Cell Death Signaling Pathway Induced by Cholix Toxin, a Cytotoxin and eEF2 ADP-Ribosyltransferase Produced by Vibrio cholerae


1
Advanced Health Care Science Research Unit, Institute for Frontier Science Initiative, Kanazawa University, Kanazawa 920-0942, Japan
2
Department of Molecular Infectiology, Graduate School of Medicine, Chiba University, Chiba 260-8670, Japan
3
Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892-1590, USA
*
Authors to whom correspondence should be addressed.
Toxins 2021, 13(1), 12; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010012
Submission received: 27 October 2020
/
Revised: 20 December 2020
/
Accepted: 22 December 2020
/
Published: 24 December 2020
(This article belongs to the Special Issue Structure and Function of Bacterial ADP-Ribosylation Toxins)
Abstract
:Pathogenic microorganisms produce various virulence factors, e.g., enzymes, cytotoxins, effectors, which trigger development of pathologies in infectious diseases. Cholera toxin (CT) produced by O1 and O139 serotypes of Vibrio cholerae (V. cholerae) is a major cytotoxin causing severe diarrhea. Cholix cytotoxin (Cholix) was identified as a novel eukaryotic elongation factor 2 (eEF2) adenosine-diphosphate (ADP)-ribosyltransferase produced mainly in non-O1/non-O139 V. cholerae. The function and role of Cholix in infectious disease caused by V. cholerae remain unknown. The crystal structure of Cholix is similar to Pseudomonas exotoxin A (PEA) which is composed of an N-terminal receptor-recognition domain and a C-terminal ADP-ribosyltransferase domain. The endocytosed Cholix catalyzes ADP-ribosylation of eEF2 in host cells and inhibits protein synthesis, resulting in cell death. In a mouse model, Cholix caused lethality with severe liver damage. In this review, we describe the mechanism underlying Cholix-induced cytotoxicity. Cholix-induced apoptosis was regulated by mitogen-activated protein kinase (MAPK) and protein kinase C (PKC) signaling pathways, which dramatically enhanced tumor necrosis factor-α (TNF-α) production in human liver, as well as the amount of epithelial-like HepG2 cancer cells. In contrast, Cholix induced apoptosis in hepatocytes through a mitochondrial-dependent pathway, which was not stimulated by TNF-α. These findings suggest that sensitivity to Cholix depends on the target cell. A substantial amount of information on PEA is provided in order to compare/contrast this well-characterized mono-ADP-ribosyltransferase (mART) with Cholix.
Keywords:
bacterial cytotoxin; ADP-ribosyltransferase; mono-ADP-ribosylation; cell death; apoptosis; hepatocytesKey Contribution: This review describes how mono-ADP-ribosyltransferase activity of Cholix toxin induces cytotoxicity of targeted cells.
1. Introduction
Vibrio cholerae (V. cholerae), a gram-negative bacterium with a curved rod shape, belongs to the Vibrionaceae family and colonizes shellfish [1,2,3] and fish [4,5] in aquatic environments such as coastal salt waters and estuaries. V. cholerae is categorized into more than 206 serogroups [6]. Serotypes O1 and O139 of V. cholerae strains possess two main virulence gene sets: those of cholera toxin (CT) and the toxin-coregulated pilus, which cause an acute watery diarrheal disease. Recent reports showed that non-O1/O139 V. cholerae (NOVC) strains also cause diarrhea [7]. The gene for CT is encoded by a filamentous bacteriophage [8]. Some NOVC strains produce CT, which is implicated in cholera-like illnesses [9,10,11]. Vezzuli et al. noted an increasing number of case reports showing bacteremia and fatalities involving sepsis and necrotizing fasciitis apart from the diarrhea associated with NOVC strains [7,12]. