Multi-Toxin Quantitative Analysis of Paralytic Shellfish Toxins and Tetrodotoxins in Bivalve Mollusks with Ultra-Performance Hydrophilic Interaction LC-MS/MS—An In-House Validation Study


Abstract
:1. Introduction
2. Results and Discussion
2.1. Specificity
2.2. Calibration and Linearity
2.3. Recovery in Sample Preparation and Matrix Effects in ESI-MS
2.4. Quantitative Analysis
- Conc (µg STXeq/kg): concentration of the toxin in the homogenate sample, µg STX eq/kg
- Conc (nmol/L): concentration of the toxin found in the sample solution, nmol/L
- d: dilution factor throughout the whole extraction procedure in ready-to-inject sample (20)
- Vol: total volume of the extract, L (5 mL of 1% acetic acid)
- MW: molecular weight of toxin, g/mol
- TEF: toxicity equivalency factor of toxin. For C1 a TEF of 0.01 was applied [33]
- m: mass of the sample homogenate, g (5 ± 0.1 g)
2.5. Repeatibilty
2.6. LOD and LOQ
3. Conclusions
4. Materials and Methods
4.1. Standards and Reagents
4.2. Materials
4.3. Sample Preparation
4.3.1. Homogenization of Shellfish Samples
4.3.2. Shellfish Extraction and Clean Up
4.4. Calibration Curve
4.5. UP-HILIC-MS/MS
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Ito, K.; Kajihara, H. Highly toxic ribbon worm Cephalothrix simula containing tetrodotoxin in Hiroshima Bay, Hiroshima Prefecture, Japan. Toxins 2013, 5, 376–395. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). Panel on contaminants in the food chain. Scientific opinion on the risks for public health related to the presence of tetrodotoxin (TTX) and TTX analogues in marine bivalves and gastropods. EFSA J. 2017, 15, 4752. [Google Scholar]
- Göransson, U.; Jacobsson, E.; Strand, M.; Andersson, H.S. The toxins of nemertean worms. Toxins 2019, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Arakawa, O. Tetrodotoxin—Distribution and accumulation in aquatic organisms, and cases of human intoxication. Mar. Drugs 2008, 6, 220–242. [Google Scholar] [CrossRef] [Green Version]
- Pratheepa, V.; Vasconcelos, V. Microbial diversity associated with tetrodotoxin production in marine organisms. Environ. Toxicol. Pharmacol. 2013, 36, 1046–1054. [Google Scholar] [CrossRef]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [Green Version]
- How, C.K.; Chern, C.H.; Huang, Y.C.; Wang, L.M.; Lee, C.H. Tetrodotoxin poisoning. Am. J. Emerg. Med. 2003, 21, 51–54. [Google Scholar] [CrossRef]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef]
- Biessy, L.; Boundy, M.J.; Smith, K.F.; Harwood, D.T.; Hawes, I.; Wood, S.A. Tetrodotoxin in marine bivalves and edible gastropods: A mini-review. Chemosphere 2019, 236, 124404. [Google Scholar] [CrossRef]
- Finch, S.C.; Boundy, M.J.; Harwood, D.T. The acute toxicity of tetrodotoxin and tetrodotoxin-saxitoxin mixtures to mice by various routes of administration. Toxins 2018, 10, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlamis, A.; Katikou, P.; Rodriguez, I.; Rey, V.; Alfonso, A.; Papazachariou, A.; Zacharaki, T.; Botana, A.M.; Botana, L.M. First detection of Tetrodotoxin in Greek shellfish by UPLC-MS/MS potentially linked to the presence of the Dinoflagellate Prorocentrum minimum. Toxins 2015, 7, 1779–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.; Alfonso, A.; Vale, C.; Alfonso, C.; Vale, P.; Tellez, A.; Botana, L.M. First toxicity report of tetrodotoxin and 5,6,11-trideoxyTTX in the trumpet shell Charonia lampas in Europe. Anal. Chem. 2008, 80, 5622–5629. [Google Scholar] [CrossRef]
- Turner, A.D.; Powell, A.; Schofield, A.; Lees, D.N.; Baker-Austin, C. Detection of the pufferfish toxin tetrodotoxin in European bivalves, England, 2013 to 2014. Eurosurveillance 2015, 20. [Google Scholar] [CrossRef] [PubMed]
- Gerssen, A.; Bovee, T.H.F.; Klijnstra, M.D.; Poelman, M.; Portier, L.; Hoogenboom, R. First report on the occurrence of tetrodotoxins in bivalve mollusks in The Netherlands. Toxins 2018, 10, 450. [Google Scholar] [CrossRef] [Green Version]
- Berdalet, E.; Fleming, L.E.; Gowen, R.; Davidson, K.; Hess, P.; Backer, L.C.; Moore, S.K.; Hoagland, P.; Enevoldsen, H. Marine harmful algal blooms, human health and wellbeing: Challenges and opportunities in the 21st century. J. Mar. Biol. Assoc. UK 2015. [Google Scholar] [CrossRef] [Green Version]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: A formidable predictive challenge. J. Phycol. 2010, 46, 220–235. [Google Scholar] [CrossRef]
- Wells, M.L.; Karlson, B.; Wulff, A.; Kudela, R.; Trick, C.; Asnaghi, V.; Berdalet, E.; Cochlan, W.; Davidson, K.; De Rijcke, M.; et al. Future HAB science: Directions and challenges in a changing climate. Harmful Algae 2020, 91, 101632. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Pannel on contaminants in the food chain (CONTAM). Scientific opinion on marine biotoxins in shellfish—Emerging toxins: Brevetoxin group. EFSA J. 2010, 8, 1–29. [Google Scholar]
- García-Altares, M.; Casanova, A.; Bane, V.; Diogène, J.; Furey, A.; de la Iglesia, P. Confirmation of pinnatoxins and spirolides in shellfish and passive samplers from Catalonia (Spain) by liquid chromatography coupled with triple quadrupole and high-resolution hybrid tandem mass spectrometry. Mar. Drugs 2014, 12, 3706–3732. [Google Scholar] [CrossRef]
- Otero, P.; Pérez, S.; Alfonso, A.; Vale, C.; Rodríguez, P.; Gouveia, N.N.; Gouveia, N.; Delgado, J.; Vale, P.; Hirama, M.; et al. First toxin profile of ciguateric fish in Madeira Arquipelago (Europe). Anal. Chem. 2010, 82, 6032–6039. [Google Scholar] [CrossRef] [PubMed]
- Rambla-Alegre, M.; Miles, C.O.; de la Iglesia, P.; Fernandez-Tejedor, M.; Jacobs, S.; Sioen, I.; Verbeke, W.; Samdal, I.A.; Sandvik, M.; Barbosa, V.; et al. Occurrence of cyclic imines in European commercial seafood and consumers risk assessment. Environ. Res. 2018, 161, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gago-Martinez, A. Europen Union Reference Laboratory for Marine Biotoxins, Universitario de Vigo, Spain—Natioanl Reference Laboratories (EURLMB-NRLs) Network Experience in Proficiency Tests until 2019; EURLMB: Vigo, Spain, 2019; Unpublished work. [Google Scholar]
- Lawrence, J.F.; Niedzwiadek, B.; Menard, C. Quantitative determination of paralytic shellfish poisoning toxins in shellfish using prechromatographic oxidation and liquid chromatography with fluorescence detection: Collaborative study. J. AOAC Int. 2005, 88, 1714–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Riet, J.; Gibbs, R.S.; Muggah, P.M.; Rourke, W.A.; MacNeil, J.D.; Quilliam, M.A. Liquid chromatography post-column oxidation (PCOX) method for the determination of paralytic shellfish toxins in mussels, clams, oysters, and scallops: Collaborative study. J. AOAC Int. 2011, 94, 1154–1176. [Google Scholar] [CrossRef] [Green Version]
- Anon. Paralytic Shellfish Toxins (PSTs) in Shellfish. Receptor Binding Assay. AOAC Official Method Standard Number 2011.27. 2011. Available online: http://www.aoacofficialmethod.org/index.php?main_page=product_info&cPath=1&products_id=2975 (accessed on 8 July 2020).
