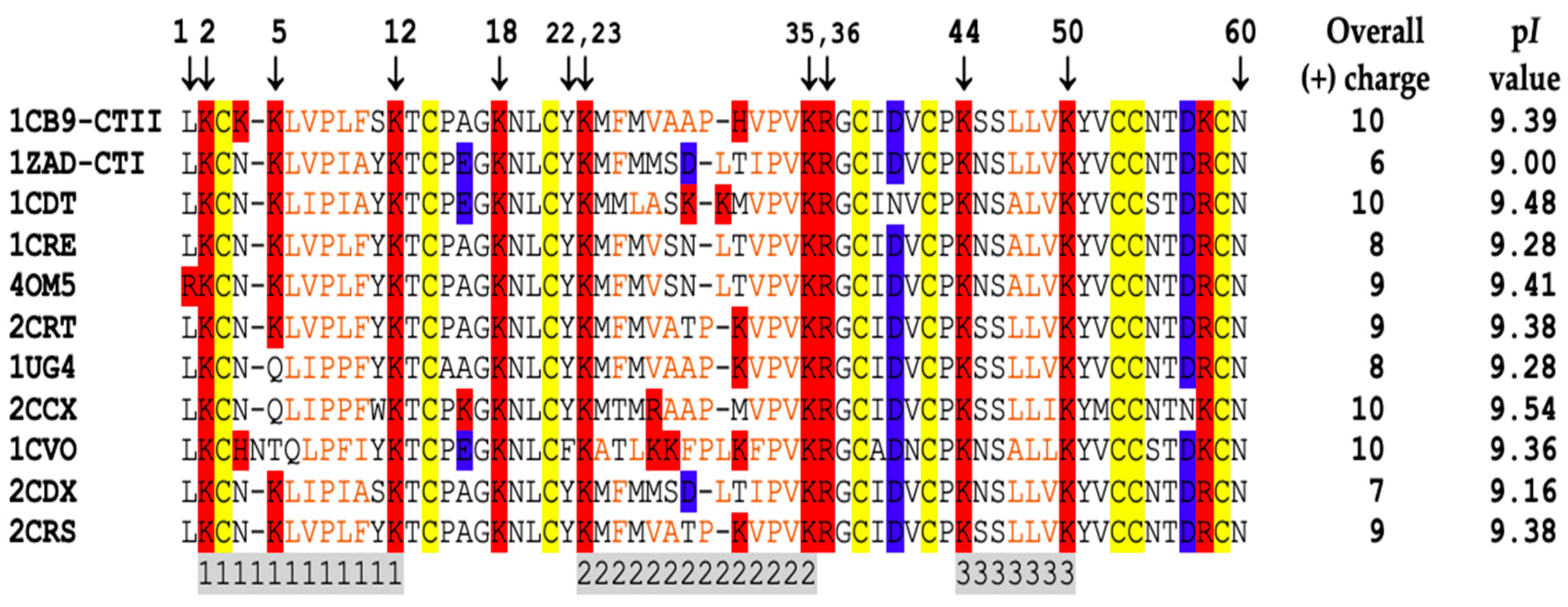
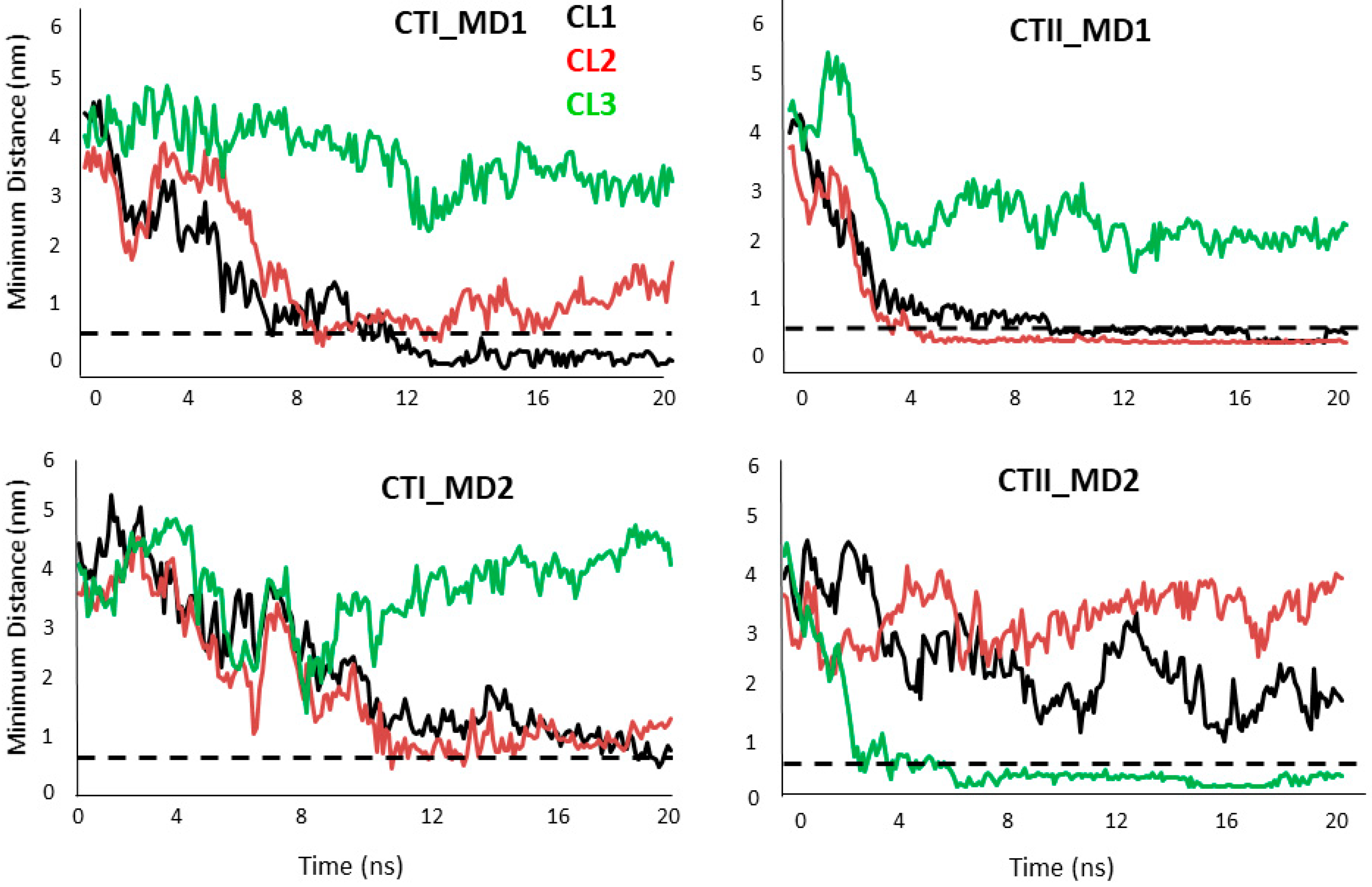
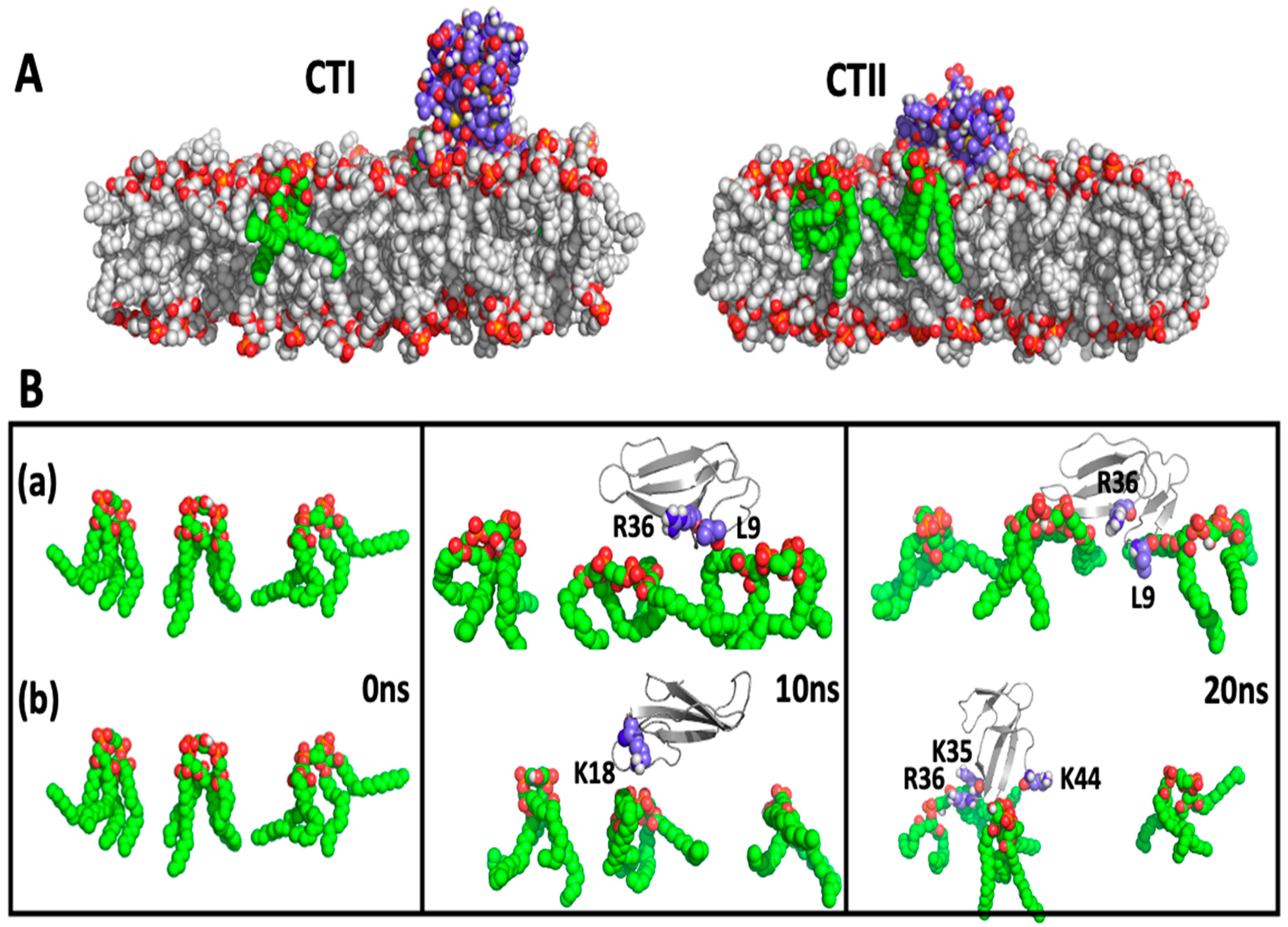
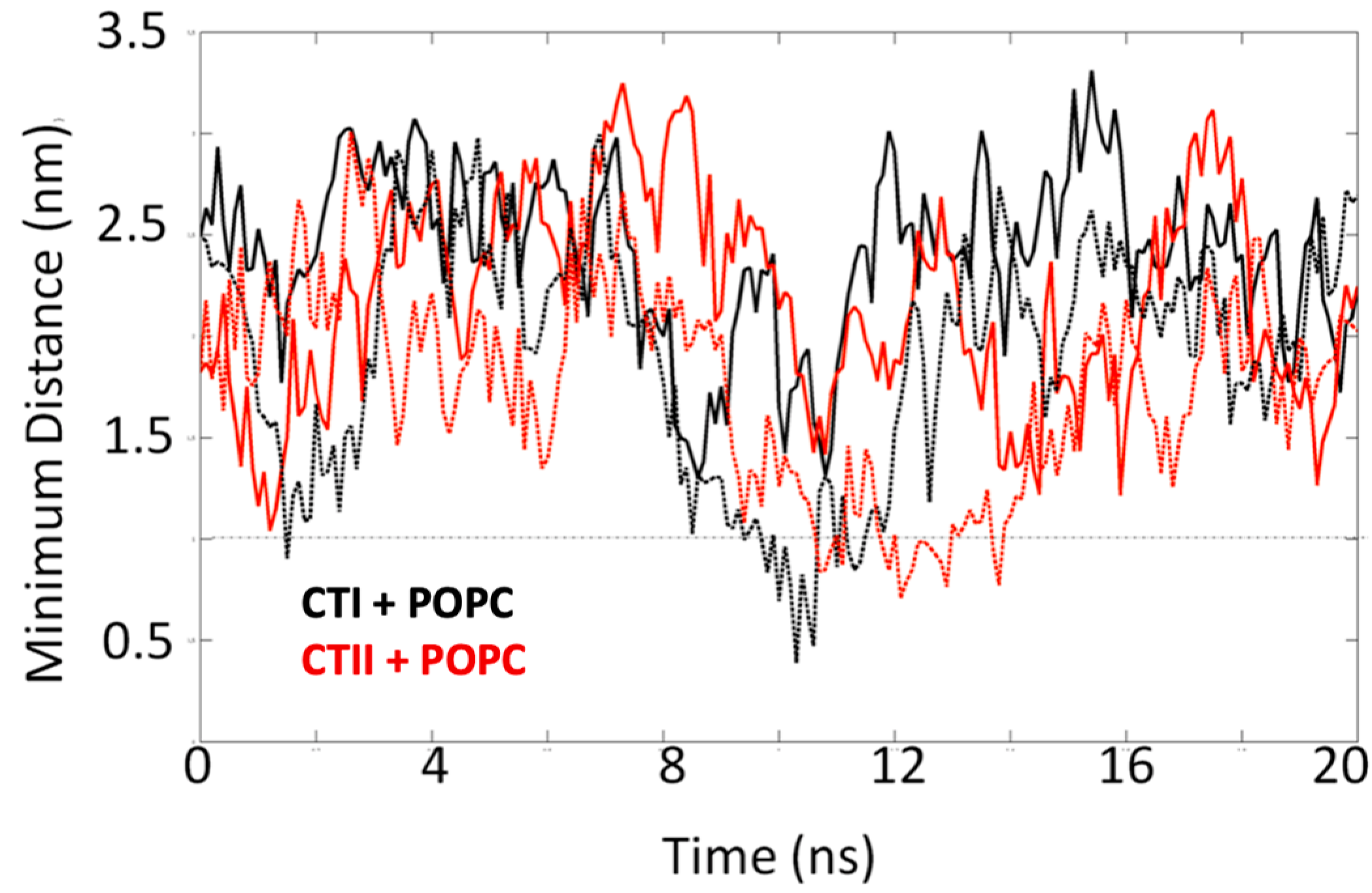
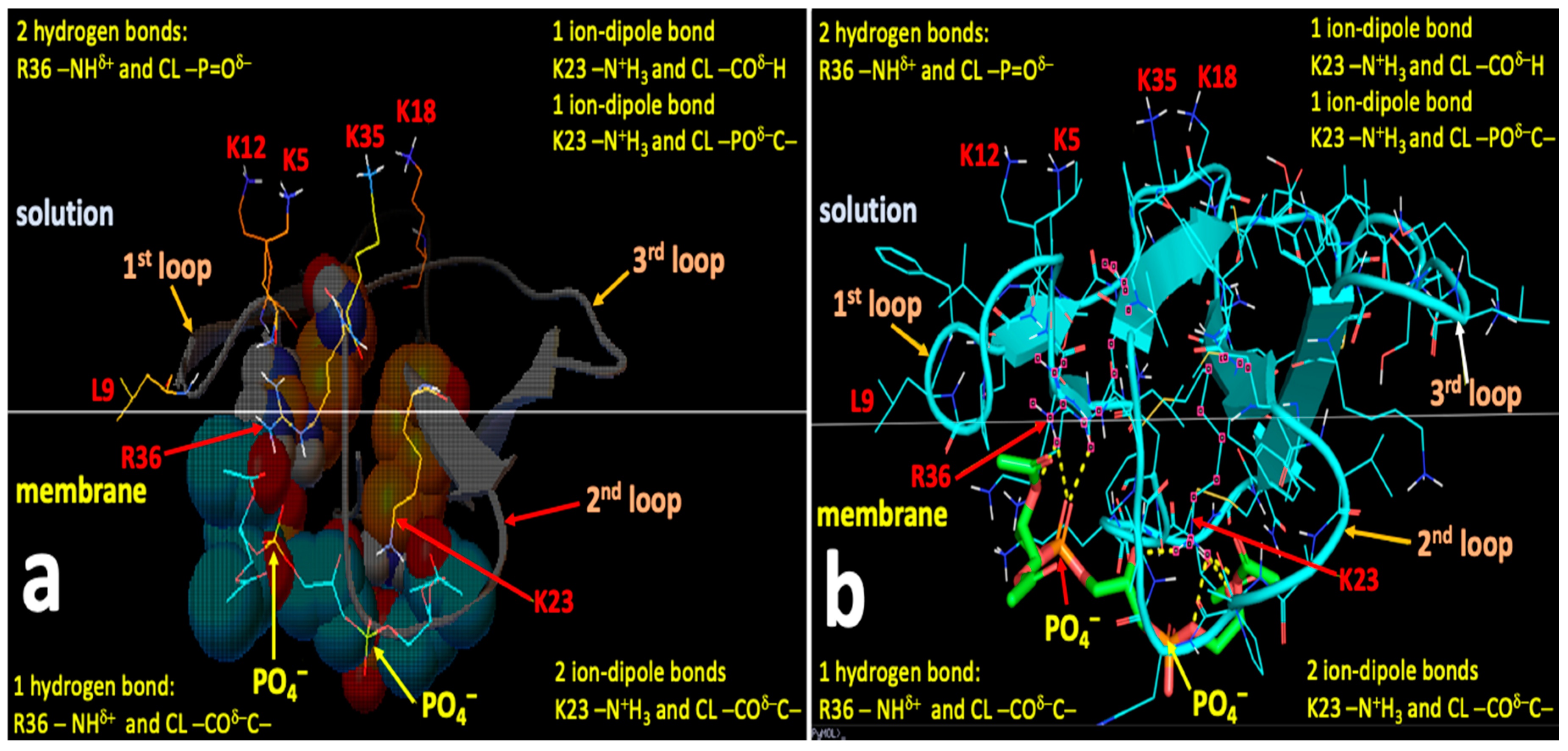
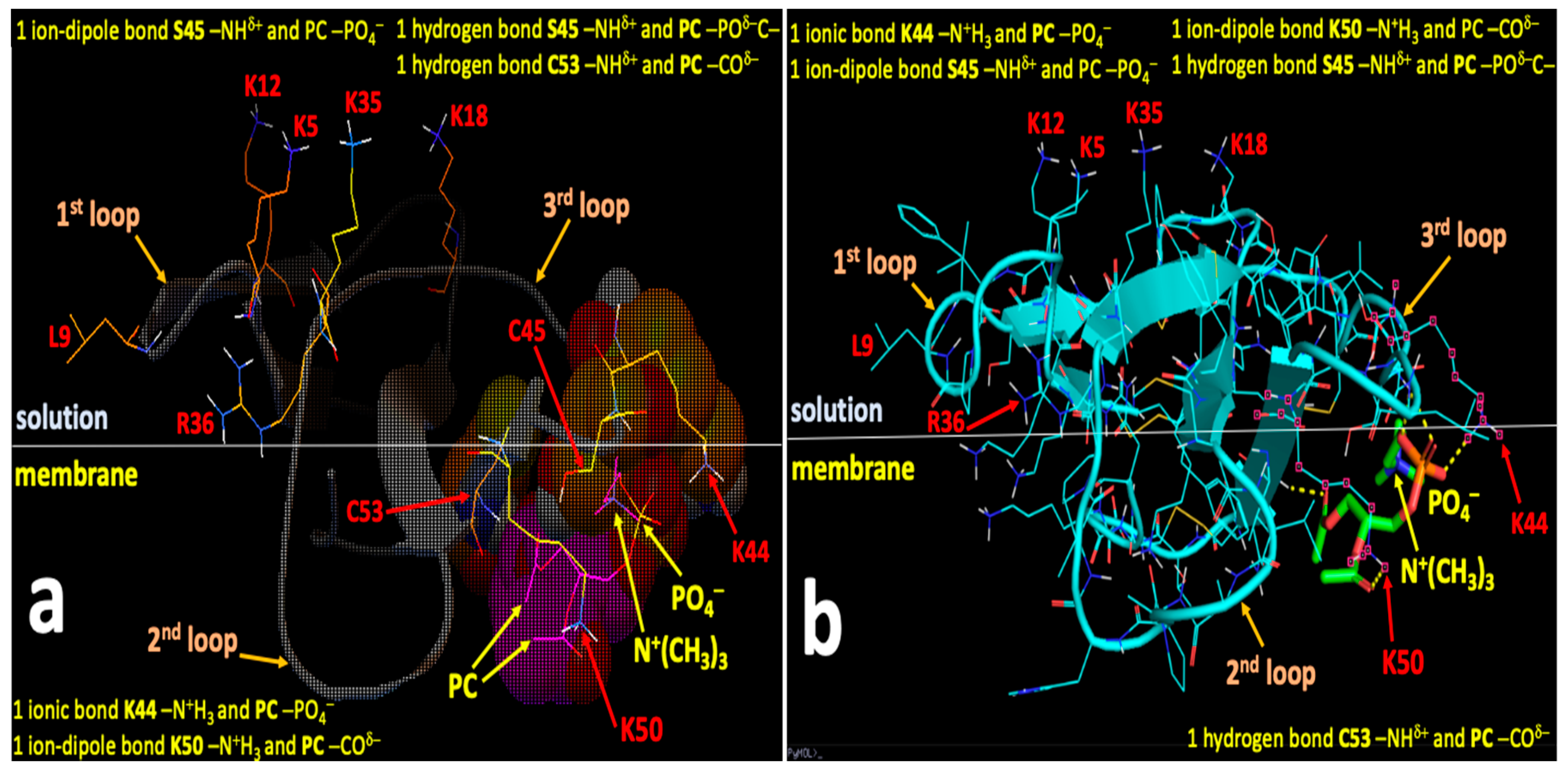
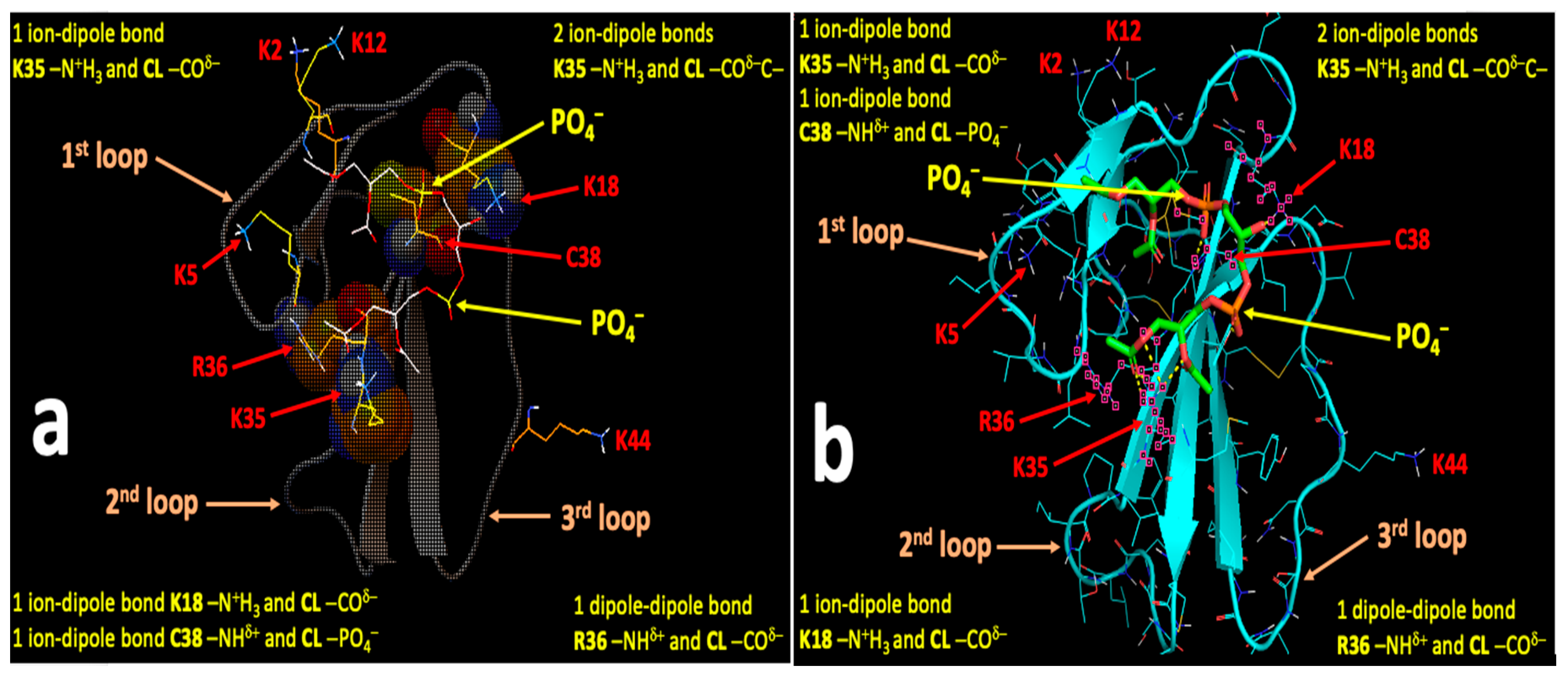
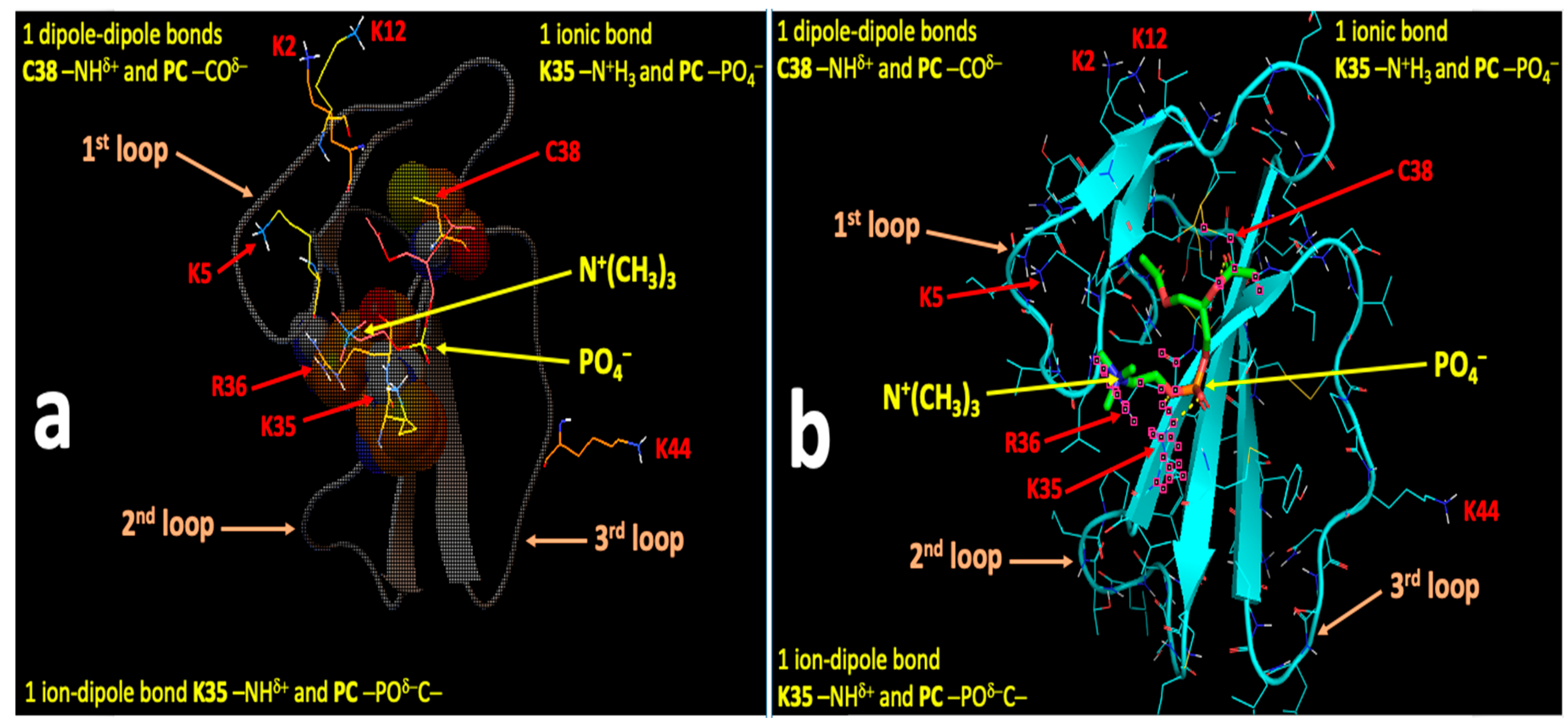
2.4. Computational Modeling Identified Key Residues that Mediate Long-Distance and Short-Distance Interactions of CTI and CTII with the OMM
3. Discussion
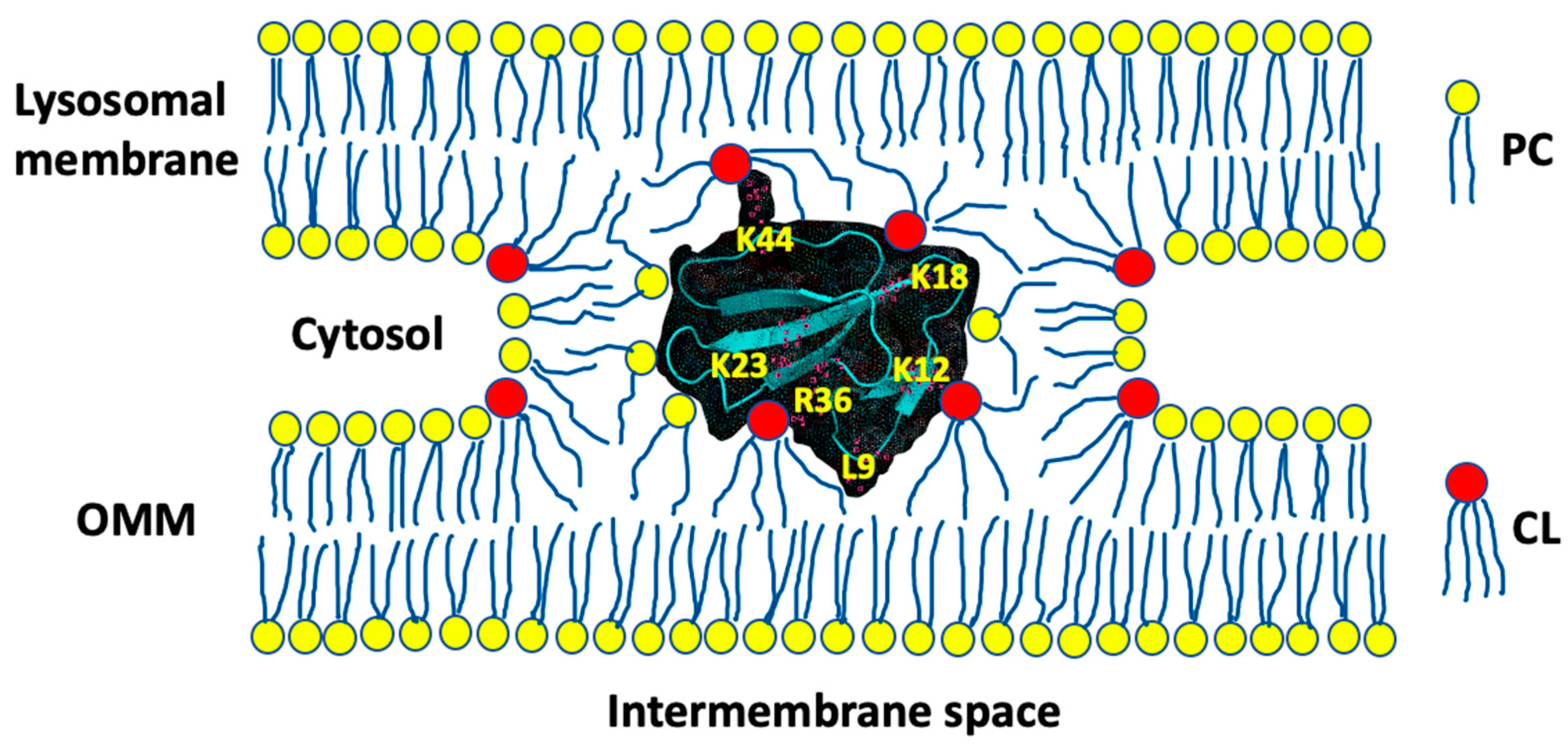
3.1. A Converging Mechanism by Which Snake Venom Cardiotoxins Bind to the OMM
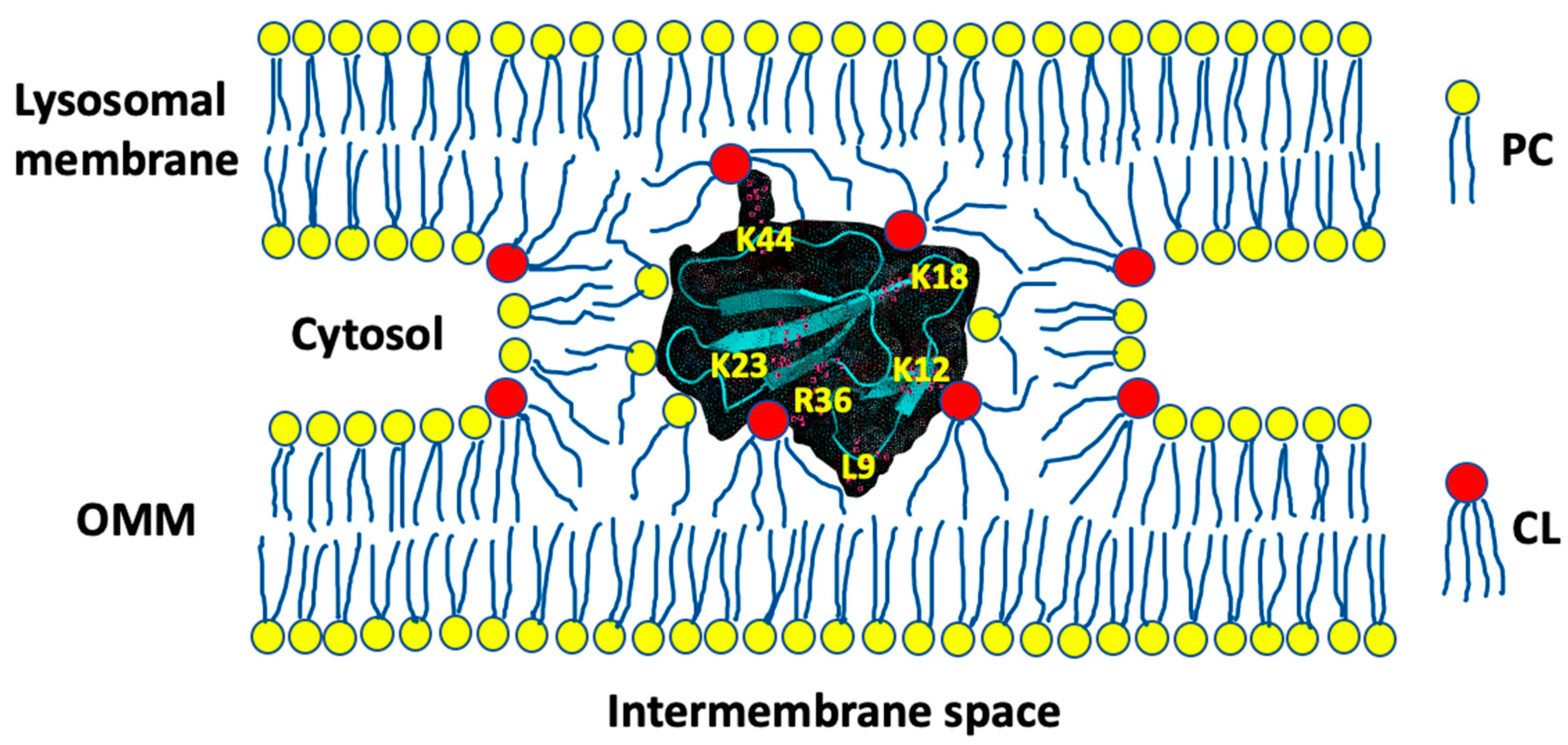
3.2. Proposed Molecular Mechanism by Which CTI and CTII Interact with OMM and Penetrate the OMM
4. Conclusions
5. Materials and Methods
5.1. Molecular Reagents
5.2. Preparation of Isolated Mitochondria
5.3. Preparation of Multilamellar Liposomes
5.4. Preparation of Unilamellar Liposomes
5.5. 31P-NMR and 1H-NMR Studies
5.6. Molecular Dynamics
5.7. Molecular Docking
5.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gasanov, S.E.; Shrivastava, I.H.; Israilov, F.S.; Kim, A.A.; Rylova, K.A.; Zhang, B.; Dagda, R.K. Naja naja oxiana Cobra Venom Cytotoxins CTI and CTII Disrupt Mitochondrial Membrane Integrity: Implications for Basic Three-Fingered Cytotoxins. PLoS ONE 2015, 10, e0129248. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Yaguzhinsky, L.S.; Dagda, R.K. Non-bilayer structures in mitochondrial membranes regulate ATP synthase activity. Biochim. Biophys. Acta Biomembr. 