Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies


1
Centre of New Technologies, University of Warsaw, ul. S. Banacha 2c, 02-097 Warsaw, Poland
2
Department of Bacterial Genetics, Institute of Microbiology, Faculty of Biology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland
*
Authors to whom correspondence should be addressed.
Toxins 2020, 12(9), 568; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12090568
Submission received: 19 August 2020
/
Revised: 31 August 2020
/
Accepted: 31 August 2020
/
Published: 4 September 2020
(This article belongs to the Special Issue Toxin-Antitoxin Systems in Pathogenic Bacteria)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The identification of novel targets for antimicrobial agents is crucial for combating infectious diseases caused by evolving bacterial pathogens. Components of bacterial toxin–antitoxin (TA) systems have been recognized as promising therapeutic targets. These widespread genetic modules are usually composed of two genes that encode a toxic protein targeting an essential cellular process and an antitoxin that counteracts the activity of the toxin. Uncontrolled toxin expression may elicit a bactericidal effect, so they may be considered “intracellular molecular bombs” that can lead to elimination of their host cells. Based on the molecular nature of antitoxins and their mode of interaction with toxins, TA systems have been classified into six groups. The most prevalent are type II TA systems. Due to their ubiquity among clinical isolates of pathogenic bacteria and the essential processes targeted, they are promising candidates for the development of novel antimicrobial strategies. In this review, we describe the distribution of type II TA systems in clinically relevant human pathogens, examine how these systems could be developed as the targets for novel antibacterials, and discuss possible undesirable effects of such therapeutic intervention, such as the induction of persister cells, biofilm formation and toxicity to eukaryotic cells.
Key Contribution: Bacterial toxin–antitoxin (TA) systems are promising targets for the development of novel antimicrobial strategies.
1. Introduction
The emergence of antibiotic-resistant bacteria is a critical issue in modern medicine [1]. Considerable efforts are being made to identify novel antimicrobials to combat infectious diseases caused by evolving multi-resistant pathogens. Bacterial toxin–antitoxin (TA) systems represent promising targets for such compounds. These small genetic modules encode two components: a stable toxin (always a protein) that recognizes a specific cellular target, and a labile antitoxin (protein or RNA molecule), produced in excess, that counteracts the activity of the toxin [2]. TAs have been classified into several groups based on the molecular nature of the antitoxin and their mode of interaction with their cognate toxin. The most prevalent and most extensively studied are type II TA systems, encoding proteic antitoxins which neutralize the toxin by forming TA complexes under optimal growth conditions [3,4]. These two-gene loci are organized in operons, whose expression is tightly regulated by both the antitoxin and toxin–antitoxin complexes [5,6]. Degradation of the antitoxin by cellular proteases liberates the toxin, which elicits a bacteriostatic or bactericidal effect [7,8,9,10].
TA loci were initially identified within bacterial plasmids, where they function as post-segregational cell killing systems (PSK), providing stable maintenance for their carrier replicon in a bacterial population due to the elimination of plasmid-less cells [11]. A surprising observation was that TA systems are also highly abundant and widespread in the chromosomes of free-living bacteria (both Gram-negative and Gram-positive), as well as in archaea [12]. More detailed studies have revealed that these TAs may be involved in important biological processes, such as (i) the stringent response, helping cells to survive stressful conditions by limiting various metabolic activities [13], (ii) programmed cell death [14] and (iii) biofilm formation [15]. In the light of these observations, it seems likely that chromosomal TA loci have been transferred to extrachromosomal replicons and adopted as plasmid stabilization systems.
The toxins of TA systems target essential cellular processes of bacteria; therefore, they may be considered intracellular “molecular time bombs”, which are activated under certain environmental conditions [16]. Compounds that can artificially activate TA toxins may form a new class of antimicrobials that could represent an alternative to antibiotics [17]. Type II TA systems are ubiquitous in bacteria [18], and their mechanisms are fairly well characterized [18], so they would appear to be excellent candidates for testing the merits of this idea.
The potential use of TA systems in combatting bacterial infections has been examined in several valuable reviews [19,20,21,22]. Here, we describe the most promising antibacterial strategies employing type II TA systems and discuss possible undesirable effects of their application, such as the induction of persister cells, biofilm formation and toxicity to eukaryotic cells. In addition, the distribution of type II TA systems among clinically relevant human pathogens is described, which may help to identify appropriate TA candidates for fighting particular infections.
2. Type II TA Systems in Pathogenic Bacteria
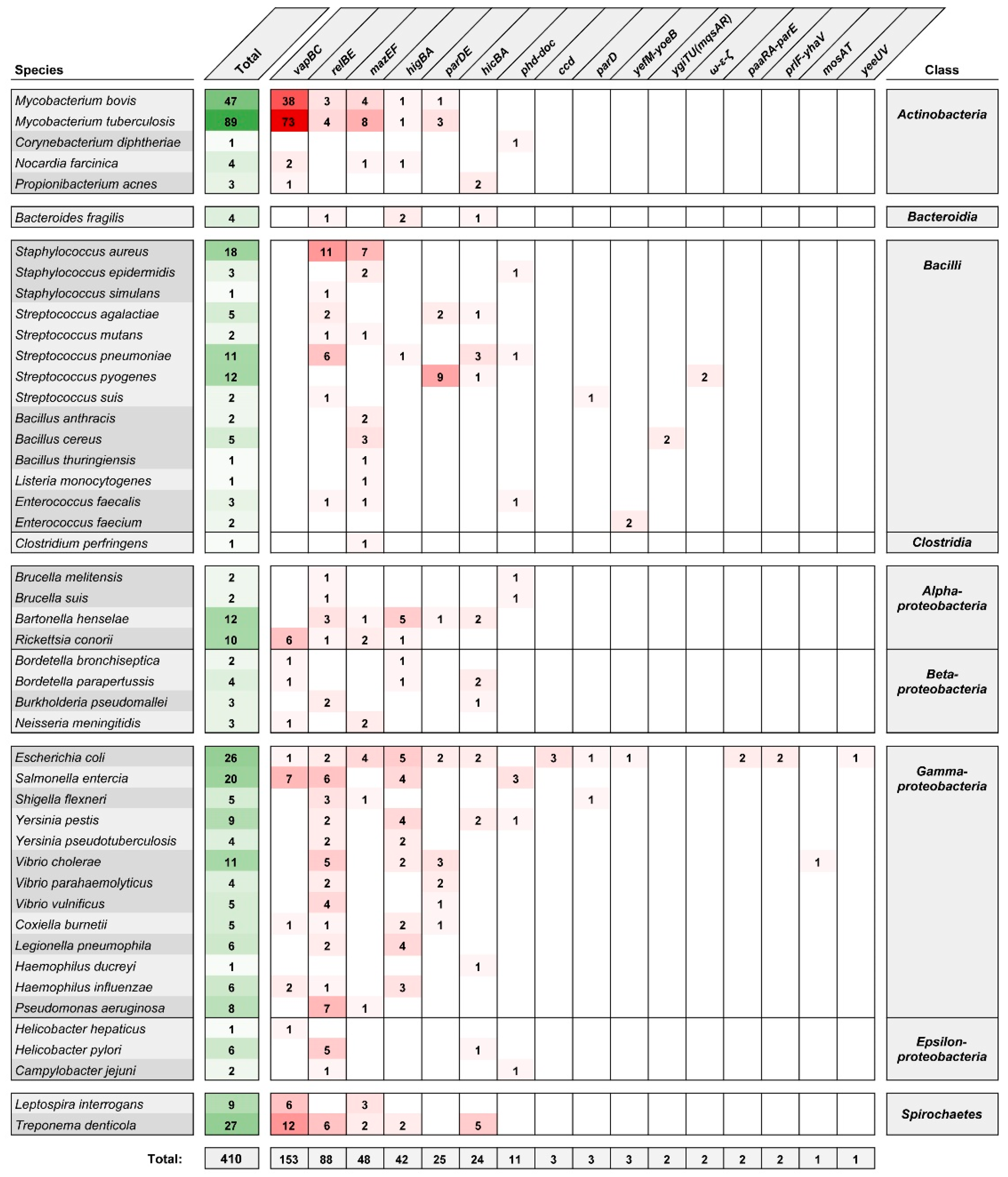
Since 2011, the studies of TA systems have been assisted by the Toxin–Antitoxin Database (TADB) [23], updated to version 2.0 in 2017 [24]. Currently, TADB 2.0 lists 6194 type II TA loci [24] categorized, where possible, in two partially interdependent ways. The first classification scheme uses a system based on the structural and functional characteristics of the toxin proteins [25,26]. There are 11 families of two-component type II TA systems: ccd, hicBA, hipBA, mazEF, parD(PemKI), parDE, phd-doc, relBE (with 5 subfamilies—relBE, higBA, yfeM-yoeB, ygiTU(mqsAR) and prlF-yhaV), vapBC, mosAT and yeeU. The less numerous three-component type II TA systems are ω-ε-ζ, pasABC and paaR-paaA-parE. The second classification scheme is based on a set of 44 conserved toxin–antitoxin protein domain pairings and better reflects the versatility and modularity of TA systems [27]. Figure 1 summarizes the variety of type II TA systems identified in bacterial species commonly associated with pathogenesis in humans. Among type II TA system (sub)families, four are most frequently encountered: vapBC, relBE, mazEF and higBA, and these account for ~80% of the listed loci. (An expanded list of type II TA systems of bacterial pathogens, organized into toxin–antitoxin domain pair groupings, is given in Supplementary Figure S1.)