- Quilliam, M.A.; Thomson, B.A.; Scott, G.J.; Siu, K.W. Ion-spray mass spectrometry of marine neurotoxins. Rapid Commun. Mass Spectrom. 1989, 3, 145–150. [Google Scholar] [CrossRef]
- Sayfritz, S.J.; Aasen, J.A.; Aune, T. Determination of paralytic shellfish poisoning toxins in Norwegian shellfish by liquid chromatography with fluorescence and tandem mass spectrometry detection. Toxicon 2008, 52, 330–340. [Google Scholar] [CrossRef]
- Turrell, E.; Stobo, L.; Lacaze, J.P.; Piletsky, S.; Piletska, E. Optimization of hydrophilic interaction liquid chromatography/mass spectrometry and development of solid-phase extraction for the determination of paralytic shellfish poisoning toxins. J. AOAC Int. 2008, 91, 1372–1386. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Matsushima, R.; Harada, T.; Oikawa, H.; Murata, M.; Suzuki, T. Quantitative determination of paralytic shellfish toxins in cultured toxic algae by LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2013, 30, 1351–1357. [Google Scholar] [CrossRef]
- Zhuo, L.; Yin, Y.; Fu, W.; Qiu, B.; Lin, Z.; Yang, Y.; Zheng, L.; Li, J.; Chen, G. Determination of paralytic shellfish poisoning toxins by HILIC-MS/MS coupled with dispersive solid phase extraction. Food Chem. 2013, 137, 115–121. [Google Scholar] [CrossRef]
- Boundy, M.J.; Selwood, A.I.; Harwood, D.T.; McNabb, P.S.; Turner, A.D. Development of a sensitive and selective liquid chromatography-mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 2015, 1387, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nordlander, I.; Persson, M.; Hallström, H.; Simonsson, M.; Karlsson, B. Kontrollprogrammet för Tvåskaliga Blötdjur—Årsrapport 2009–2011. Rapport 14-2011. 2011. Available online: https://www.livsmedelsverket.se/bestall-ladda-ner-material/sok-publikationer/artiklar/2011/2011-nr-14-arsrapport-2009-2010-kontrollprogrammet-for-tvaskaliga-blotdjur (accessed on 7 July 2020).
- Turner, A.D.; Dhanji-Rapkova, M.; Coates, L.; Bickerstaff, L.; Milligan, S.; O’Neill, A.; Faulkner, D.; McEneny, H.; Baker-Austin, C.; Lees, D.N.; et al. Detection of Tetrodotoxin Shellfish Poisoning (TSP) toxins and causative factors in Bivalve Molluscs from the UK. Mar. Drugs 2017, 15, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anon. Paralytic Shellfish Poisoning Toxins in Shellfish—Prechromatographic Oxidation and Liquid Chromatography with Fluorescence Detection. AOAC Official Method 2005.06. 2005. Available online: http://www.aoacofficialmethod.org/index.php?main_page=product_info&cPath=1&products_id=261 (accessed on 8 July 2020).
- Anon. DIN EN 14526 Foodstuffs—Determination of Saxitoxin-Group Toxins in Shellfish—HPLC Method Using Pre-Column Derivatization with Peroxide or Periodate Oxidation; European Standard: CEN—European Committee for Standardization: Brussels, Belgium, 2016. [Google Scholar]
- Turner, A.D.; Boundy, M.J.; Rapkova, M.D. Development and single-laboratory validation of a liquid chromatography tandem mass spectrometry method for quantitation of tetrodotoxin in mussels and oysters. J. AOAC Int. 2017, 100, 1469–1482. [Google Scholar] [CrossRef]
- Turner, A.D.; McNabb, P.S.; Harwood, D.T.; Selwood, A.I.; Boundy, M.J. Single-laboratory validation of a multitoxin ultra-performance LC-hydrophilic interaction LC-MS/MS method for quantitation of paralytic shellfish toxins in bivalve shellfish. J. AOAC Int. 2015, 98, 609–621. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Marine biotoxins in shellfish—Saxitoxin group. Scientific opinion of the panel on contaminants in the food chain. EFSA J. 2009, 1–76. [Google Scholar] [CrossRef]
- European Commission. Commission Decision (2002/657/EC) Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results.; Publications Office of the EU: Brussels, Belgium, 2002.
