In Vivo Evaluation of the Chronic Oral Toxicity of the Marine Toxin Palytoxin

 , , ,
, , , 

Abstract
:1. Introduction
2. Results
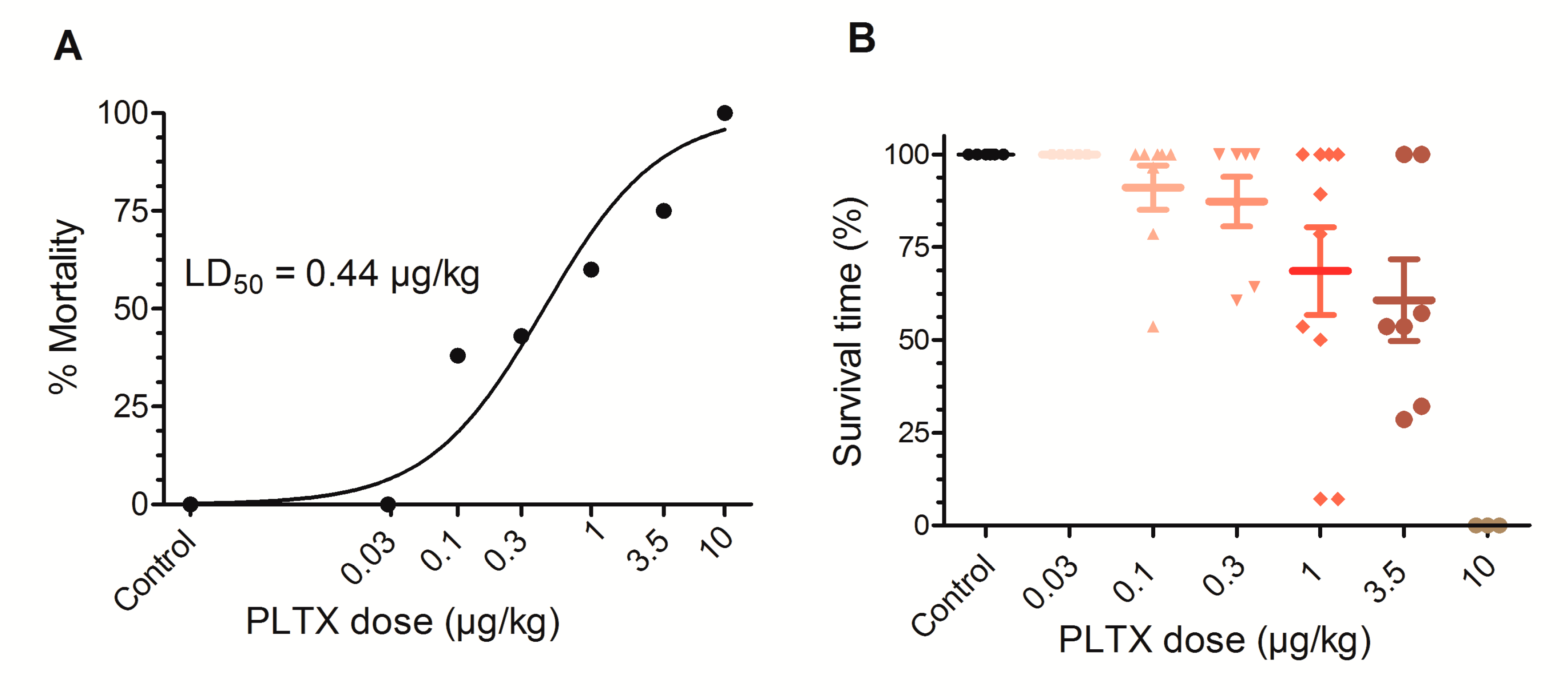
2.1. Chronic Toxicity Elicited by Daily Oral PLTX
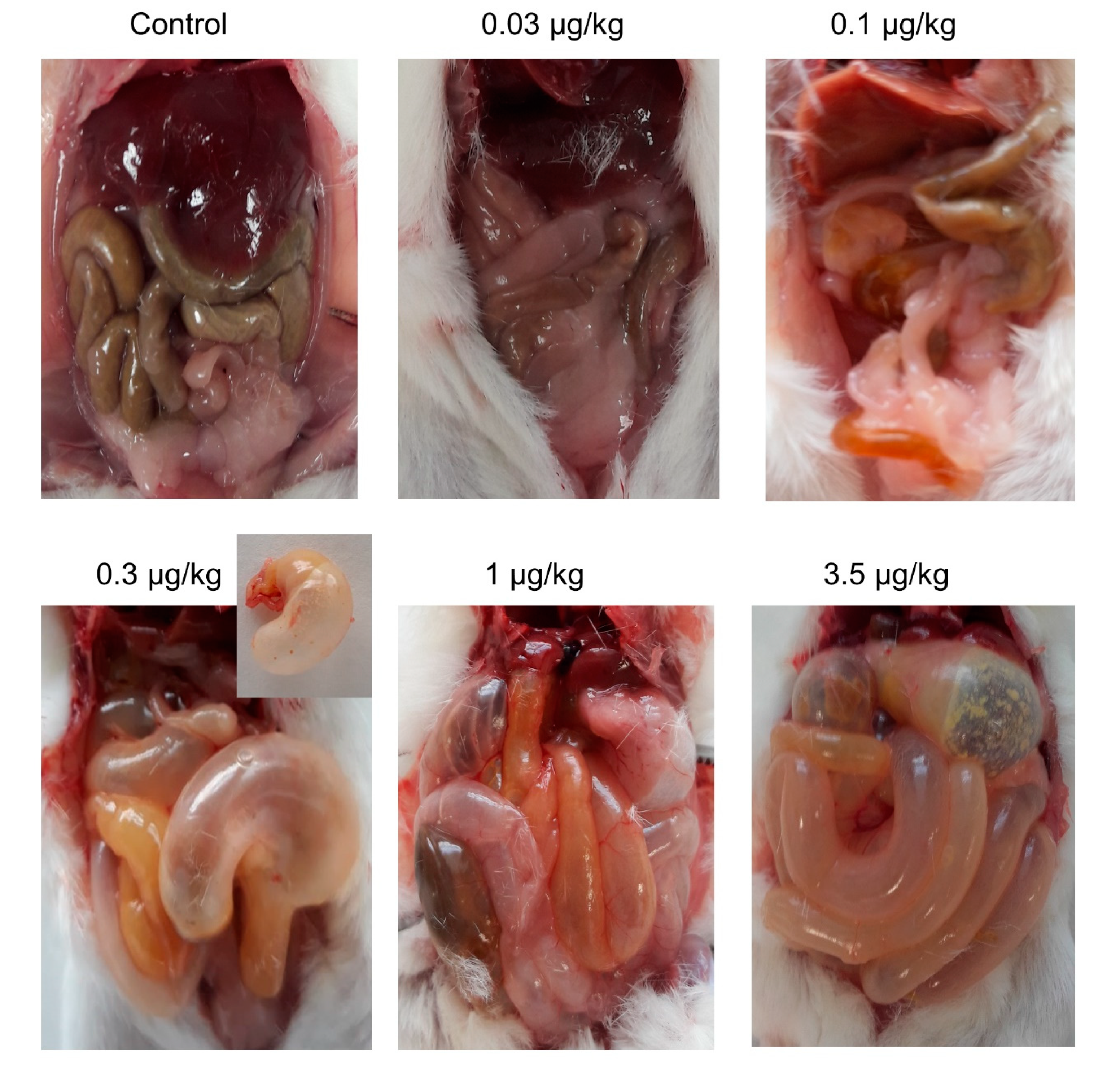
2.2. Chronic Oral PLTX Induced Macroscopic Changes in the Intestines and Stomachs of Treated Animals
2.3. Chronic Oral PLTX at Doses of 1 and 3.5 µg/kg Decreased Urine Production After the 28 Days Treatment Period
2.4. Urine Parameters in Control Mice and in Mice Treated Daily with PLTX
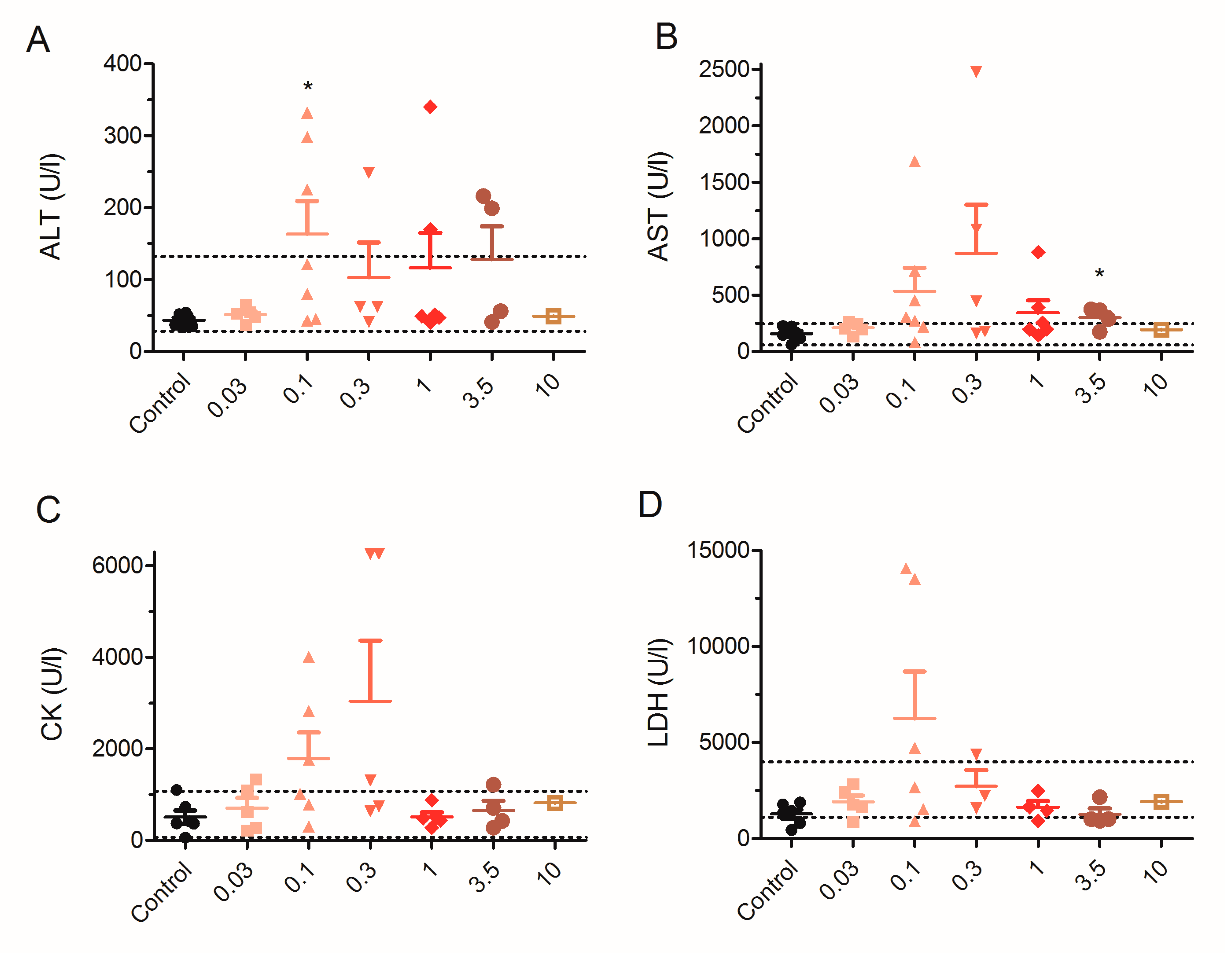
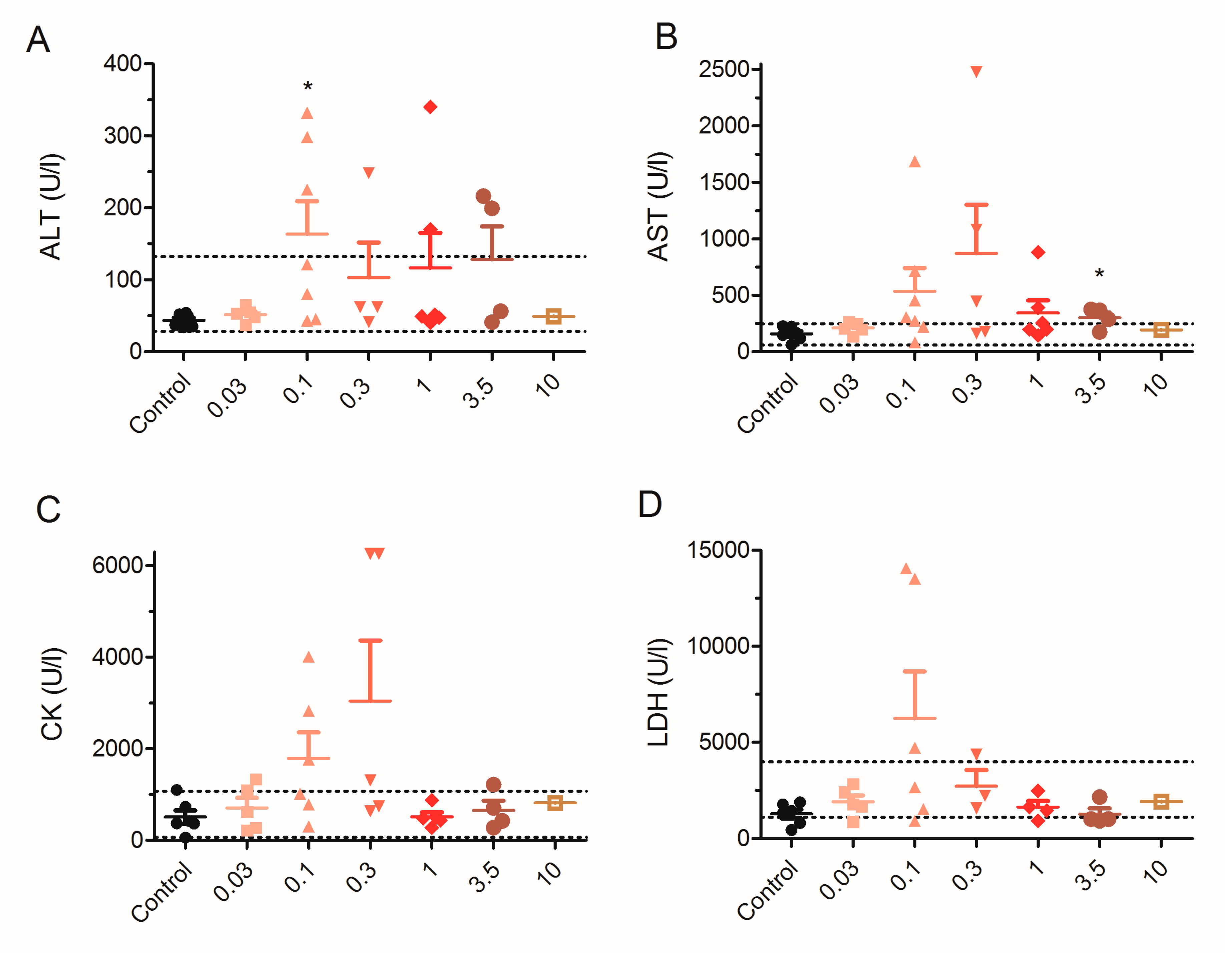
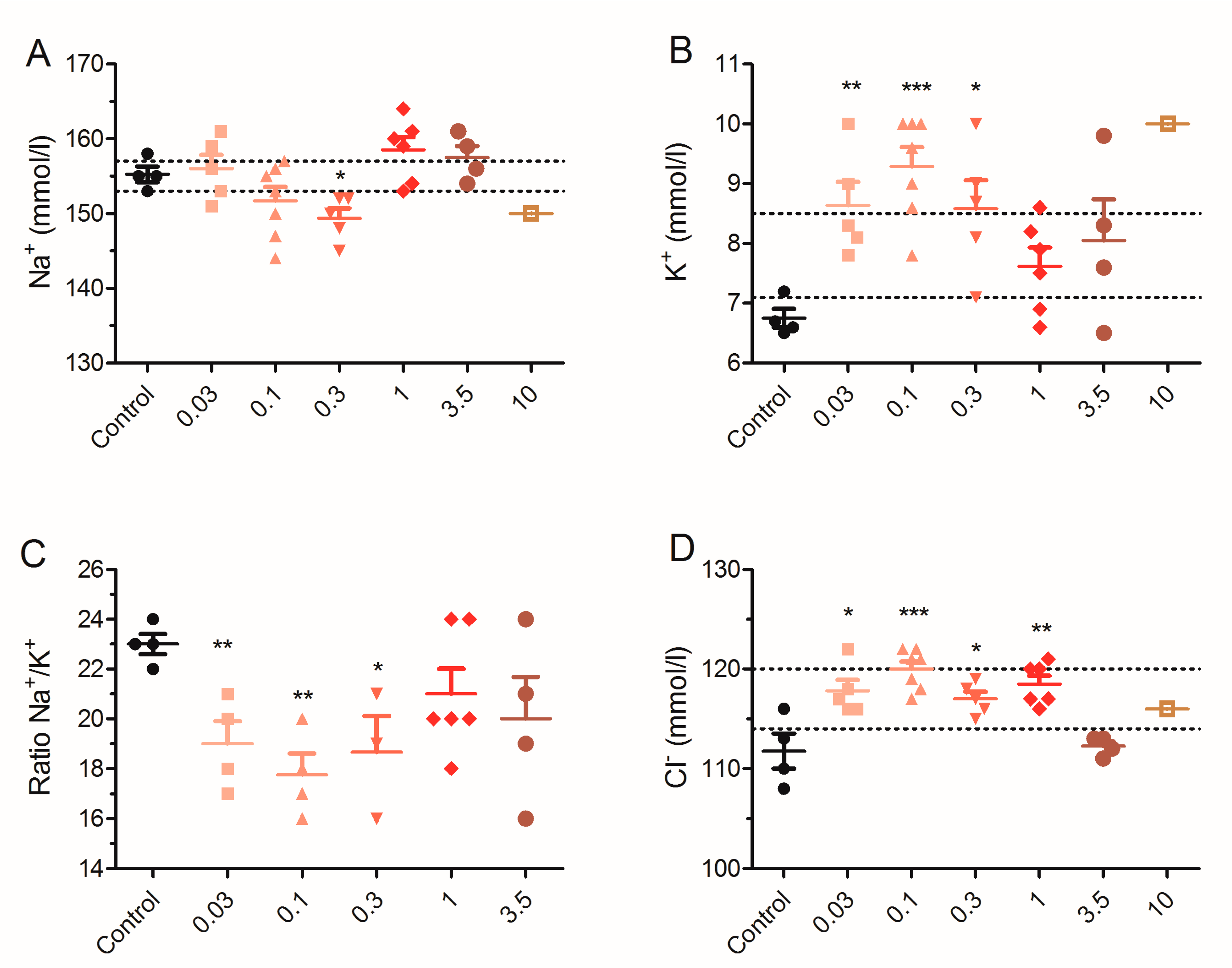
2.5. Effect of 28 Day Repeated Exposure of Mice to Low Doses of PLTX on Blood Biochemical Parameters
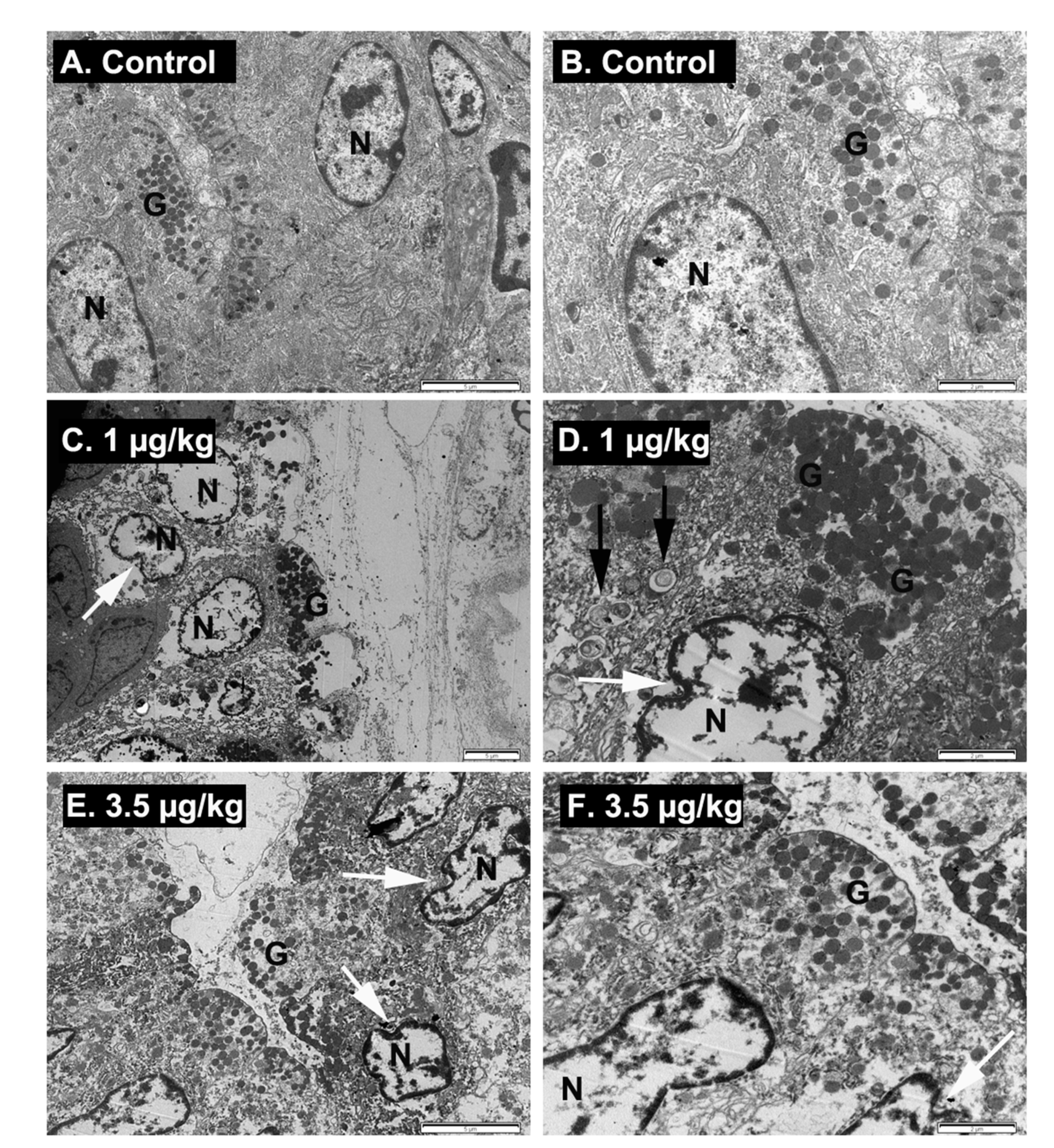
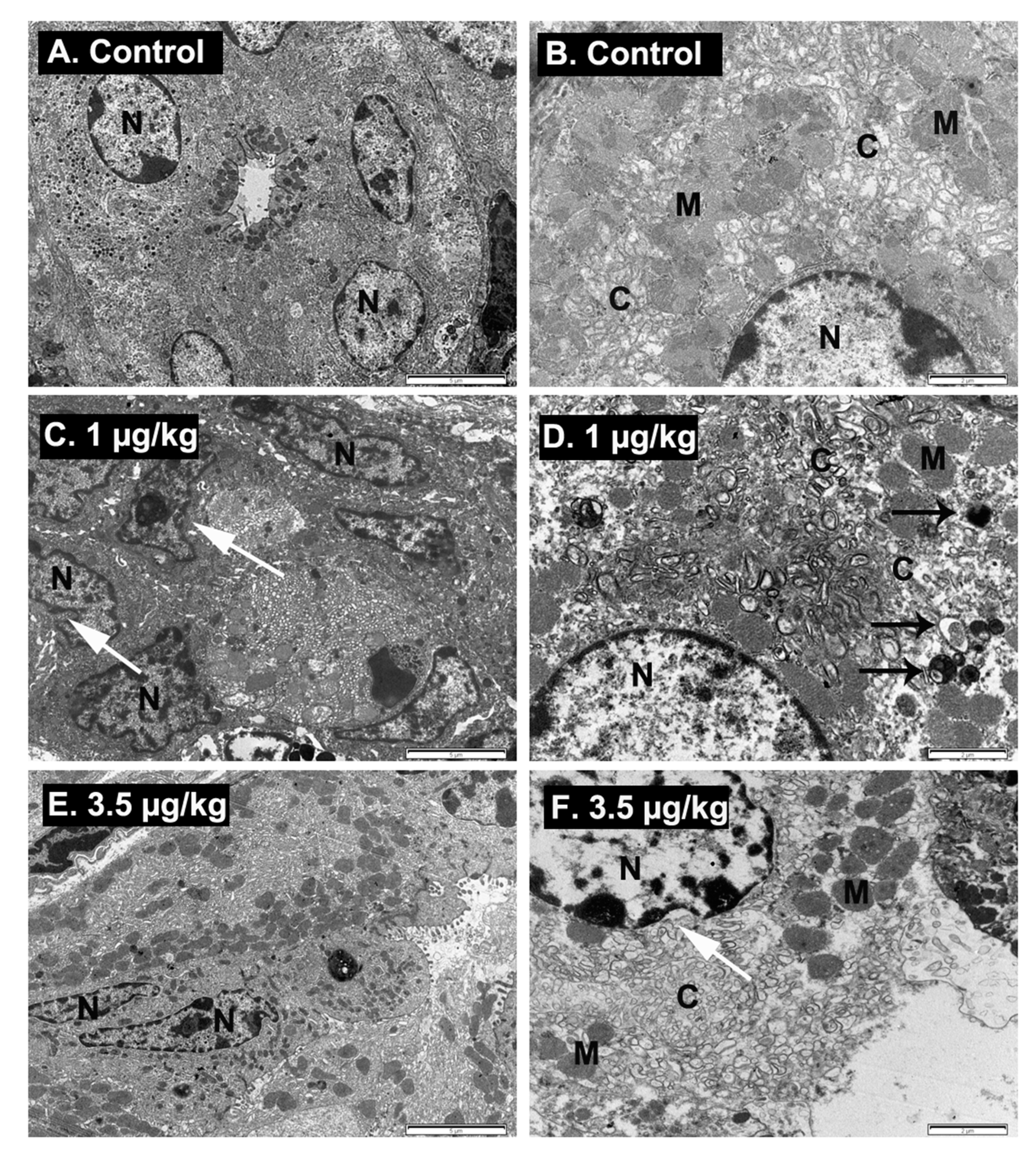
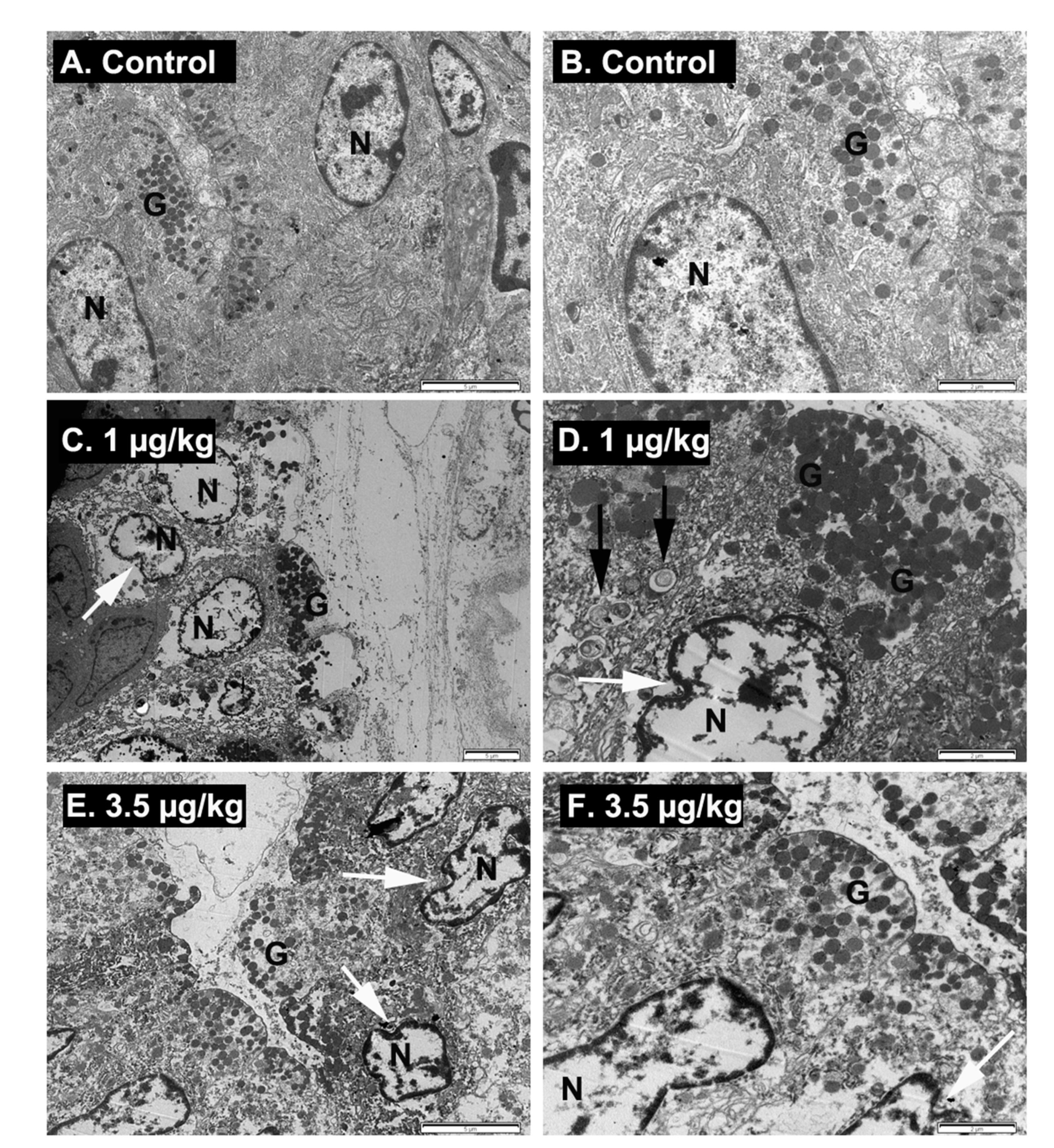
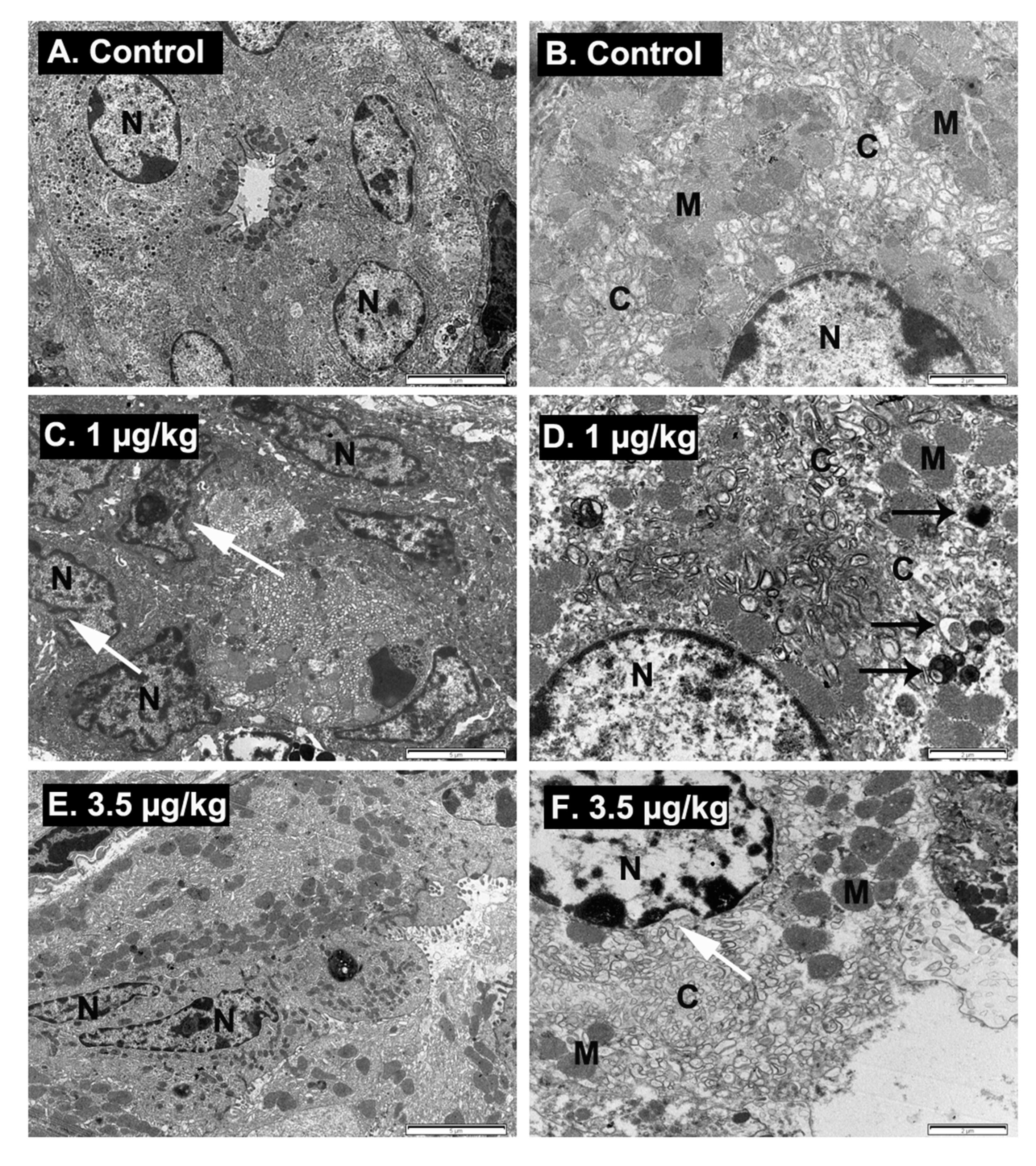
2.6. Ultrastructural Examination of Stomach Damage Induced by Repeated Oral Dosing of Mice with PLTX
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. In Vivo Experimental Procedure
5.2. Evaluation of Enzyme and Electrolyte Levels in Plasma of Control and PLTX-Treated Mice
5.3. Urine Analysis
5.4. Ultraestructural Analysis
5.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore, R.E.; Scheuer, P.J. Palytoxin: A new marine toxin from a coelenterate. Science 1971, 172, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, G.S.; Tartaglione, L.; Grillo, C.; Melchiorre, N. The Genoa 2005 outbreak. Determination of putative palytoxin in mediterranean Ostreopsis ovata by a new liquid chromatography tandem mass spectrometry method. Anal. Chem. 2006, 78, 6153–6159. [Google Scholar] [CrossRef]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Tartaglione, L.; Grillo, C.; Melchiorre, N. Putative palytoxin and its new analogue, ovatoxin-a, in Ostreopsis ovata collected along the Ligurian coasts during the 2006 toxic outbreak. J. Am. Soc. Mass Spectrom. 