Due to their widespread occurrence within the accessory genomes of human pathogens, their probable role in pathogenicity and potential as therapeutic targets, type II TA systems have been a subject of growing interest in recent years. Several systematic reviews have focused on the distribution and roles of the TA systems in clinically relevant pathogenic bacteria, including Escherichia coli, Mycobacterium tuberculosis, Neisseria gonorrheae, Streptococcus spp., Burkholderia spp. and species of the ESKAPE group (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) [19,28,29,30,31,32]. An increasing number of studies are being undertaken to describe the repertoire of TA systems in pathogenic bacteria at the level of a given strain or of a whole taxon. Not only do such approaches shed light on the importance of overall TA system networks with respect to the physiology, virulence and evolution of pathogens, but thanks to the large-scale genome comparisons involved, they also lead to the discovery of novel TA loci.
M. tuberculosis is notable for its abundance of TA systems, especially of the vapBC, mazEF and relBE families (Figure 1), and their activity has been linked to the regulation of adaptive responses to stress caused by interaction with the host and drug treatment [33]. Predictably, M. tuberculosis has been the object of studies on the common expression patterns of TA loci [34,35,36], as well as cross-activation between homologous systems [37,38], and putative interaction between non-cognate toxin–antitoxin pairs, which remains controversial [39,40,41]. Furthermore, comparative studies of the distribution of TA systems in mycobacteria have demonstrated the variability of TA loci between different M. tuberculosis lineages, and led to the discovery of putative novel TA systems [42,43]. Similarly, the body of knowledge about the TA systems of S. aureus [44] was recently expanded by several studies providing insights into the association between the TA systems landscape and strain phenotypes [45,46,47,48]. A recent in-depth analysis of the diversity and distribution of type II and type IV TA systems in the genomes of K. pneumoniae species complex identified several novel toxins and demonstrated the co-occurrence of TA loci and clinically relevant genes [49].
Comprehensive strategies for the identification of novel TA loci are required in order to gain a greater understanding of the role of TA networks in the biology of bacterial cells. In response to this need, Akarsu and colleagues created the “discovery-oriented” database TASmania in 2019 [50]. Using this newly developed pipeline, they annotated a set of over 41,000 assemblies from the EnsemblBacteria database, resulting in the identification of >2 × 106 candidate TA loci [50]. The greater flexibility of TASmania compared to the TAFinder search tool in TADB 2.0 allowed for the identification of a higher number of putative TA loci, thus providing a starting point for experimental analyses [50]. Moreover, the “guilt-by-association” strategy used throughout the annotation process, i.e., targeting loci directly neighboring orphan toxin or antitoxin genes, facilitated the discovery of new TA protein families. In the case of Listeria monocytogenes, TASmania-assisted large-scale genomic comparisons led to the identification of 14 putative TA genes [51].
3. Strategies for the Artificial Activation of Toxin–Antitoxin Systems
The growth-inhibitory and lethal consequences of the activity of TA systems led to the proposal that artificial activation of the toxins could provide an effective antibacterial strategy [17,52]. Type II TA systems seem to be most convenient for developing such strategies because many toxins of these systems have been thoroughly characterized and their cellular targets are known [53,54,55]. Moreover, as shown in Figure 1, the presence and conservation of type II TAs have been confirmed in major human-associated bacterial pathogens. Importantly, TA systems have no human homologs, and no pre-existing resistance against TA toxins has been observed. To date, several TA-based antibacterial strategies have been proposed [19,20], but in most cases they are not supported by significant experimental data.
In this review, we focus our attention on the most promising approaches: (i) the direct activation of TA systems by the use of specific molecules that interfere with TAs and imbalance the delicate stoichiometry of active toxin and antitoxin in bacterial cells, and (ii) the indirect activation of TAs by triggering other cellular components whose functions are interconnected with the TA system. Another novel and interesting approach that we consider involves the use of engineered species-specific toxins that can selectively kill selected strains of pathogenic bacteria.
3.1. Direct Activation of TA Systems
3.1.1. Disruption or Preventing the Formation of TA Complexes
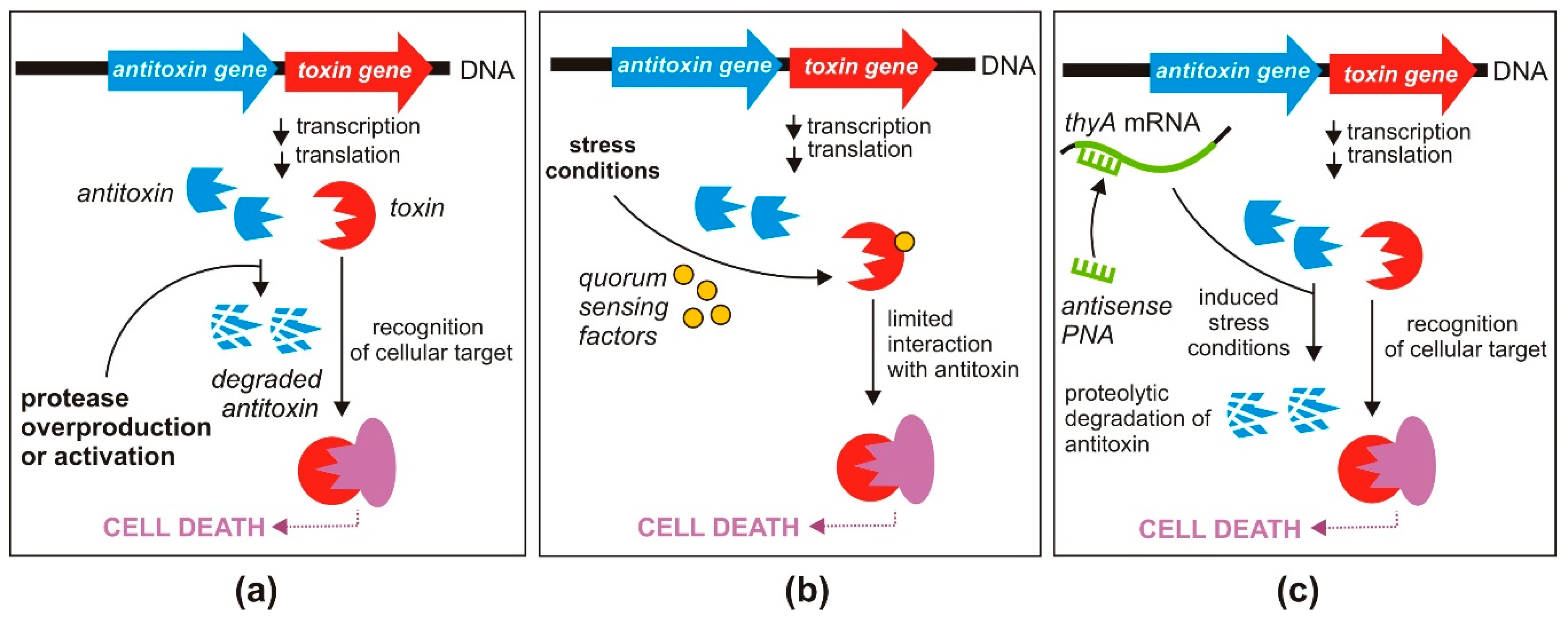
The most straightforward strategy for direct activation of the antibacterial effect of type II TA systems requires disruption of the protein toxin and antitoxin complexes, leading to toxin liberation (Figure 2a). This strategy has been validated by several research groups who focused their efforts on identifying high affinity peptide inhibitors that can efficiently displace toxins from their cognate antitoxins.