- Estevez, P.; Castro, D.; Pequeño-Valtierra, A.; Giraldez, J.; Gago-Martinez, A. Emerging marine biotoxins in seafood from European coasts: Incidence and analytical challenges. Foods 2019, 8, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]



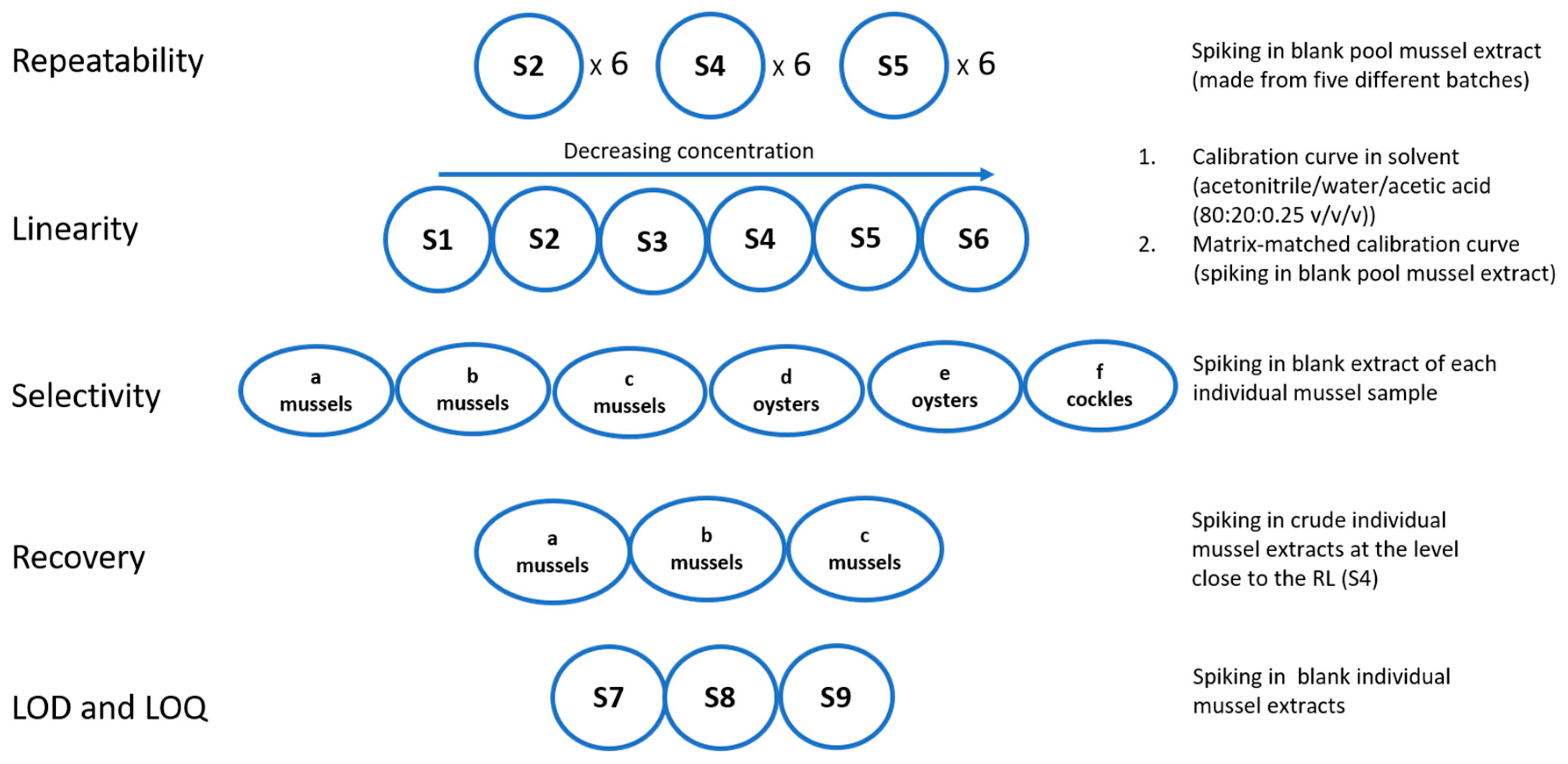{kind=link}
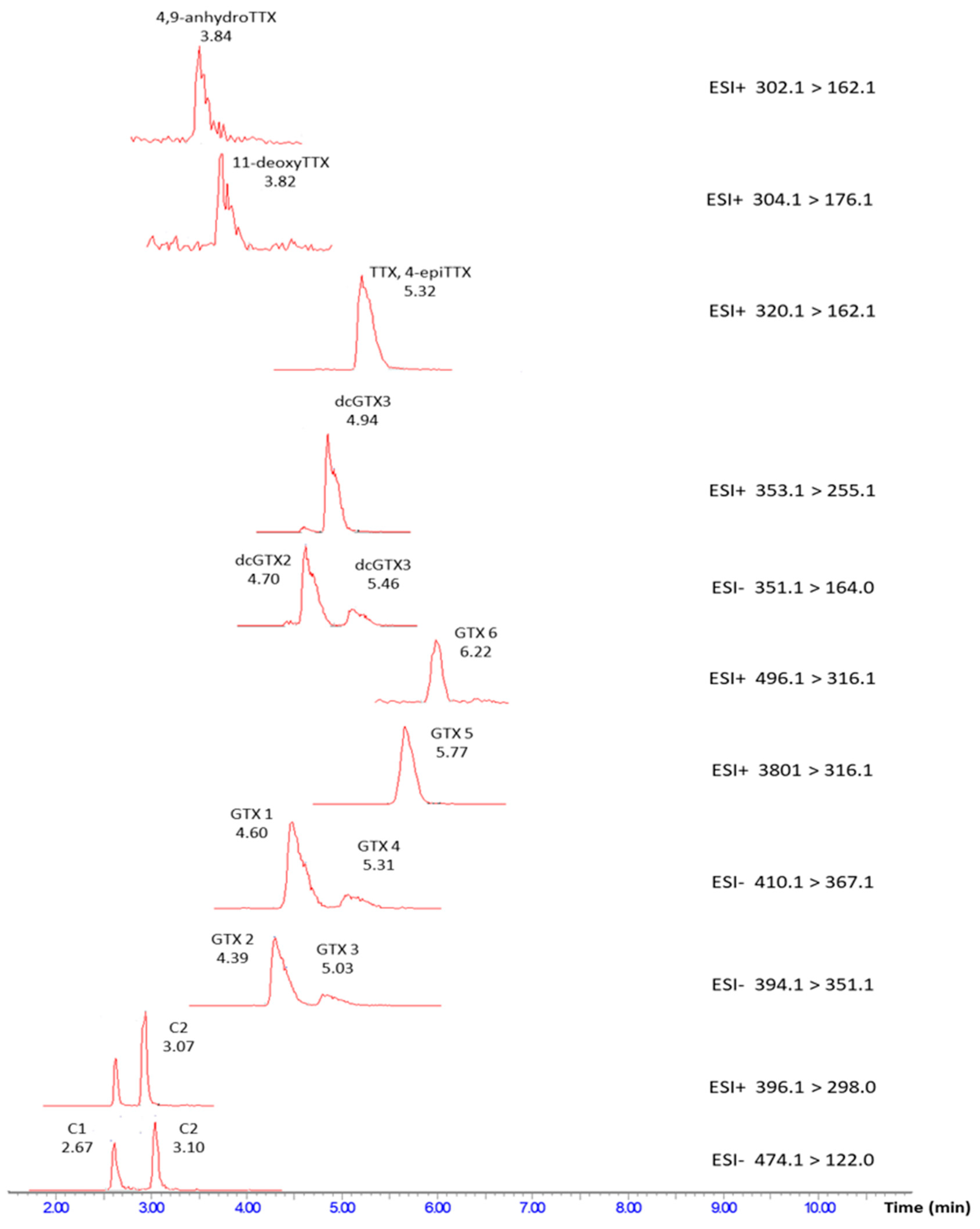{kind=link}
| Table | Calibration Range | Correlation Coefficient (R2) | Matrix Effect (ME) | ||
|---|---|---|---|---|---|
| nmol/L | µg STX eq/kg µgTTX/kg | Solvent | Matrix-Match | %ME | |
| dcSTX | 4.1–130.0 | 21.0–671.5 | 0.995 | 0.998 | 59 |
| dcNEO | 1.9–60.8 | 5.25–167.9 | 0.991 | 0.995 | 69 |
| STX | 4.1–132.6 | 24.8–793.7 | 0,996 | 0.997 | 65 |
| NEO | 4.1–130.2 | 25.7–821.2 | 0.991 | 0.996 | 81 |
| TTX, 4-epiTTX | 4.9–157.2 | 31.4–1003.3 | 0.993 | 0.999 | 45 |
| 11-deoxyTTX | 0.2–5.0 | 0.9–30.3 | 0.997 | 0.998 | 182 |
| 4,9-anhydroTTX | 0.6–19.8 | 3.4–119.2 | 0.996 | 0.998 | 169 |
| dcGTX3 | 0.7–21.6 | 3.8–120.1 | 0.998 | 0.999 | 75 |
| GTX6 | 2.0–64.5 | 0.6–19.8 | 0.996 | 0.997 | 76 |
| GTX5 | 6.2–200.2 | 2.8–88.8 | 0.998 | 0.999 | 86 |
| C2 | 1.8–58.8 | 2.0–64.5 | 0.997 | 0.999 | 59 |
| GTX2 | 3.6–114.4 | 20.2–649.0 | 0.998 | 1.000 | 98 |
| GTX3 | 1.1–36.0 | 12.9–412.7 | 0.998 | 0.999 | 81 |
| GTX1 | 6.4–205.2 | 29.4–942.2 | 0.997 | 0.999 | 102 |
| GTX4 | 2.7–87.0 | 6.5–207.3 | 0.990 | 0.998 | 93 |
| dcGTX2 | 3.7–117.0 | 20.3–649.0 | 0.996 | 0.999 | 73 |
| C1 | 0.8–25.0 | 0.67–21.6 | 0.999 | 0.999 | 51 |
| Toxins | Conc. in Spiked Sample | Mean Recovery (n = 9) (%) | RSDr (%) | RSDR (%) | |