2008, 19, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuma, Y.; Satake, M.; Ukena, T.; Roux, J.; Chanteau, S.; Rasolofonirina, N.; Ratsimaloto, M.; Naoki, H.; Yasumoto, T. Identification of putative palytoxin as the cause of clupeotoxism. Toxicon 1999, 37, 55–65. [Google Scholar] [CrossRef]
- Nincevic Gladan, Z.; Arapov, J.; Casabianca, S.; Penna, A.; Honsell, G.; Brovedani, V.; Pelin, M.; Tartaglione, L.; Sosa, S.; Dell’Aversano, C.; et al. Massive occurrence of the harmful benthic dinoflagellate Ostreopsis cf. ovata in the eastern Adriatic Sea. Toxins 2019, 11, 300. [Google Scholar]
- Accoroni, S.; Romagnoli, T.; Penna, A.; Capellacci, S.; Ciminiello, P.; Dell’Aversano, C.; Tartaglione, L.; Abboud-Abi Saab, M.; Giussani, V.; Asnaghi, V.; et al. Ostreopsis fattorussoi sp. Nov. (dinophyceae), a new benthic toxic Ostreopsis species from the eastern Mediterranean Sea. J. Phycol. 2016, 52, 1064–1084. [Google Scholar]
- Aligizaki, K.; Katikou, P.; Nikolaidis, G.; Panou, A. First episode of shellfish contamination by palytoxin-like compounds from Ostreopsis species (Aegean Sea, Greece). Toxicon 2008, 51, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Amzil, Z.; Sibat, M.; Chomerat, N.; Grossel, H.; Marco-Miralles, F.; Lemee, R.; Nezan, E.; Sechet, V. Ovatoxin-a and palytoxin accumulation in seafood in relation to Ostreopsis cf. ovata blooms on the French Mediterranean coast. Mar. Drugs 2012, 10, 477–496. [Google Scholar]
- David, H.; Moita, M.T.; Laza, A.; Mateus, M.; Pablo, H.; Orive, E. First bloom of Ostreopsis cf. ovata in the continental Portuguese coast. Harmful Algae News 2012, 45, 12–13. [Google Scholar]
- Solino, L.; Garcia-Altares, M.; Godinho, L.; Costa, P.R. Toxin profile of Ostreopsis cf. ovata from Portuguese continental coast and Selvagens Islands (Madeira, Portugal). Toxicon 2020, 181, 91–101. [Google Scholar] [CrossRef]