2018, 1860, 586–599. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Dagda, R.K. The Possible Role of Nonbilayer Structures in Regulating ATP Synthase Activity in Mitochondrial Membranes. Biophysics 2016, 61, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Gasanov, S.E.; Kim, A.A.; Dagda, R.K. Possible role of non-bilayer structures in regulating the activity of ATP synthase in mitochondria. Biofizika 2016, 61, 705–710. [Google Scholar]
- Zhang, B.; Li, F.; Chen, Z.; Srivastava, I.H.; Gasanoff, E.S.; Dagda, R.K. Naja mosambica mossambica cobra cardiotoxin targets mitochondria to disrupt mitochondrial membrane structure and function. Toxins 2019, 11, 152. [Google Scholar] [CrossRef] [Green Version]
- Cullis, P.; De Kruijff, B.; Hope, M.; Nayar, R.; Rietveld, A.; Verkleij, A. Structural properties of phospholipids in the rat liver inner mitochondrial membrane. A 31P NMR study. Biochim. Biophys. Acta Biomembr. 1980, 600, 625–635. [Google Scholar] [CrossRef]
- De Kruijff, B.; Cullis, P.; Verkleij, A.; Hope, M.; Van Echteld, C.; Taraschi, T.; Van Hoogevest, P.; Killian, J.; Rietveld, A.; Van der Steen, A. Modulation of lipid polymorphism by lipid-protein interactions. Prog. Protein Lipid Interact. 1985, 89–142. [Google Scholar]
- Aripov, T.F.; Gasanov, S.E.; Salakhutdinov, B.A.; Rozenshtein, I.A.; Kamaev, F.G. Central Asian cobra venom cytotoxins-induced aggregation, permeability and fusion of liposomes. Gen. Physiol. Biophys. 1989, 8, 459–473. [Google Scholar]