|---|---|---|---|---|---|
| nmol/L | µg STX eq/kg µg TTX/kg | ||||
| dcSTX | 16.25 | 83.94 | 66 | 7 | 18 |
| dcNEO | 7.60 | 20.99 | 62 | 10 | 22 |
| STX | 16.58 | 99.21 | 65 | 5 | 15 |
| NEO | 16.28 | 102.63 | 65 | 8 | 19 |
| TTX, 4-epiTTX | 19.65 | 125.41 | 68 | 20 | 21 |
| 11-deoxyTTX | 0.63 | 3.79 | 71 | 18 | 18 |
| 4,9-anhydroTTX | 2.48 | 14.90 | 72 | 12 | 13 |
| dcGTX3 | 7.35 | 15.01 | 68 | 6 | 14 |
| GTX6 | 14.62 | 2.47 | 34 | 24 | 52 |
| GTX5 | 3.13 | 11.09 | 61 | 7 | 18 |
| C2 | 8.48 | 8.06 | 54 | 20 | 23 |
| GTX2 | 25.65 | 81.13 | 62 | 19 | 24 |
| GTX3 | 10.88 | 51.59 | 66 | 11 | 16 |
| GTX1 | 14.30 | 117.64 | 64 | 18 | 25 |
| GTX4 | 4.50 | 25.91 | 74 | 12 | 30 |
| dcGTX2 | 25.03 | 34.07 | 70 | 10 | 16 |
| C1 | 28.35 | 2.70 | 49 | 30 | 31 |
| Toxins | High-Level | Mid-Level | Low-Level | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Conc (µg STX eq/kg) * | %RSDr (n = 6) | %RSDR (n = 18) | Conc (µg STX eq/kg) * | %RSDr (n = 6) | %RSDR (n = 18) | Conc (µg STX eq/kg) * | %RSDr (n = 6) | %RSDR (n = 18) | ||
| dcSTX | 335.66 | 2 | 9 | 86.06 | 3 | 5 | 43.44 | 4 | 11 | |
| dcNEO | 84.31 | 3 | 8 | 21.98 | 6 | 7 | 11.93 | 6 | 10 | |
| STX | 395.35 | 4 | 7 | 102.47 | 2 | 4 | 53.87 | 2 | 10 | |
| NEO | 411.77 | 3 | 10 | 105.75 | 4 | 6 | 54.29 | 4 | 9 | |
| TTX, 4-epiTTX | 501.89 | 2 | 7 | 124.71 | 3 | 6 | 64.94 | 4 | 5 | |
| 11-deoxyTTX | 14.56 | 4 | 9 | 3.82 | 8 | 14 | 2.00 | 9 | 12 | |
| 4,9-anhydroTTX | 55.01 | 3 | 12 | 14.22 | 12 | 13 | 7.41 | 10 | 16 | |
| dcGTX3 | 74.82 | 2 | 3 | 19.52 | 3 | 3 | 10.65 | 3 | 3 | |