- Accoroni, S.; Tartaglione, L.; Dello Iacovo, E.; Pichierri, S.; Marini, M.; Campanelli, A.; Dell’Aversano, C.; Totti, C. Influence of environmental factors on the toxin production of Ostreopsis cf. ovata during bloom events. Mar. Pollut. Bull. 2017, 123, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Botana, L.M. Toxicological perspective on climate change: Aquatic toxins. Chem. Res. Toxicol. 2016, 29, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.; Pratheepa, V.K.; Botana, L.M.; Vasconcelos, V. Emergent toxins in North Atlantic temperate waters: A challenge for monitoring programs and legislation. Toxins 2015, 7, 859–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcala, A.C.; Alcala, L.C.; Garth, J.S.; Yasumura, D.; Yasumoto, T. Human fatality due to ingestion of the crab Demania reynaudii that contained a palytoxin-like toxin. Toxicon 1988, 26, 105–107. [Google Scholar] [CrossRef]
- Kodama, A.M.; Hokama, Y.; Yasumoto, T.; Fukui, M.; Manea, S.J.; Sutherland, N. Clinical and laboratory findings implicating palytoxin as cause of ciguatera poisoning due to Decapterus macrosoma (mackerel). Toxicon 1989, 27, 1051–1053. [Google Scholar] [CrossRef]
- Okano, H.; Masuoka, H.; Kamei, S.; Seko, T.; Koyabu, S.; Tsuneoka, K.; Tamai, T.; Ueda, K.; Nakazawa, S.; Sugawa, M.; et al. Rhabdomyolysis and myocardial damage induced by palytoxin, a toxin of blue humphead parrotfish. Intern. Med. 1998, 37, 330–333. [Google Scholar] [CrossRef] [Green Version]
- Taniyama, S.; Mahmud, Y.; Terada, M.; Takatani, T.; Arakawa, O.; Noguchi, T. Occurrence of a food poisoning incident by palytoxin from a serranid Epinephelus sp. In japan. J. Natl. Toxins 2002, 11, 277–282. [Google Scholar]
- Murphy, L.T.; Charlton, N.P. Prevalence and characteristics of inhalational and dermal palytoxin exposures reported to the national poison data system in the U.S. Envon. Toxicol. Pharm. 2017, 55, 107–109. [Google Scholar] [CrossRef]