Lioy and co-workers selected the ε-ζ TA system from Streptococcus pyogenes as their research model because the free ζ toxin was shown to trigger a loss of cell proliferation similar to that caused by known antimicrobials [56]. Using high-throughput methods, they screened several oligopeptide libraries for the ability to impair the assembly of ε–ζ complexes. This led to the identification of a library containing a mixture of 17-amino acid-long oligopeptides that interfered with the TA interaction. However, further subfractionation of this library resulted in a diminished effect. The authors suggested that the disruption of ε–ζ complexes that was originally observed might have been a consequence of the concerted action of several weak-binding oligopeptides. Nevertheless, this study provided a proof-of-concept for the antimicrobial potential of this strategy [56].
A similar approach was applied in the case of two related TA systems—pemIK and moxXT of Bacillus anthracis. The former module encodes the toxin PemK, a ribonuclease whose overexpression exerts a toxic effect in B. anthracis cells characterized by the drastic inhibition of protein synthesis [57]. Based on in silico protein structural modeling, several peptides were designed to mimic the C-terminal domain of the antitoxin PemI, which is involved in toxin binding. In vitro experiments indicated the effectiveness of the designed molecules and demonstrated the feasibility of disrupting the TA interaction using octapeptides [58]. Similar results were obtained in studies on the related TA system moxXT. Based on the crystal structure of the MoxX–MoxT complex, Verma and colleagues [59] designed a series of peptides that effectively disturbed the interaction of MoxT with antitoxin MoxX and stimulated MoxT ribonuclease activity [60].
3.1.2. Inhibition of Antitoxin Translation
Toxin release can also be caused by a reduction in the amount of antitoxin molecules in a bacterial cell. One way to achieve such an effect is by blocking antitoxin translation (Figure 2b) [20]. The toxin and antitoxin genes, co-transcribed as a single mRNA, possess separate Shine–Dalgarno sequences [62]. Therefore, inhibition of antitoxin translation should not influence the translation of the toxin protein. This strategy was tested by Równicki and co-workers who designed a peptide nucleic acid (PNA)-based treatment to inhibit translation of the antitoxins of the mazEF and hipBA TA systems of E. coli [63].
The sequence-specific antisense PNAs targeted either mazE or hipB antitoxin mRNAs and as a result lowered the cellular levels of these transcripts, which caused an effective inhibition of E. coli growth [63]. Importantly, the PNA treatment did not change the relative levels of the mazF or hipA toxin mRNAs. The crucial role of the MazF and HipA toxins in the observed growth inhibition was confirmed by showing that E. coli mutants lacking the genes encoding these proteins were “resistant” to treatment with the antitoxin-specific PNAs.
Another PNA-based strategy, described in the same report, used PNA oligomers directed at the cellular target of the toxin, thus bypassing the involvement of the TA system. It was demonstrated that PNAs can mimic the action of the HipA toxin by silencing the gltX gene encoding its cellular target, glutamyl-tRNA synthase [63]. For these experiments, the PNA oligomers were conjugated with a cell penetrating peptide—(KFF)3K, which indicated the importance of employing an efficient carrier to introduce these antisense oligonucleotides into bacterial cells [63].
The above results confirmed experimentally that TA systems are susceptible to sequence-specific antisense agents and provided a basis for their further exploitation in antimicrobial strategies. Importantly, PNA oligomers exhibit nuclease and protease resistance, high binding affinity to natural nucleic acids, and negligible toxicity to eukaryotic cells [64,65].
3.2. Indirect Activation of TA Systems
3.2.1. Enhanced Expression of Proteases Degrading Antitoxins
As previously mentioned, the antitoxins of type II TA systems are more susceptible to degradation by host cytoplasmic proteases than their cognate toxins. In most cases, bacterial Lon protease is involved in antitoxin degradation; however, the two-component protease ClpP, acting in cooperation with the chaperones ClpA or ClpX, is used by some TA systems [66,67,68].
The depletion of antitoxin molecules results in the liberation of the toxin proteins from TA complexes and relieves transcriptional repression of TA operons, causing increased production of TA transcripts. These events alter the stoichiometry of TA molecules in a cell and ultimately have a lethal effect [69]. Therefore, increasing the level of cellular proteases or designing specific molecules that activate these proteases could be a promising strategy for the indirect activation of toxins (Figure 3a).
Increased protease expression can be achieved by introducing a plasmid carrying a cloned protease gene. Overproduction of the Lon protease is known to be lethal for E. coli cells [70]. However, by employing an inducible Lon overproduction system, Christensen and colleagues overcame this problem to demonstrate that the lethality is partially dependent on the yefM-yoeB TA system [66]. They showed that overproduction of Lon triggered YoeB-dependent mRNA cleavage, leading to translation inhibition. This, in turn, activated the YoeB toxin by preventing the synthesis of its unstable YefM antidote, which was eventually lethal to the host cells [66].
Proteolysis is a tightly controlled process which can be significantly influenced by specific molecules targeting proteases. For example, acyldepsipeptides (ADEPs) are compounds with antibiotic properties that specifically activate the bacterial protease ClpP. Uncontrolled proteolysis induced in this way inhibits bacterial cell division and results in cell death, possibly with the participation of activated toxins [71].
3.2.2. Triggering of TA Systems by Quorum-Sensing Factors
Another interesting antimicrobial strategy was developed by Kumar and Engelberg-Kulka [72,73]. Their approach targeted mazEF TA systems, which are among the most abundant bacterial TA loci (Figure 1). Toxin MazF is a sequence-specific endoribonuclease that initiates a programmed cell death pathway in response to environmental stress conditions [74], while MazE is a labile antitoxin that is preferentially degraded by the serine protease ClpAP [14].
The proposed strategy involves the use of a newly discovered group of pentapeptides secreted by bacteria, called extracellular death factors (EDFs), which act in quorum sensing and enhance MazF activity under stressful conditions (Figure 3b) [72]. Interestingly, it was shown that EDFs bind directly to MazF in a sequence-specific manner, and this binding is likely to limit interaction of the toxin with its cognate antitoxin MazE [73,75]. EDFs can also stimulate activation of mazEF in heterologous hosts, which might broaden the potential application of this strategy [73,75]. Although an early study on MazF classified its toxicity as lethal to cells [14], this statement has since been revisited, suggesting that MazF is involved in growth arrest rather than cell death [76,77].