- Gasanov, S.E.; Salakhutdinov, B.A.; Aripov, T.F. Formation of nonbilayer structures in phospholipid membranes induced by cationic polypeptides. Biol. Membr. 1990, 7, 1045–1055. [Google Scholar]
- Gasanov, S.E.; Kamaev, F.G.; Salakhutdinov, B.A.; Aripov, T.F. The fusogenic properties of the cytotoxins of cobra venom in a model membrane system. Nauchnye Dokl. Vysshei Shkoly Biol. Nauki 1990, 2, 42–50. [Google Scholar]
- Gasanov, S.E.; Aripov, T.F.; Salakhutdinov, B.A. Intermembrane exchange of lipids induced by cobra venom cytotoxins. Biofizika 1990, 35, 958–962. [Google Scholar] [PubMed]
- Gasanov, S.E.; Vernon, L.P.; Aripov, T.F. Modification of phospholipid membrane structure by the plant toxic peptide Pyrularia thionin. Arch. Biochem. Biophys. 1993, 301, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Gasanov, S.E.; Alsarraj, M.A.; Gasanov, N.E.; Rael, E.D. Cobra venom cytotoxin free of phospholipase A2 and its effect on model membranes and T leukemia cells. J. Membr. Biol. 1997, 155, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Vuister, G.W.; Fogh, R.H.; Hendrickx, P.M.; Doreleijers, J.F.; Gutmanas, A. An overview of tools for the validation of protein NMR structures. J. Biomol. NMR 2014, 58, 259–285. [Google Scholar] [CrossRef]
- Daum, G. Lipids of mitochondria. Biochim. Biophys. Acta 1985, 822, 1–42. [Google Scholar] [CrossRef]