- Patocka, J.; Nepovimova, E.; Wu, Q.; Kuca, K. Palytoxin congeners. Arch. Toxicol. 2018, 92, 143–156. [Google Scholar] [CrossRef]
- Thakur, L.K.; Jha, K.K. Palytoxin-induced acute respiratory failure. Respir. Med. Case Rep. 2016, 20, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, K.; Hermanns-Clausen, M.; Buhl, C.; Büchler, M.W.; Schemmer, P.; Mebs, D.; Kauferstein, S. A case of palytoxin poisoning due to contact with zoanthid corals through a skin injury. Toxicon 2008, 51, 1535–1537. [Google Scholar] [CrossRef] [PubMed]
- Tartaglione, L.; Pelin, M.; Morpurgo, M.; Dell’Aversano, C.; Montenegro, J.; Sacco, G.; Sosa, S.; Reimer, J.D.; Ciminiello, P.; Tubaro, A. An aquarium hobbyist poisoning: Identification of new palytoxins in Palythoa cf. toxica and complete detoxification of the aquarium water by activated carbon. Toxicon 2016, 121, 41–50. [Google Scholar] [PubMed]
- Artigas, P.; Gadsby, D.C. Na+/K+-pump ligands modulate gating of palytoxin-induced ion channels. Proc. Natl. Acad. Sci. USA 2003, 100, 501–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artigas, P.; Gadsby, D.C. Ion occlusion/deocclusion partial reactions in individual palytoxin-modified Na/K pumps. Ann. N. Y. Acad. Sci. 2003, 986, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Artigas, P.; Gadsby, D.C. Large diameter of palytoxin-induced na/k pump channels and modulation of palytoxin interaction by Na/K pump ligands. J. Gen. Physiol. 2004, 123, 357–376. [Google Scholar] [CrossRef] [Green Version]
- Poli, M.; Ruiz-Olvera, P.; Nalca, A.; Ruiz, S.; Livingston, V.; Frick, O.; Dyer, D.; Schellhase, C.; Raymond, J.; Kulis, D. , et al. Toxicity and pathophysiology of palytoxin congeners after intraperitoneal and aerosol administration in rats. Toxicon 2018, 150, 235–250. [Google Scholar] [CrossRef]