| GTX6 | 9.91 | 3 | 10 | 2.53 | 5 | 12 | 1.25 | 10 | 15 | |
| GTX5 | 43.70 | 1 | 6 | 11.15 | 2 | 7 | 5.88 | 3 | 9 | |
| C2 | 29.85 | 3 | 15 | 7.86 | 5 | 13 | 4.26 | 8 | 9 | |
| GTX2 | 313.05 | 5 | 8 | 81.00 | 4 | 7 | 40.55 | 5 | 7 | |
| GTX3 | 198.16 | 6 | 8 | 52.38 | 6 | 6 | 27.90 | 7 | 9 | |
| GTX1 | 470.33 | 5 | 6 | 117.73 | 5 | 4 | 58.58 | 6 | 8 | |
| GTX4 | 105.04 | 8 | 11 | 27.93 | 9 | 10 | 14.22 | 15 | 18 | |
| dcGTX2 | 133.61 | 3 | 5 | 34.21 | 5 | 6 | 17.28 | 6 | 6 | |
| C1 | 9.99 | 3 | 5 | 2.67 | 5 | 10 | 1.57 | 6 | 18 | |
| Toxins | LOQ Conc | LOD Conc |
|---|---|---|
| (µg STX eq/kg or µg TTX/kg) | ||
| dcSTX | 20.99 | 2.62 |
| dcNEO | 5.25 | 2.62 |
| STX | 24.80 | 3.10 |
| NEO | 25.66 | 6.41 |
| TTX, 4-epiTTX | 15.68 | 7.84 |
| 11-deoxyTTX | 1.89 * | 0.95 |
| 4,9-anhydroTTX | 7.45 * | 3.73 |
| dcGTX3 | 3.75 | 0.94 |
| GTX6 | 0.62 | 0.31 |
| GTX5 | 2.77 | 0.35 |
| C2 | 2.01 | 0.50 |
| GTX2 | 20.28 | 2.54 |
| GTX3 | 12.90 | 6.45 |
| GTX1 | 29.41 | 14.71 |
| GTX4 | 12.96 * | 6.48 |
| dcGTX2 | 8.52 | 4.26 |
| C1 | 0.67 | 0.08 |
| Certified Reference Standard | Containing Toxins | Concentration (µmol/L) | Commercial Provider |
|---|---|---|---|
| CRM-dcGTX2&3-c | dcGTX2 | 100.1 | NRCC, NS, Canada |
| dcGTX3 | 29.4 | ||
| CRM-dcSTX-b | dcSTX | 65 | |
| CRM-GTX1&4-d | GTX1 | 57.2 | |
| GTX4 | 18 | ||
| CRM-GTX2&3-d | GTX2 | 102.6 | |
| GTX3 | 43.5 | ||
| CRM-NEO-d | NEO | 65.1 | |
| CRM-STX-f | STX | 66.3 | |
| CRM-C1&2-b | C1 | 113.4 | |
| C2 | 33.9 | ||
| CRM-dcNEO-d | dcNEO | 30.4 | |