- Levtsova, O.V.; Antonov, M.Y.; Mordvintsev, D.Y.; Utkin, Y.N.; Shaitan, K.V.; Kirpichnikov, M.P. Steered molecular dynamics simulations of cobra cytotoxin interaction with zwitterionic lipid bilayer: No penetration of loop tips into membranes. Comput. Biol. Chem. 2009, 33, 29–32. [Google Scholar] [CrossRef]
- Konshina, A.G.; Boldyrev, I.A.; Utkin, Y.N.; Omel’kov, A.V.; Efremov, R.G. Snake cytotoxins bind to membranes via interactions with phosphatidylserine head groups of lipids. PLoS ONE 2011, 6, e19064. [Google Scholar] [CrossRef] [Green Version]
- Konshina, A.G.; Boldyrev, I.A.; Omelkov, A.V.; Utkin, Y.N.; Efremov, R.G. Anionic lipids: Determinants of binding cytotoxins from snake venom on the surface of cell membranes. Acta Nat. 2010, 2, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Grishin, E.V.; Sukhikh, A.P.; Adamovich, T.B.; Ovchinnikov, Y.A. The isolation and sequence determination of a cytotoxin from the venom of the Middle-Asian cobra Naja naja oxiana. FEBS Lett. 1974, 48, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Yukelson, L.Y.; Sadikov, E.; Sorokin, V.M. Isolation and characteristics of a “direct” haemolytic factor of Central Asian cobra venom. Biokhimiya 1974, 39, 310–313. [Google Scholar]
- Feofanov, A.V.; Sharonov, G.V.; Astapova, M.V.; Rodionov, D.I.; Utkin, Y.N.; Arseniev, A.S. Cancer cell injury by cytotoxins from cobra venom is mediated through lysosomal damage. Biochem. J. 2005, 390, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ming, W.; Tang, Y.; Zhou, S.; Kong, T.; Dong, W. The anticancer effect of cytotoxin 1 from Naja atra