- Rouzaire-Dubois, B.; Dubois, J.M. Characterization of palytoxin-induced channels in mouse neuroblastoma cells. Toxicon 1990, 28, 1147–1158. [Google Scholar] [CrossRef]
- Del Favero, G.; Beltramo, D.; Sciancalepore, M.; Lorenzon, P.; Coslovich, T.; Poli, M.; Testai, E.; Sosa, S.; Tubaro, A. Toxicity of palytoxin after repeated oral exposure in mice and in vitro effects on cardiomyocytes. Toxicon 2013, 75, 3–15. [Google Scholar] [CrossRef]
- Del Favero, G.; Sosa, S.; Poli, M.; Tubaro, A.; Sbaizero, O.; Lorenzon, P. In vivo and in vitro effects of 42-hydroxy-palytoxin on mouse skeletal muscle: Structural and functional impairment. Toxicol. Lett. 2014, 225, 285–293. [Google Scholar] [CrossRef]
- Fernandez, D.A.; Louzao, M.C.; Vilarino, N.; Espina, B.; Fraga, M.; Vieytes, M.R.; Roman, A.; Poli, M.; Botana, L.M. The kinetic, mechanistic and cytomorphological effects of palytoxin in human intestinal cells (Caco-2) explain its lower-than-parenteral oral toxicity. FEBS J. 2013, 280, 3906–3919. [Google Scholar] [CrossRef]
- Pelin, M.; Sosa, S.; Pacor, S.; Tubaro, A.; Florio, C. The marine toxin palytoxin induces necrotic death in HaCaT cells through a rapid mitochondrial damage. Toxicol. Lett. 2014, 229, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Alfonso, A.; Sunol, C.; Vieytes, M.R.; Botana, L.M. Modulation of calcium entry and glutamate release in cultured cerebellar granule cells by palytoxin. J. Neurosci. Res. 2006, 83, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Gomez-Limia, B.; Vieytes, M.R.; Botana, L.M. Mitogen-activated protein kinases regulate palytoxin-induced calcium influx and cytotoxicity in cultured neurons. Br. J. Pharm. 2007, 152, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vale-Gonzalez, C.; Gomez-Limia, B.; Vieytes, M.R.; Botana, L.M. Effects of the marine phycotoxin palytoxin on neuronal pH in primary cultures of cerebellar granule cells. J. Neurosci. Res. 2007, 85, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Boente-Juncal, A.; Vale, C.; Camina, M.; Cifuentes, J.M.; Vieytes, M.R.; Botana, L.M. Reevaluation of the acute toxicity of palytoxin in mice: Determination of lethal dose 50 (LD50) and no-observed-adverse-effect level (NOAEL). Toxicon 2020, 177, 16–24. [Google Scholar] [CrossRef]