| CRM-GTX5-c | GTX5 | 55.7 | |
| CRM-GTX6 | GTX6 | 12.5 | |
| GTX5 | 2.79 | ||
| CRM-03-TTXs | TTX | 78.6 | CIFGA; Lugo, Spain |
| 4,9-anhydro TTXs | 9.9 | ||
| 11-deoxyTTX * | 2.50 |
| Time Windows Group | Analytes | Precursor Ion, m/z | Product Ion, m/z | Cone, V | Dwell, ms | CE, eV |
|---|---|---|---|---|---|---|
| 1 | * dido-dcSTX | 225.1 | 166.1; 60 | 10 | 40 | 15 |
| 2 | STX | 300.1 | 204.1; 138 | 10 | 40 | 24; 30 |
| NEO | 316.1 | 298.1; 220.1; 126.1 | 10 | 40 | 15; 24; 26 | |
| dcSTX | 257.1 | 222; 126.1 | 10 | 40 | 22; 19 | |
| dcNEO | 273.1 | 225.1; 126.1 | 10 | 40 | 18; 20 | |
| * doSTX | 241.1 | 206.1; 60 | 10 | 40 | 22; 23 | |
| 3 | TTX, 4-epi TTX | 320.1 | 162.1; 302.1 | 40 | 25 | 38; 25 |
| 11-deoxy TTX | 304.1 | 176.1; 286.1 | 40 | 25 | 30 | |
| 4,9-Anhydro TTX | 302.1 | 162.1; 256.1 | 40 | 25 | 30 | |
| * 11-nor TTX-6-ol | 290.1 | 162.1; 272.1 | 40 | 25 | 30 | |
| * 5,6,11-Trideoxy TTX | 272.1 | 162.1; 254.1 | 40 | 25 | 30 | |
| 4 | GTX3 | 396.1 | 298.1 | 10 | 50 | 17 |
| GTX4 | 412.1 | 314.1 | 10 | 50 | 18 | |
| dcGTX3 | 353.1 | 255.1 | 10 | 50 | 18 | |
| * dcGTX4 | 369.1 | 271.1 | 10 | 50 | 18 | |
| 5 | GTX5 | 380.1 | 300.1 | 10 | 100 | 16 |
| GTX6 | 396.1 | 316.1 | 10 | 100 | 15 | |
| 6 | C1 | 396.1 | 298.1 | 18 | 40 | 20 |
| C2 | 396.1 | 298.1 | 18 | 40 | 20 | |
| * C3 | 412.1 | 332.1; 314.1 | 18 | 40 | 16; 20 | |
| * C4 | 412.1 | 332.1; 314.1 | 18 | 40 | 16; 20 |
| Time Windows Group | Analytes | Precursor Ion, m/z | Product Ion, m/z | Cone, V | Dwell, ms | CE, eV |
|---|---|---|---|---|---|---|