Cantor venom is mediated by a lysosomal cell death pathway involving lysosomal membrane permeabilization and cathepsin B release. Am. J. Chin. Med. 2013, 41, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Chiou, Y.L.; Kao, P.H.; Lin, S.R.; Chang, L.S. Taiwan cobra cardiotoxins induce apoptotic death of human neuroblastoma SK-N-SH cells mediated by reactive oxygen species generation and mitochondrial depolarization. Toxicon 2008, 51, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Gasanov, S.E.; Dagda, R.K.; Rael, E.D. Snake venom cytotoxins, phospholipase A2s, and Zn2+-dependent metalloproteinases: Mechanisms of action and pharmacological relevance. J. Clin. Toxicol. 2014, 4, 1000181. [Google Scholar] [CrossRef] [Green Version]
- Wi, S.; Kim, C. Pore structure, thinning effect, and lateral diffusive dynamics of oriented lipid membranes interacting with antimicrobial peptide protegrin-1: 31P and 2H solid-state NMR study. J. Phys. Chem. B 2008, 112, 11402–11414. [Google Scholar] [CrossRef]
- Wang, C.H.; Wu, W.G. Amphiphilic beta-sheet cobra cardiotoxin targets mitochondria and disrupts its network. FEBS Lett. 2005, 579, 3169–3174. [Google Scholar] [CrossRef]
- Chen, K.C.; Lin, S.R.; Chang, L.S. Involvement of mitochondrial alteration and reactive oxygen species generation in Taiwan cobra cardiotoxin-induced apoptotic death of human neuroblastoma SK-N-SH cells. Toxicon 2008, 52, 361–368. [Google Scholar] [CrossRef]