- Munday, R. Palytoxin toxicology: Animal studies. Toxicon 2011, 57, 470–477. [Google Scholar] [CrossRef]
- Sosa, S.; Del Favero, G.; De Bortoli, M.; Vita, F.; Soranzo, M.R.; Beltramo, D.; Ardizzone, M.; Tubaro, A. Palytoxin toxicity after acute oral administration in mice. Toxicol. Lett. 2009, 191, 253–259. [Google Scholar] [CrossRef]
- Tubaro, A.; Del Favero, G.; Beltramo, D.; Ardizzone, M.; Forino, M.; De Bortoli, M.; Pelin, M.; Poli, M.; Bignami, G.; Ciminiello, P.; et al. Acute oral toxicity in mice of a new palytoxin analog: 42-hydroxy-palytoxin. Toxicon 2011, 57, 755–763. [Google Scholar] [CrossRef]
- Pelin, M.; Brovedani, V.; Sosa, S.; Tubaro, A. Palytoxin-containing aquarium soft corals as an emerging sanitary problem. Mar. Drugs 2016, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Tubaro, A.; del Favero, G.; Pelin, M.; Bignami, G.; Poli, M. Palytoxin and Analogues: Biological Effects and Detection. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 741–772. [Google Scholar]
- EFSA. Panel on contaminants in the food chain (CONTAM). Scientific opinion on marine biotoxins in shellfish—Palytoxin group. EFSA J. 2009, 7, 1393. [Google Scholar] [CrossRef]
- Ito, E.; Yasumoto, T. Toxicological studies on palytoxin and ostreocin-D administered to mice by three different routes. Toxicon 2009, 54, 244–251. [Google Scholar] [CrossRef] [PubMed]
- OECD. Test No. 407: Repeated Dose 28-Day Oral Toxicity Study in Rodents. 2008. Available online: https://0-www-oecd--ilibrary-org.brum.beds.ac.uk/docserver/9789264070684-en.pdf?expires=1596012513&id=id&accname=guest&checksum=AA219AD249F8EB6D4AE423559A037054 (accessed on 29 July 2020).