3.2.3. Induction of the Stringent Response
Another antibacterial strategy involving the indirect activation of toxins is based on induction of the stringent response, a conserved mechanism that allows bacteria to adapt their metabolism in response to stressful environmental conditions, e.g., nutrient deprivation. Many chromosomal type II TA systems, including the aforementioned mazEF loci, are transcriptionally upregulated under stressful conditions [78], and their activity leads to remodeling of cellular metabolism and/or programmed cell death, affecting part of the bacterial community [74].
The stringent response (mediated by the alarmone guanosine 3,5 bispyrophosphate, ppGpp) is activated by different natural starvation and stress signals [79], and this can also be achieved by the application of artificial factors. Równicki and colleagues used sequence-specific PNAs targeting the thyA gene of E. coli, conjugated with a (KFF)3K peptide as a carrier, to trigger MazF toxin production by inducing thymine starvation [63] (Figure 3c). The thyA gene encodes thymidylate synthase, an enzyme involved in folic acid metabolism, which normally interferes with mazEF-mediated growth inhibition [74]. As shown for E. coli, thymine starvation leads to accumulation of ppGpp in bacterial cells, which reduces global transcription [80]. As a consequence, the inhibition of transcription of mazEF leads to activation of the MazF toxin [81]. The significantly reduced level of thyA mRNA after treatment with a complementary anti-thyA PNA and the resulting growth inhibition confirmed the effectiveness of this silencing strategy [63].
3.3. Engineered TA Systems in a Targeted Killing Strategy
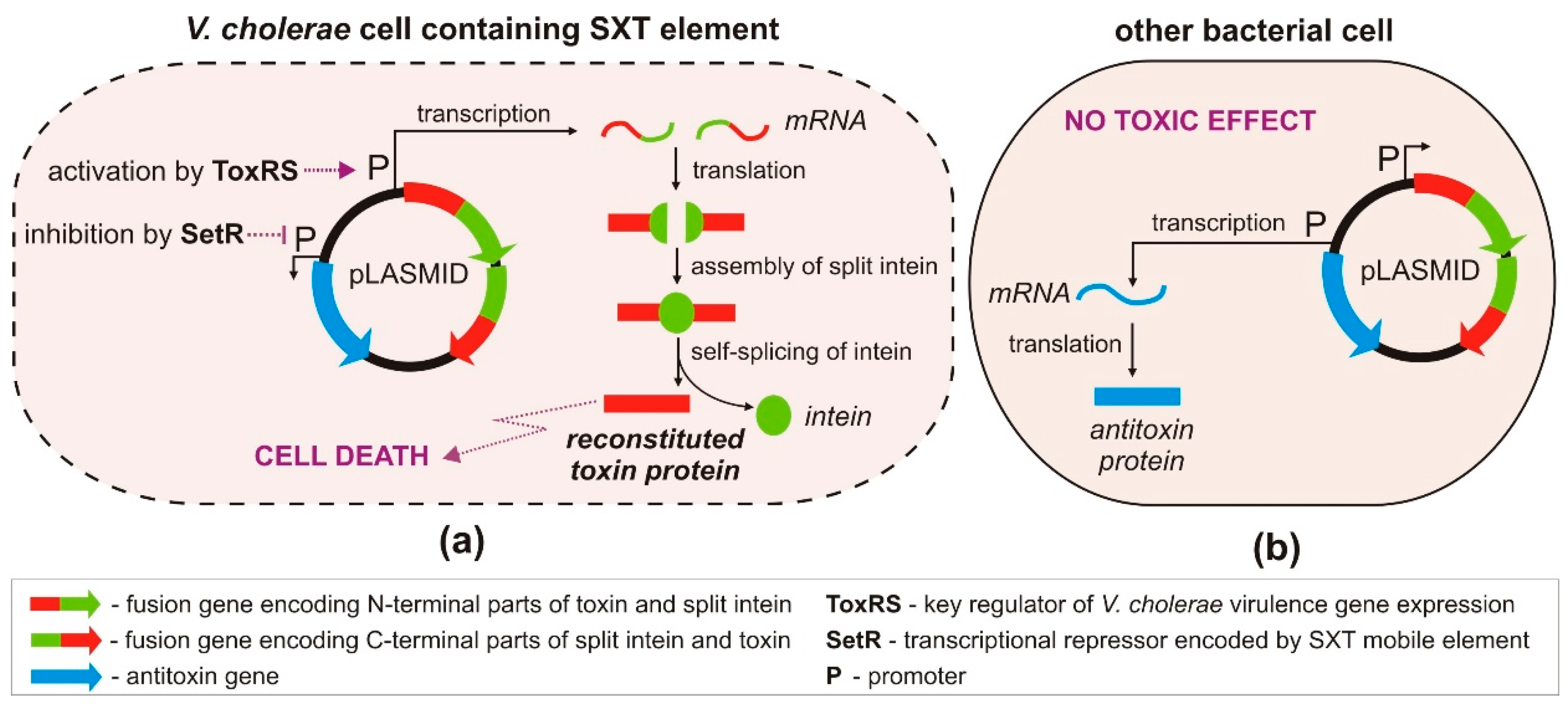
An innovative strategy for the targeted killing of selected pathogenic bacteria, without harming beneficial members of the microbiota inhabiting eukaryotic host organisms, was recently proposed by López-Igual et al. [82] (Figure 4). This strategy is based on the use of the ccdA-ccdB type II TA system, encoding the toxin CcdB that poisons DNA gyrase—an enzyme responsible for the negative supercoiling of bacterial DNA [83]. CcdB interferes with the activity of DNA gyrase, inducing it to form a covalent GyrA–DNA complex that cannot be resolved, thus promoting DNA breakage and cell death. This mechanism is closely related to the action of quinolone antibiotics, which also target DNA gyrase [84].
To better control CcdB production in vivo, the gene for this toxin was divided into two parts, and each was fused with DNA encoding split inteins. The generation of a functional toxin occurs in three stages: (i) expression of the two polypeptides, (ii) their association and ligation into a single fusion protein, and (iii) self-splicing of the intein (Figure 4). An important step in this strategy was the construction of a mobilizable plasmid containing genes encoding the antitoxin and the engineered toxin–intein, whose expression could be independently regulated in bacteria harboring specific transcription factors. Although the plasmid could be readily transferred by conjugation from E. coli to other bacteria, the toxic effect was observed exclusively in strains of Vibrio cholerae. This host specificity was achieved by cloning the engineered ccdB toxin–intein genes downstream of a promoter regulated by the transcriptional activator ToxRS, a cholera toxin-associated activator characteristic for V. cholerae [85] (Figure 4). Bacterial strains lacking ToxRS were unaffected, including the E. coli donor strain and non-pathogenic Vibrio spp.
The second component of the TA module, the antitoxin gene ccdA, was placed under the transcriptional control of the repressor SetR. The presence of setR is considered a hallmark of SXT—an integrative and conjugative mobile element (ICE) of V. cholerae that often includes various antibiotic resistance genes [86]. Therefore, expression of the antitoxin gene is repressed in pathogenic multidrug-resistant V. cholerae cells containing the setR gene, allowing the toxin to poison gyrase and cause a bactericidal effect (Figure 4). However, in cells that lack setR, the antitoxin CcdA is produced, neutralizing the effects of the toxin. Thus, the described approach allows species-specific killing of antibiotic-resistant V. cholerae strains without affecting the growth of other bacteria present in the mixed populations. The effectiveness of this strategy was confirmed in vivo, in the microbiota of zebrafish and crustacean larvae, where Vibrio spp. naturally occur [82].