- Aripov, T.F.; Gasanov, S.E.; Salakhutdinov, B.A.; Sadykov, A.S. Interaction of cobra venom cytotoxin with oriented phospholipid multi-bilayers. Dokl. Akad. Nauk SSSR 1986, 288, 728–730. [Google Scholar]
- Gasanov, S.E.; Rael, E.D.; Martinez, M.; Baeza, G.; Vernon, L.P. Modulation of phospholipase A2 activity by membrane-active peptides on liposomes of different phospholipid composition. Gen. Physiol. Biophys. 1994, 13, 275–286. [Google Scholar]
- Gasanov, S.E.; Gasanov, N.E.; Rael, E.D. Phospholipase A2 and cobra venom cytotoxin Vc5 interactions and membrane structure. Gen. Physiol. Biophys. 1995, 14, 107–123. [Google Scholar] [PubMed]
- Gasanov, S.E.; Gasanov, E.E. An asymmetric enlargement of the monolayer surfaces mechanism of membrane fusion. J. Biol. Phys. 1994, 19, 235–242. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Aripov, T.F.; Gasanov, E.E. Study on Structure of Phospholipid Membranes Modified by Membrane-Active Polypeptides; Nuclear Physics Institute UzSSR Academy of Sciences: Tashkent, USSR, 1988. [Google Scholar]
- Segal, N.K.; Gasanov, S.E.; Palamarchuk, L.A.; Ius’Kovich, A.K.; Kolesova, G.M.; Mansurova, S.E.; Iaguzhinskii, L.S. Mitochondrial proteolipids. Biokhimiia 1993, 58, 1812–1819. [Google Scholar]
- Cunningham, C.C.; George, D.T. The relationship between the bovine heart mitochondrial adenosine triphosphatase, lipophilic compounds, and oligomycin. J. Biol. Chem. 1975, 250, 2036–2044. [Google Scholar]