- Yu, S.P.; Choi, D.W. Ions, cell volume, and apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9360–9362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbany, M.; Rossell, M.; Salvador, A. Toxic corneal reaction due to exposure to palytoxin. Arch. Soc. Esp Oftalmol. 2019, 94, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Gaudchau, A.; Pfeiffer, N.; Gericke, A. Chemical burns caused by crust anemone. Ophthalmologe 2019, 116, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Levy, D.; Sattler, S. A case of palytoxin poisoning in a home aquarium enthusiast and his family. Case Rep. Emerg. Med. 2015, 621815, 26. [Google Scholar] [CrossRef]
- Wieringa, A.; Bertholee, D.; ter Horst, P.; van den Brand, I.; Haringman, J.; Ciminiello, P. Respiratory impairment in four patients associated with exposure to palytoxin containing coral. Clin. Toxicol. 2014, 52, 150–151. [Google Scholar] [CrossRef]
- Munday, R. Toxicological requirements for risk assessment of shellfish contaminants: A review. Afr. J. Mar. Sci. 2006, 28, 447–449. [Google Scholar] [CrossRef]
- Pirahanchi, Y.; Jessu, R.; Aeddula, N.R. Physiology, Sodium Potassium Pump (Na+ K+ Pump); Statpearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Yan, Y.; Shapiro, J.I. The physiological and clinical importance of sodium potassium ATPase in cardiovascular diseases. Curr. Opin. Pharm. 2016, 27, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.Y.; Krieger, E.; Schaftenaar, G.; Swarts, H.G.; Willems, P.H.; De Pont, J.J.; Koenderink, J.B. Reconstruction of the complete ouabain-binding pocket of Na,K-ATPase in gastric H,K-ATPase by substitution of only seven amino acids. J. Biol. Chem. 2005, 280, 32349–32355. [Google Scholar] [CrossRef] [Green Version]
- Rossini, G.P.; Bigiani, A. Palytoxin action on the Na(+),K(+)-ATPase and the disruption of ion equilibria in biological systems. Toxicon 2011, 57, 429–439. [Google Scholar] [CrossRef]
- Karam, S.M. A focus on parietal cells as a renewing cell population. World J. Gastroenterol. 2010, 16, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, S.M.; Forte, J.G. Inhibiting gastric H(+)-K(+)-ATPase activity by omeprazole promotes degeneration and production of parietal cells. Am. J. Physiol. 1994, 266, G745–G758. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Pareja, I.; Clemente-Postigo, M.; Tinahones, F.J. Metabolic and endocrine consequences of bariatric surgery. Front. Endocrinol. (Lausanne) 2019, 10, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boente-Juncal, A.; Vale, C.; Cifuentes, M.; Otero, P.; Camina, M.; Rodriguez-Vieytes, M.; Botana, L.M. Chronic in vivo effects of repeated exposure to low oral doses of tetrodotoxin: Preliminary evidence of nephrotoxicity and cardiotoxicity. Toxins 2019, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Abal, P.; Louzao, M.C.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Acute toxicity assessment: Macroscopic and ultrastructural effects in mice treated with oral tetrodotoxin. Toxins 2019, 11, 305. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (µg/kg) | Total Mice | Dead Mice | Individual Survival Time (Days) (Mean Survival Time ± SD) | Mortality % |
|---|---|---|---|---|
| Control | 6 | 0 | 28 | 0 |
| 0.03 | 5 | 0 | 28 | 0 |
| 0.1 | 8 | 3 | 15, 22, 27, 28, 28, 28, 28, 28 (25.5 ± 4.7) | 38 |
| 0.3 | 7 | 3 | 17, 18, 24, 28, 28, 28, 28 (24.4 ± 5.0) | 43 |
| 1 | 10 | 6 | 2, 2, 14, 15, 22, 25, 28, 28, 28, 28 (19.2 ± 10.5) | 60 |
| 3.5 | 8 | 6 | 0.3, 8, 9, 15, 15, 16, 28, 28 (14.9 ± 9.6) | 75 |
| 10 | 3 | 3 | 8, 10, 18 (12.0 ± 5.3) | 100 |
| Day 0 | Day 7 | Day 14 | Day 21 | Day 28 | |
|---|---|---|---|---|---|
| Control | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) |
| Body weight (g) | 22.2 ± 0.9 | 23.9 ± 0.6 | 25.9 ± 0.5 | 27.1 ± 0.8 | 27.3 ± 0.7 |
| Cumulative BW (g) | 0.0 | 1.7 ± 0.4 | 3.7 ± 0.6 | 4.8 ± 0.8 | 5.1 ± 0.9 |
| 0.03 µg/kg | (n = 5) | (n = 5) | (n = 5) | (n = 5) | (n = 5) |
| Body weight (g) | 22.9 ± 0.6 | 23.5 ± 0.3 | 24.5 ± 0.2 | 25.6 ± 0.4 | 25.8 ± 0.6 |
| Cumulative BW (g) | 0.0 | 0.6 ± 0.4 | 1.6 ± 0.7 | 2.7 ± 0.3 | 2.9 ± 0.3 |
| 0.1 µg/kg | (n = 8) | (n = 8) | (n = 8) | (n = 7) | (n = 5) |
| Body weight (g) | 23.4 ± 0.8 | 23.7 ± 0.7 | 25.0 ± 0.6 | 25.1 ± 0.8 | 24.2 ± 0.8 * |
| Cumulative BW (g) | 0.0 | 0.3 ± 0.6 | 1.6 ± 0.3 | 2.2 ± 0.7 | 1.4 ± 1.6 |
| 0.3 µg/kg | (n = 7) | (n = 7) | (n = 7) | (n = 5) | (n = 4) |
| Body weight (g) | 23.7 ± 0.4 | 23.3 ± 0.6 | 24.0 ± 0.8 | 25.7 ± 0.6 | 26.1 ± 1.5 |
| Cumulative BW (g) | 0.0 | −0.4 ± 0.7 | 0.3 ± 0.9 * | 1.9 ± 0.4 | 2.3 ± 1.0 |