4. Toxicity to Eukaryotic Cells
Although TA loci do not occur in eukaryotic genomes, most toxins of TA systems are endoribonucleases that cleave mRNAs irrespective of their origin, be that prokaryotic or eukaryotic. Therefore, before using these toxins in potential antibacterial strategies, it is important to consider possible cytotoxicity and side effects on human cells. Unfortunately, only a few studies have addressed this issue to date. Notably, toxin cytotoxicity was exploited in one study that employed an engineered version of the MazF toxin to reduce solid tumors in mice [87]. Similarly, the VapC toxin from a TA system of M. tuberculosis demonstrated pro-apoptotic activity in human cancer cells, regardless of the expression system used. In another study, Chono and colleagues found that the MazF toxin dosage is a critical factor in determining its activity and cytotoxicity in eukaryotic cells [88,89]. The above examples show that mammalian cells are sensitive to the ribonuclease activity of toxins. However, the ability of bacterial TA-system toxins to penetrate eukaryotic cells is currently unknown, and strategies that specifically target pathogens or utilize toxins that lack targets in human cells might prevent any deleterious effects.
5. Role of Type II TA Modules in Biofilm Formation and Bacterial Persistence
Two potentially undesirable effects of artificial activation of TAs are bacterial persistence and biofilm formation. Persister cells are a subpopulation of slow-growing or growth-arrested bacterial cells that have a decreased susceptibility to killing by bactericidal antibiotics within an otherwise susceptible clonal population [90]. It has been shown that increased tolerance of biofilms to antibiotics is due to the higher amounts of persister cells within the biofilm community [91,92]. While antibiotics kill the majority of biofilm cells, persisters remain viable and repopulate biofilms when the level of antibiotics drops [93]. Thus, persister cells appear to play a central role in the recalcitrance of chronic and biofilm-related infections [94].
One of the first items of evidence linking type II TA modules to biofilm formation comes from a well-characterized chromosomal TA system mqsRA in E. coli [95,96]. The mqsRA is a unique type II TA system where the toxin gene msqR precedes the antitoxin msqA [97]. It has been shown that a Tn5 insertion mutant of the toxin mqsR formed less adherent biomass [96]. On the contrary, later reanalysis of the msqRA revealed that this TA module does not affect biofilm formation in nutrient-rich conditions [98]. The authors identified two new promoters located in the toxin coding sequence that allow the constitutive expression of mqsA, thereby allowing a constant and steady level of the MqsA antitoxin compared to the MqsR toxin. This work, quite understandably, has opened up the debate about the role of TA systems in persistence and biofilm formation [78,99]. This result disproves the role of mqsRA (and nine others) TA modules in spontaneous biofilm formation, but does not address the question of a role of TA systems in growth control and biofilm formation under stress conditions. The first persistence-related gene to be identified was hipA, encoding the toxin of the E. coli hipBA TA system—a serine/threonine kinase that inhibits cell growth by inactivating the glutamyl-tRNA synthetase GltX [100]. This discovery linked TA modules and bacterial persistence for the first time. Since then, correlations between antibiotic persistence and TA systems have been extensively studied. Evidence both for and against the participation of TA systems in persister cell formation has been accumulated, making their contribution to this phenomenon unclear [99,101,102,103,104]. For example, the deletion of 10 ribonuclease-encoding TA systems from the E. coli genome was found to decrease the number of persisters. However, it was subsequently shown that this result was influenced by the presence of φ80 bacteriophage contamination [105]. Moreover, reconstruction of this mutant strain demonstrated that deletion of the 10 TA systems did not affect susceptibility to ofloxacin or ampicillin [106]. These contradictory findings have given rise to considerable debate [78,107,108,109], and the relevance of TA systems to bacterial persistence remains unclear.
In contrast, the results of several other studies support the contribution of TA systems to bacterial persistence. For example, overexpression of the toxins RelE or MazF was shown to increase the survival of E. coli under antibiotic exposure [9,110]. In another study, the dinJ/yafQ TA module was found to be involved in tolerance to cephalosporin and aminoglycoside antibiotics [111]. Further evidence linking type II TA modules and bacterial persistence was obtained in a study on uropathogenic isolates of E. coli [112]. TA systems have also been linked to bacterial persistence in Salmonella, where the shpB1 allele, carrying a mutation in the antitoxin of the shpAB TA module, was associated with the salmonella high persister phenotype [113]. In addition to the shpAB system, 13 other type II TA modules were shown to contribute to persistence in Salmonella triggered by stresses encountered during macrophage infection [114]. Furthermore, the overexpression of three acetyltransferase toxins (TacT, TacT2, TacT3—acetylating tRNA) in Salmonella enterica was found to increase the level of persisters in the population, and their deletion resulted in a decrease in the proportion of persister cells [115,116]. Since the general mechanism of persistence is still unclear, it is crucial to determine whether the activation of TAs influences persister or biofilm formation before they are considered for use as antimicrobial targets. Besides the type II TA modules, other TA systems have been shown to be involved in persistence in E. coli. For further information see important recent publications by Fisher et al. [117], Ronneau and Helaine [78], Wilmaerts et al. [118], Dorr et al. [119], and papers from the Wood group [120,121].
While persistence is among the possible side effects of using TAs as antibacterial targets, these systems can also serve in antipersister strategies [122]. Conlon and colleagues [123] reasoned that a compound capable of striking a target in dormant cells will kill persisters. They used an acyldepsipeptide antibiotic (ADEP4) to globally activate the protease ClpP and showed that this becomes fairly non-specific and kills persisters by degrading over 400 proteins, which forces cells to self-digest. Subsequently, this strategy was used to eradicate a biofilm in an animal model, which confirmed its potential as the basis of therapies to treat chronic infections. In a different approach, Li and co-workers [124] identified a novel inhibitor of the E. coli HipA toxin which interfered with persister formation in an antibiotic-independent manner. A comprehensive review describing current antipersister strategies was recently published by Defraine et al. [122].
6. Conclusions
Although TA-based antimicrobial strategies have great potential, it is still too early to assess the therapeutic value of toxin activation in clinical settings. Drug discovery and development are time consuming and costly processes, so selecting the right approach will be very important if efforts to produce new antimicrobials based on TA activators are to progress. It is also crucial to select the correct TA targets, and such decisions should be based on data concerning their clinical relevance (i.e., prevalence in antibiotic-resistant clinical isolates), effect exerted on bacterial cells (bactericidal, bacteriostatic), influence on persister cell and biofilm formation, cytotoxicity to human cells and lastly the necessary mode of drug delivery. It would appear advantageous to combine the use of TA activators with conventional antibiotics, since such a strategy could be more effective against a broader spectrum of multidrug-resistant strains [63]. The use of engineered toxins is another promising avenue of research [82,125]. According to the type and properties of such recombinant proteins, they might be applied in targeted antimicrobial strategies or directed against human cancer cells in novel anti-cancer therapies.
Supplementary Materials
The following is available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6651/12/9/568/s1, Figure S1: The number of the type II TA systems identified in human pathogenic bacteria according to the toxin-antitoxin domain pair system [27] as collected in TADB 2.0 [24].