- Rogers, G.W.; Brand, M.D.; Petrosyan, S.; Ashok, D.; Elorza, A.A.; Ferrick, D.A.; Murphy, A.N. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 2011, 6, e21746. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.; Leeuwenburgh, C. Method for measuring ATP production in isolated mitochondria: ATP production in brain and liver mitochondria of Fischer-344 rats with age and caloric restriction. Am. J. Phys. Regul. Integr. Comp. Physiol. 2003, 285, R1259–R1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.C.; Ekiel, I.H. Phosphorus-31 NMR of Phospholipids in Membranes. In Phosphorous-31 NMR: Principles and Applications; Academic Press: Orlando, FL, USA, 1984; pp. 447–475. [Google Scholar]
- Peitsch, M.C. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans. 1996, 24, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Soares, T.A.; Van Der Vegt, N.F.A.; Van Gunsteren, W.F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Van Aalten, D.M.F.; Bywater, R.; Findlay, J.B.C.; Hendlich, M.; Hooft, R.W.W.; Vriend, G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.; Khaniya, U.; Chan, C.K.; Dehez, F.; Shekhar, M.; Gunner, M.R.; Sazanov, L.; Chipot, C.; Singharoy, A. Charge transfer and chemo-mechanical coupling in respiratory complex I. J. Am. Chem. Soc. 2020, 142, 9220–9230. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiotoxin and Liposomes System | Percentage of Non-Bilayer Immobilized PC Molecules | Immobilized PC over CTI or CTII Molar Ratio |
|---|---|---|
| CTI + PC | 0 | 0 |
| CTI + PC + 2.5 mol% CL | 2.56 ± 0.14 | 2 |
| CTI + PC + 5.0 mol% CL | 2.58 ± 0.15 | 2 |