| 1 µg/kg | (n = 10) | (n = 8) | (n = 8) | (n = 6) | (n = 4) |
| Body weight (g) | 24.5 ± 0.8 | 21.8 ± 1.0 | 21.1 ± 1.0 ** | 20.1 ± 0.9 *** | 21.8 ± 1.2 ** |
| Cumulative BW (g) | 0.0 | −2.4 ± 0.8 *** | −3.6 ± 0.7 *** | −4.2 ± 1.2 *** | −2.6 ± 1.4 *** |
| 3.5 µg/kg | (n = 8) | (n = 7) | (n = 5) | (n = 2) | (n = 2) |
| Body weight (g) | 23.6 ± 0.9 | 20.4 ± 1.3 * | 18.6 ± 2.0 ** | 20.0 ± 3.8 * | 21.8 ± 3.0 * |
| Cumulative BW (g) | 0.0 | −3.6 ± 0.7 *** | −5.1 ± 2.1 *** | −3.6 ± 3.5 *** | −1.9 ± 2.7 * |
| 10 µg/kg | (n = 3) | (n = 3) | (n = 1) | (n = 1) | |
| Body weight (g) | 22.2 ± 1.5 | 20.0 ± 0.7 | 21.5 | 21.9 | |
| Cumulative BW (g) | 0.0 | −2.2 ± 1.2 * | −3.4 *** | −3.0 *** |
| Symptoms | 0.03 µg/kg | 0.1 µg/kg | 0.3 µg/kg | 1 µg/kg | 3.5 µg/kg | 10 µg/kg |
|---|---|---|---|---|---|---|
| Lethargy | 0/5 | 1/8 | 3/7 | 8/10 | 6/8 | 3/3 |
| Piloerection | 0/5 | 3/8 | 3/7 | 8/10 | 7/8 | 3/3 |
| Dyspnea | 0/5 | 2/8 | 0/7 | 6/10 | 4/8 | 2/3 |
| Cyanosis | 0/5 | 1/8 | 0/7 | 1/10 | 0/8 | 0/3 |
| Abdominal pain | 0/5 | 2/8 | 2/7 | 4/10 | 7/8 | 2/3 |
| Kyphosis | 0/5 | 1/8 | 2/7 | 4/10 | 4/8 | 3/3 |
| Circling | 0/5 | 0/8 | 0/7 | 1/10 | 2/8 | 0/3 |
| Ataxia | 0/5 | 0/8 | 0/7 | 1/10 | 2/8 | 1/3 |
| Vocalization | 0/5 | 0/8 | 0/7 | 0/10 | 2/8 | 1/3 |
| Facial swelling | 0/5 | 1/8 | 3/7 | 3/10 | 1/8 | 3/3 |
| Group/Analysed Parameters | |
|---|---|
| Control (n = 6) | |
| Food consumption (g) | 4.9 ± 0.6 |
| Faeces (g) | 1.3 ± 0.2 |
| Urine (mL) | 2.5 ± 1.0 |
| 0.03 µg/kg PLTX (n = 5) | |
| Food consumption (g) | 4.4 ± 0.3 |
| Faeces (g) | 1.0 ± 0.4 |
| Urine (mL) | 1.1 ± 0.1 |
| 0.1 µg/kg PLTX (n = 5) | |
| Food consumption (g) | 4.4 ± 0.2 |
| Faeces (g) | 1.3 ± 0.1 |
| Urine (mL) | 1.2 ± 0.2 |
| 0.3 µg/kg PLTX (n = 4) | |
| Food consumption (g) | 3.8 ± 0.6 |
| Faeces (g) | 1.5 ± 0.1 |
| Urine (mL) | 1.3 ± 0.5 |
| 1 µg/kg PLTX (n = 4) | |
| Food consumption (g) | 4.3 ± 0.3 |
| Faeces (g) | 1.0 ± 0.2 |
| Urine (mL) | 0.3 ± 0.1 |
| 3.5 µg/kg PLTX (n = 2) | |
| Food consumption (g) | 3.4 ± 0.8 |
| Faeces (g) | 0.9 ± 0.4 |
| Urine (mL) | 0.3 ± 0.2 |
| Group/Analyzed Parameters | Control (n = 6) | 0.03 µg/kg (n = 5) | 0.1 µg/kg (n = 5) | 0.3 µg/kg (n = 4) | 1 µg/kg (n = 3) | 3.5 µg/kg (n = 2) |
|---|---|---|---|---|---|---|
| Colour | (2) Pale Yellow (3) Amber (1) Dark Yellow | (1) Pale Yellow (1) Amber (3) Dark Yellow | (3) Amber (2) Dark Yellow | (4) Amber | (3) Amber | (2) Amber |
| Turbididy | (1) Clear (1) Slightly cloudy (4) Cloudy | (3) Clear (2) Cloudy | (1) Slightly cloudy (4) Cloudy | (4) Very Cloudy | (3) Cloudy | (2) Cloudy |
| Specific Gravity | (1) 1020 (1) 1032 (4) >1050 | (2) 1030 (1) 1032 (2) >1050 | (1) 1028 (4) >1050 | (1) 1038 (1) 1050 (2) >1050 | (3) >1050 | (2) >1050 |
| Urine protein (g/L) | 0.4 ± 0.2 (3) 0 (1) Trace (2) 1 | 0 ± 0 (4) 0 (1) Trace | 0.4 ± 0.2 (1) 0 (3) 0.3 (1) 1 | 0.3 ± 0 (1) Trace (3) 0.3 | 2.1 ± 1.5 (1) 0.3 (1) 1 (1) 5 | 0.7 ± 0.4 (1) 0.3 (1) 1 |
| Glucose (mmol/L) | 1.5 ± 0.7 (3) 0 (3) 3 | 3 ± 0 (5) 3 | 5.8 ± 2.8 (4) 3 (1) 17 | 3 ± 0 (4) 3 | 0 ± 0 (2) 0 | 0 ± 0 (2) 0 |
| Ketones (mmol/L) | 0.8 ± 0.4 (3) 0 (3) 1.5 | 0.3 ± 0.3 (4) 0 (1) 1.5 | 1.2 ± 0.3 (1) 0 (4) 1.5 | 0.4 ± 0.4 (3) 0 (1) 1.5 | 0.8 ± 0.8 (1) 0 (1) 1.5 | 0 ± 0 (2) 0 |
| Blood/Haemoglobin (Ery/µL) | 1.6 ± 1.6 (5) 0 (1) 10 | 0 ± 0 (5) 0 | 12 ± 9.7 (3) 0 (1) 10 (1) 50 | 2.5 ± 2.5 (3) 0 (1) 10 | 0 ± 0 (2) 0 | 5 ± 5 (1) 0 (1) 10 |
| Bilirubin (µmol/L) | 11.2 ± 8.3 (4) 0 (1) 17 (1) 50 | 10.2 ± 4.2 (2) 0 (3) 17 | 43.4 ± 6.6 * (1) 17 (4) 50 | 25.3 ± 8.3 (3) 17 (1) 50 | 17 ± 0 (2) 17 | 50 ± 0 * (2) 50 |
| Urobilinogen (µmol/L) | 2.8 ± 2.8 (5) 0 (1) 17 | 10.2 ± 4.2 (2) 0 (3) 17 | 6.8 ± 4.2 (3) 0 (2) 17 | 0 ± 0 (4) 0 | 8.5 ± 8.5 (1) 0 (1) 17 | 8.5 ± 8.5 (1) 0 (1) 17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boente-Juncal, A.; Raposo-García, S.; Vale, C.; Louzao, M.C.; Otero, P.; Botana, L.M. In Vivo Evaluation of the Chronic Oral Toxicity of the Marine Toxin Palytoxin. Toxins 2020, 12, 489. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12080489
Boente-Juncal A, Raposo-García S, Vale C, Louzao MC, Otero P, Botana LM. In Vivo Evaluation of the Chronic Oral Toxicity of the Marine Toxin Palytoxin. Toxins. 2020; 12(8):489. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12080489
Chicago/Turabian StyleBoente-Juncal, Andrea, Sandra Raposo-García, Carmen Vale, M. Carmen Louzao, Paz Otero, and Luis M. Botana. 2020. "In Vivo Evaluation of the Chronic Oral Toxicity of the Marine Toxin Palytoxin" Toxins 12, no. 8: 489. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12080489