Funding
We acknowledge support from the National Science Centre, Poland (PRELUDIUM 2017/25/N/NZ1/01578 to M.R. and J.T.; UMO-2016/23/B/NZ1/03198 to J.T).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ventola, C.L. The antibiotic resistance crisis: Causes and threats. Pharm. Ther. J. 2015, 40, 277–283. [Google Scholar]
- Gerdes, K.; Nielsen, A.; Thorsted, P.; Wagner, E.G.H. Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable Hok, SrnB and PndA effector messenger RNAs. J. Mol. Biol. 1992, 226, 637–649. [Google Scholar] [CrossRef]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II toxin-antitoxin systems: Evolution and revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Coussens, N.P.; Daines, D.A. Wake me when it’s over-Bacterial toxin-antitoxin proteins and induced dormancy. Exp. Biol. Med. 2016, 241, 1332–1342. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Piscotta, F.J.; Jeffrey, P.D.; James Link, A. ParST is a widespread toxin–antitoxin module that targets nucleotide metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin–antitoxin systems. Biology, identification, and application. Mob. Genet. Elem. 2013, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180. [Google Scholar] [CrossRef] [Green Version]
- Robson, J.; McKenzie, J.L.; Cursons, R.; Cook, G.M.; Arcus, V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009, 390, 353–367. [Google Scholar] [CrossRef]
- Dziewit, L.; Jazurek, M.; Drewniak, L.; Baj, J.; Bartosik, D. The SXT conjugative element and linear prophage N15 encode toxin-antitoxin-stabilizing systems homologous to the tad-ata module of the Paracoccus aminophilus plasmid pAMI2. J. Bacteriol. 2007, 189, 1983–1997. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef] [Green Version]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by 3′,5′-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin-antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef]
- Kȩdzierska, B.; Lian, L.Y.; Hayes, F. Toxin-antitoxin regulation: Bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 2007, 35, 325–339. [Google Scholar] [CrossRef]
- Williams, J.; Hergenrother, P. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2014, 20, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Lee, B.J. Structure, biology, and therapeutic application of toxin-antitoxin systems in pathogenic bacteria. Toxins (Basel) 2016, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.T.; Balsa, D.; Espinosa, M. One cannot rule them all: Are bacterial toxins-antitoxins druggable? FEMS Microbiol. Rev. 2015, 39, 522–540. [Google Scholar] [CrossRef] [Green Version]
- Bravo, A.; Ruiz-Cruz, S.; Alkorta, I.; Espinosa, M. When humans met superbugs: Strategies to tackle bacterial resistances to antibiotics. Biomol. Concepts 2018, 9, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Carlos, L.; Barbosa, B.; Sânder, A.; Cangussu, R.; Garrido, S.S.; Marchetto, R. Toxin-antitoxin systems and its biotechnological applications. Afr. J. Biotechnol. 2014, 13, 11–17. [Google Scholar]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; De Bast, M.S. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of Type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, L.; Blasco, L.; Lopez, M.; Bou, G.; Garcia-Contreras, R.; Wood, T.; Tomas, M. Toxin-antitoxin systems in clinical pathogens. Toxins (Basel) 2016, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [Green Version]
- Kędzierska, B.; Hayes, F. Emerging roles of toxin-antitoxin modules in bacterial pathogenesis. Molecules 2016, 21, 790. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.E.; Walsh, T.R. Toxin-antitoxin systems and their role in disseminating and maintaining antimicrobial resistance. FEMS Microbiol. Rev. 2017, 41, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Kim, D.H.; Jin, C.; Lee, B.J. A systematic overview of type II and III toxin-antitoxin systems with a focus on druggability. Toxins (Basel) 2018, 10, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slayden, R.A.; Dawson, C.C.; Cummings, J.E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018, 76, fty039. [Google Scholar] [CrossRef] [PubMed]
- Korch, S.B.; Malhotra, V.; Contreras, H.; Clark-Curtiss, J.E. The Mycobacterium tuberculosis relBE toxin:antitoxin genes are stress-responsive modules that regulate growth through translation inhibition. J. Microbiol. 2015, 53, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Venkataraman, B.; Vasudevan, M.; Gopinath Bankar, K. Co-expression network analysis of toxin-antitoxin loci in Mycobacterium tuberculosis reveals key modulators of cellular stress. Sci. Rep. 2017, 7, 5868. [Google Scholar] [CrossRef] [PubMed]
- Thakur, Z.; Saini, V.; Arya, P.; Kumar, A.; Mehta, P.K. Computational insights into promoter architecture of toxin-antitoxin systems of Mycobacterium tuberculosis. Gene 2018, 641, 161–171. [Google Scholar] [CrossRef]
- Yang, M.; Gao, C.; Wang, Y.; Zhang, H.; He, Z.G. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PLoS ONE 2010, 5, e10672. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Tiwari, P.; Deep, A.; Kidwai, S.; Gupta, S.; Thakur, K.G.; Singh, R. System-wide analysis unravels the differential regulation and in vivo essentiality of virulence-associated proteins B and C toxin-antitoxin systems of Mycobacterium tuberculosis. J. Infect. Dis. 2018, 217, 1809–1820. [Google Scholar] [CrossRef]
- Zhu, L.; Sharp, J.D.; Kobayashi, H.; Woychik, N.A.; Inouye, M. Noncognate Mycobacterium tuberculosis toxin-antitoxins can physically and functionally interact. J. Biol. Chem. 2010, 285, 39732–39738. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.V.; Dawson, C.C.; Crew, R.; England, K.; Slayden, R.A. MazF6 toxin of Mycobacterium tuberculosis demonstrates antitoxin specificity and is coupled to regulation of cell growth by a Soj-like protein. BMC Microbiol. 2013, 13, 240. [Google Scholar] [CrossRef] [Green Version]
- Tandon, H.; Melarkode Vattekatte, A.; Srinivasan, N.; Sandhya, S. Molecular and structural basis of cross-reactivity in M. tuberculosis toxin–antitoxin systems. Toxins (Basel) 2020, 12, 481. [Google Scholar] [CrossRef] [PubMed]
- Solano-Gutierrez, J.S.; Pino, C.; Robledo, J. Toxin-antitoxin systems shows variability among Mycobacterium tuberculosis lineages. FEMS Microbiol. Lett. 2019, 366, fny276. [Google Scholar] [CrossRef] [PubMed]
- Tandon, H.; Sharma, A.; Wadhwa, S.; Varadarajan, R.; Singh, R.; Srinivasan, N.; Sandhya, S. Bioinformatic and mutational studies of related toxin–antitoxin pairs in Mycobacterium tuberculosis predict and identify key functional residues. J. Biol. Chem. 2019, 294, 9048–9063. [Google Scholar] [CrossRef] [Green Version]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems of Staphylococcus aureus. Toxins (Basel) 2016, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, M.; Hyz, K.; Janczak, M.; Hydzik, M.; Dubin, G.; Wladyka, B. Identification of novel mazEF/pemIK family toxin-antitoxin loci and their distribution in the Staphylococcus genus. Sci. Rep. 2017, 7, 13462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, G.; Zhu, Q.; Sun, B. Bioinformatics and functional assessment of toxin-antitoxin systems in Staphylococcus aureus. Toxins (Basel) 2018, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Kato, F.; Yoshizumi, S.; Yamaguchi, Y.; Inouye, M. Genome-wide screening for identification of novel toxin-antitoxin systems in Staphylococcus aureus. Appl. Environ. Microbiol. 2019, 85, e00915-19. [Google Scholar] [CrossRef]
- Sierra, R.; Viollier, P.; Renzoni, A. Linking toxin-antitoxin systems with phenotypes: A Staphylococcus aureus viewpoint. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 742–751. [Google Scholar] [CrossRef]
- Horesh, G.; Fino, C.; Harms, A.; Dorman, M.J.; Parts, L.; Gerdes, K.; Heinz, E.; Thomson, N.R. Type II and type IV toxin-antitoxin systems show different evolutionary patterns in the global Klebsiella pneumoniae population. Nucleic Acids Res. 2020, 48, 4357–4370. [Google Scholar] [CrossRef] [Green Version]
- Akarsu, H.; Bordes, P.; Mansour, M.; Bigot, D.J.; Genevaux, P.; Falquet, L. TASmania: A bacterial toxin-antitoxin systems database. PLoS Comput. Biol. 2019, 15, e1006946. [Google Scholar] [CrossRef] [Green Version]