| CTII + PC | 0 | 0 |
| CTII + PC + 2.5 mol% CL | 5.26 ± 0.29 | 4 |
| CTII + PC + 5.0 mol% CL | 5.27 ± 0.29 | 4 |
| Amino Acid Residues on the Molecular Surfaces of CTII and CTI that Bind to CL and PC | ||||
|---|---|---|---|---|
| # | CTII + CL | CTII + PC | CTI + CL | CTI + PC |
| 1 | K23, R36 | K44, S45, K50, C53 | K18, K35, R36, C38 | K35, C38 |
| 2 | K23 | K18, K35, C38 | K12, K35 | Y22, R36, C38 |
| 3 | K5, K12, K35 | K35 | L9, Y22, C38 | |
| 4 | K5, K18, K35 | |||
| Total number of binding sites for CL and PC available on CTII | Total number of binding sites for CL and PC available on CTI | |||
| 7 (4 CL + 3 PC) | 4 (3 CL + 1 PC) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Shrivastava, I.H.; Hanlon, P.; Dagda, R.K.; Gasanoff, E.S. Molecular Mechanism by Which Cobra Venom Cardiotoxins Interact with the Outer Mitochondrial Membrane. Toxins 2020, 12, 425. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070425
Li F, Shrivastava IH, Hanlon P, Dagda RK, Gasanoff ES. Molecular Mechanism by Which Cobra Venom Cardiotoxins Interact with the Outer Mitochondrial Membrane. Toxins. 2020; 12(7):425. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070425
Chicago/Turabian StyleLi, Feng, Indira H. Shrivastava, Paul Hanlon, Ruben K. Dagda, and Edward S. Gasanoff. 2020. "Molecular Mechanism by Which Cobra Venom Cardiotoxins Interact with the Outer Mitochondrial Membrane" Toxins 12, no. 7: 425. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070425