| 1 | GTX2 | 394.1 | 351.1; 333.1 | 10 | 60 | 16, 22 |
| GTX3 | 394.1 | 351.1; 333.1 | 10 | 60 | 16; 22 | |
| GTX1 | 410.1 | 367.1; 349.1 | 10 | 60 | 15; 22 | |
| GTX4 | 410.1 | 367.1; 349.1 | 10 | 60 | 15; 22 | |
| dcGTX2 | 351.1 | 333.1; 164.0 | 10 | 60 | 17; 30 | |
| dcGTX3 | 351.1 | 333.1; 164.0 | 10 | 60 | 17; 30 | |
| 2 | GTX5 | 378.1 | 122 | 10 | 100 | 25 |
| GTX6 | 394.1 | 122 | 10 | 100 | 25 | |
| 3 | C1 | 474.1 | 351.1; 122.0 | 10 | 40 | 25; 30 |
| C2 | 474.1 | 351.1; 122.0 | 10 | 40 | 25; 30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patria, F.P.; Pekar, H.; Zuberovic-Muratovic, A. Multi-Toxin Quantitative Analysis of Paralytic Shellfish Toxins and Tetrodotoxins in Bivalve Mollusks with Ultra-Performance Hydrophilic Interaction LC-MS/MS—An In-House Validation Study. Toxins 2020, 12, 452. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070452
Patria FP, Pekar H, Zuberovic-Muratovic A. Multi-Toxin Quantitative Analysis of Paralytic Shellfish Toxins and Tetrodotoxins in Bivalve Mollusks with Ultra-Performance Hydrophilic Interaction LC-MS/MS—An In-House Validation Study. Toxins. 2020; 12(7):452. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070452
Chicago/Turabian StylePatria, Fadillah Putri, Heidi Pekar, and Aida Zuberovic-Muratovic. 2020. "Multi-Toxin Quantitative Analysis of Paralytic Shellfish Toxins and Tetrodotoxins in Bivalve Mollusks with Ultra-Performance Hydrophilic Interaction LC-MS/MS—An In-House Validation Study" Toxins 12, no. 7: 452. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070452