- Agüero, J.A.; Akarsu, H.; Aguilar-Bultet, L.; Oevermann, A.; Falquet, L. Large-scale comparison of toxin and antitoxins in listeria monocytogenes. Toxins (Basel) 2020, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S. Speculative strategies for new antibacterials: All roads should not lead to Rome. J. Antibiot. (Tokyo) 2013, 66, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Castro-Roa, D.; Zenkin, N.; Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 2013, 52, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-P.; Wang, Q.; Quan, S.-W.; Yu, X.-Q.; Wang, Y.; Guo, D.-D.; Peng, L.; Feng, H.-Y.; Yong-Xing, H. Type II toxin–antitoxin system in bacteria: Activation, function, and mode of action. Biophys. Rep. 2020, 6, 68–79. [Google Scholar] [CrossRef]
- Ramisetty, B.C.M.; Santhosh, R.S. Endoribonuclease type II toxin-antitoxin systems: Functional or selfish? Microbiology 2017, 163, 931–939. [Google Scholar] [CrossRef]
- Lioy, V.S.; Rey, O.; Balsa, D.; Pellicer, T.; Alonso, J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid 2010, 63, 31–39. [Google Scholar] [CrossRef]
- Agarwal, S.; Agarwal, S.; Bhatnagar, R. Identification and characterization of a novel toxin-antitoxin module from Bacillus anthracis. FEBS Lett. 2007, 581, 1727–1734. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK toxin of Bacillus anthracis is a ribonuclease: An insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Kumar, S.; Gupta, V.P.; Gourinath, S.; Bhatnagar, S.; Bhatnagar, R. Structural basis of Bacillus anthracis MoxXT disruption and the modulation of MoxT ribonuclease activity by rationally designed peptides. J. Biomol. Struct. Dyn. 2015, 33, 606–624. [Google Scholar] [CrossRef]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided. Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef]
- Lee, I.G.; Lee, S.J.; Chae, S.; Lee, K.Y.; Kim, J.H.; Lee, B.J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015, 43, 7624–7637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.T.; Moreno-Córdoba, I.; Yeo, C.C.; Espinosa, M. Toxin-antitoxin genes of the Gram-positive pathogen Streptococcus pneumoniae: So few and yet so many. Microbiol. Mol. Biol. Rev. 2012, 76, 773–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Równicki, M.; Pieńko, T.; Czarnecki, J.; Kolanowska, M.; Bartosik, D.; Trylska, J. Artificial activation of Escherichia coli mazEF and hipBA toxin–antitoxin systems by antisense peptide nucleic acids as an antibacterial strategy. Front. Microbiol. 2018, 9, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, M.; Równicki, M.; Mieczkowski, A.; Miszkiewicz, J.; Trylska, J. Antibacterial peptide nucleic acids—Facts and perspectives. Molecules 2020, 25, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.K.; Maenhaut-Michel, G.; Mine, N.; Gottesman, S.; Gerdes, K.; Van Melderen, L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: Involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 2004, 51, 1705–1717. [Google Scholar] [CrossRef]
- Brzozowska, I.; Zielenkiewicz, U. The ClpXP protease is responsible for the degradation of the Epsilon antidote to the Zeta toxin of the streptococcal pSM19035 plasmid. J. Biol. Chem. 2014, 289, 7514–7523. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; Thi, M.H.D.; Lecchi, P.; Gottesman, S.; Couturier, M.; Maurizi, M.R. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996, 271, 27730–27738. [Google Scholar] [CrossRef] [Green Version]
- Muthuramalingam, M.; White, J.C.; Bourne, C.R. Toxin-antitoxin modules are pliable switches activated by multiple protease pathways. Toxins (Basel) 2016, 8, 214. [Google Scholar] [CrossRef]
- Goff, S.A.; Goldberg, A.L. An increased content of protease La, the lon gene product, increases protein degradation and blocks growth in Escherichia coli. J. Biol. Chem. 1987, 262, 4508–4515. [Google Scholar]
- Brötz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Engelberg-Kulka, H. Quorum sensing peptides mediating interspecies bacterial cell death as a novel class of antimicrobial agents. Curr. Opin. Microbiol. 2014, 21, 22–27. [Google Scholar] [CrossRef]
- Kumar, S.; Kolodkin-Gal, I.; Engelberg-Kulka, H. Novel quorum-sensing peptides mediating interspecies bacterial cell death. MBio 2013, 4, e00314-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelberg-Kulka, H.; Hazan, R.; Amitai, S. mazEF: A chromosomal toxin-antitoxin module that triggers programmed cell death in bacteria. J. Cell Sci. 2005, 118, 4327–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, A.; Kumar, S.; Engelberg-Kulka, H. Quorum sensing extracellular death peptides enhance the endoribonucleolytic activities of mycobacterium tuberculosis MazF toxins. MBio 2018, 9, e00685-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramisetty, B.C.M.; Raj, S.; Ghosh, D. Escherichia coli MazEF toxin-antitoxin system does not mediate programmed cell death. J. Basic Microbiol. 2016, 56, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tamber, S.; Memmi, G.; Donegan, N.P.; Cheung, A.L. Overexpression of MazF in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J. Bacteriol. 2009, 191, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Ronneau, S.; Helaine, S. Clarifying the link between toxin–antitoxin modules and bacterial persistence. J. Mol. Biol. 2019, 431, 3462–3471. [Google Scholar] [CrossRef]
- Jimmy, S.; Saha, C.K.; Kurata, T.; Stavropoulos, C.; Oliveira, S.R.A.; Koh, A.; Cepauskas, A.; Takada, H.; Rejman, D.; Tenson, T.; et al. A widespread toxin–antitoxin system exploiting growth control via alarmone signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 10500–10510. [Google Scholar] [CrossRef]
- Török, I.; Kari, C. Accumulation of ppGpp in a relA mutant of Escherichia coli during amino acid starvation. J. Biol. Chem. 1980, 255, 3838–3840. [Google Scholar]
- Ramisetty, B.C.M.; Natarajan, B.; Santhosh, R.S. MazEF-mediated programmed cell death in bacteria: “What is this?”. Crit. Rev. Microbiol. 2015, 41, 89–100. [Google Scholar] [CrossRef] [PubMed]
- López-Igual, R.; Bernal-Bayard, J.; Rodríguez-Patón, A.; Ghigo, J.M.; Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. 2019, 37, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, P.; Kézdy, K.E.; Van Melderen, L.; Steyaert, J.; Wyns, L.; Pato, M.L.; Higgins, P.N.; Couturier, M. The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. J. Mol. Biol. 1993, 234, 534–541. [Google Scholar] [CrossRef]
- Childers, B.M.; Klose, K.E. Regulation of virulence in Vibrio cholerae: The ToxR regulon. Future Microbiol. 2007, 2, 335–344. [Google Scholar] [CrossRef]
- Beaber, J.W.; Hochhut, B.; Waldor, M.K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 2004, 427, 72–74. [Google Scholar] [CrossRef]
- Shimazu, T.; Mirochnitchenko, O.; Phadtare, S.; Inouye, M. Regression of solid tumors by induction of MazF, a bacterial mRNA endoribonuclease. J. Mol. Microbiol. Biotechnol. 2014, 24, 228–233. [Google Scholar] [CrossRef]
- Chono, H.; Saito, N.; Tsuda, H.; Shibata, H.; Ageyama, N.; Terao, K.; Yasutomi, Y.; Mineno, J.; Kato, I. In vivo safety and persistence of endoribonuclease gene-transduced CD4+ t cells in cynomolgus macaques for HIV-1 gene therapy model. PLoS ONE 2011, 6, e23585. [Google Scholar] [CrossRef]
- Chono, H.; Matsumoto, K.; Tsuda, H.; Saito, N.; Lee, K.; Kim, S.; Shibata, H.; Ageyama, N.; Terao, K.; Yasutomi, Y.; et al. Acquisition of HIV-1 resistance in T lymphocytes using an ACA-specific E. coli mRNA interferase. Hum. Gene Ther. 2011, 22, 35–43. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry 2005, 70, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [PubMed]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- Soo, V.W.C.; Wood, T.K. Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 2013, 3, 3186. [Google Scholar] [CrossRef] [Green Version]
- González Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 2006, 188, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Kasari, V.; Kurg, K.; Margus, T.; Tenson, T.; Kaldalu, N. The Escherichia coli mqsR and ygiT genes encode a new toxin-antitoxin pair. J. Bacteriol. 2010, 192, 2908–2919. [Google Scholar] [CrossRef] [Green Version]
- Fraikin, N.; Rousseau, C.J.; Goeders, N.; Van Melderen, L. Reassessing the role of the type II MqsRA toxin-antitoxin system in stress response and biofilm formation: MqsA is transcriptionally uncoupled from mqsR. MBio 2019, 10, e02678-19. [Google Scholar] [CrossRef] [Green Version]
- Ramisetty, B.C.M.; Ghosh, D.; Chowdhury, M.R.; Santhosh, R.S. Corrigendum: What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2017, 8, 458. [Google Scholar] [CrossRef]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; Wood, T.K. Commentary: What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2017, 14, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Y.; Gandt, A.B.; Rowe, S.E.; Deisinger, J.P.; Conlon, B.P.; Lewis, K. ATP-Dependent persister formation in Escherichia coli. MBio 2017, 8, e02267-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramisetty, B.C.M.; Ghosh, D.; Chowdhury, M.R.; Santhosh, R.S. What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2016, 7, 1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 16, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. Retraction notice to: (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2018, 172, 1135. [Google Scholar] [CrossRef] [Green Version]
- Goormaghtigh, F.; Fraikin, N.; Putrinš, M.; Hallaert, T.; Hauryliuk, V.; Garcia-Pino, A.; Sjödin, A.; Kasvandik, S.; Udekwu, K.; Tenson, T.; et al. Reassessing the role of type II toxin-antitoxin systems in formation of escherichia coli type II persister cells. MBio 2018, 9, e00640-18. [Google Scholar] [CrossRef] [Green Version]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel) 2013, 6, 304–324. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L. Toxin-antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; Ambroa, A.; López, M.; Bou, G.; Cantón, R.; Garcia-Contreras, R.; Wood, T.K.; et al. (p)ppGpp and its role in bacterial persistence: New challenges. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Tripathi, A.; Dewan, P.C.; Siddique, S.A.; Varadarajan, R. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 2014, 289, 4191–4205. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.J.; Wade, W.D.; Akierman, S.; Vacchi-Suzzi, C.; Stremick, C.A.; Turner, R.J.; Ceri, H. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 2009, 53, 2253–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.P.; Mulvey, M.A. Toxin-Antitoxin Systems Are Important for Niche-Specific Colonization and Stress Resistance of Uropathogenic Escherichia coli. PLoS Pathog. 2012, 8, e1002954. [Google Scholar] [CrossRef] [PubMed]
- Slattery, A.; Victorsen, A.H.; Brown, A.; Hillman, K.; Phillips, G.J. Isolation of highly persistent mutants of Salmonella enterica serovar typhimurium reveals a new toxin-antitoxin module. J. Bacteriol. 2013, 195, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Cheverton, A.M.; Gollan, B.; Przydacz, M.; Wong, C.T.; Mylona, A.; Hare, S.A.; Helaine, S. A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell 2016, 63, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rycroft, J.A.; Gollan, B.; Grabe, G.J.; Hall, A.; Cheverton, A.M.; Larrouy-Maumus, G.; Hare, S.A.; Helaine, S. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat. Commun. 2018, 9, 1993. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Wilmaerts, D.; Dewachter, L.; De Loose, P.-J.; Bollen, C.; Verstraeten, N.; Michiels, J. HokB Monomerization and Membrane Repolarization Control Persister Awakening. Mol. Cell 2019, 75, 1031–1042. [Google Scholar] [CrossRef]
- Dörr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.Y.; Soo, V.W.C.; Islam, S.; McAnulty, M.J.; Benedik, M.J.; Wood, T.K. Toxin GhoT of the GhoT/GhoS toxin/antitoxin system damages the cell membrane to reduce adenosine triphosphate and to reduce growth under stress. Environ. Microbiol. 2014, 16, 1741–1754. [Google Scholar] [CrossRef]
- Wang, X.; Lord, D.M.; Hong, S.H.; Peti, W.; Benedik, M.J.; Page, R.; Wood, T.K. Type II toxin/antitoxin MqsR/MqsA controls type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013, 15, 1734–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defraine, V.; Fauvart, M.; Michiels, J. Fighting bacterial persistence: Current and emerging anti-persister strategies and therapeutics. Drug Resist. Updates 2018, 38, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016, 1, 16051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yin, N.; Liu, H.; Pei, J.; Lai, L. Novel inhibitors of toxin HipA reduce multidrug tolerant persisters. ACS Med. Chem. Lett. 2016, 7, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Solecki, O.; Mosbah, A.; Baudy Floc’h, M.; Felden, B. Converting a Staphylococcus aureus toxin into effective cyclic pseudopeptide antibiotics. Chem. Biol. 2015, 22, 329–335. [Google Scholar] [CrossRef]
Figure 1.
Type II toxin–antitoxin (TA) systems identified in human pathogenic bacteria according to the toxin family classification system [25,26] as collected in the Toxin–Antitoxin Database (TADB) 2.0 [24]. The number given for an individual species is the sum of the values for all strains of a given taxon. This list was manually curated to represent strains and/or species frequently associated with infections in humans (including opportunistic pathogens).
Figure 1.
Type II toxin–antitoxin (TA) systems identified in human pathogenic bacteria according to the toxin family classification system [25,26] as collected in the Toxin–Antitoxin Database (TADB) 2.0 [24]. The number given for an individual species is the sum of the values for all strains of a given taxon. This list was manually curated to represent strains and/or species frequently associated with infections in humans (including opportunistic pathogens).

Figure 2.
Proposed antibacterial strategies based on the direct activation of toxins of TA systems; (a) disruption of the protein toxin and antitoxin complex and/or prevention of protein complex formation; (b) inhibition of antitoxin translation (see text for details).
Figure 2.
Proposed antibacterial strategies based on the direct activation of toxins of TA systems; (a) disruption of the protein toxin and antitoxin complex and/or prevention of protein complex formation; (b) inhibition of antitoxin translation (see text for details).

Figure 3.
Proposed antibacterial strategies based on the indirect activation of toxins of TA systems: (a) activation of the Lon or ClpP proteases that degrade antitoxins; (b) triggering TA systems by quorum sensing factors; (c) triggering TAs by artificial induction of the stringent response (see text for details).
Figure 3.
Proposed antibacterial strategies based on the indirect activation of toxins of TA systems: (a) activation of the Lon or ClpP proteases that degrade antitoxins; (b) triggering TA systems by quorum sensing factors; (c) triggering TAs by artificial induction of the stringent response (see text for details).

Figure 4.
Engineered CcdB toxin as a V. cholerae-specific antimicrobial agent. pLASMID—carrier plasmid molecule containing engineered genes of the ccdAB TA system. (a) In V. cholerae cells, by producing ToxRS and SetR transcription factors, transcription of the antitoxin gene is repressed, and expression of engineered toxin genes is activated, leading to cell death; (b) in other bacterial cells (lacking both transcription factors), the antitoxin gene is preferentially expressed.
Figure 4.
Engineered CcdB toxin as a V. cholerae-specific antimicrobial agent. pLASMID—carrier plasmid molecule containing engineered genes of the ccdAB TA system. (a) In V. cholerae cells, by producing ToxRS and SetR transcription factors, transcription of the antitoxin gene is repressed, and expression of engineered toxin genes is activated, leading to cell death; (b) in other bacterial cells (lacking both transcription factors), the antitoxin gene is preferentially expressed.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Równicki, M.; Lasek, R.; Trylska, J.; Bartosik, D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins 2020, 12, 568. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12090568
AMA Style
Równicki M, Lasek R, Trylska J, Bartosik D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins. 2020; 12(9):568. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12090568
Chicago/Turabian StyleRównicki, Marcin, Robert Lasek, Joanna Trylska, and Dariusz Bartosik. 2020. "Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies" Toxins 12, no. 9: 568. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12090568
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.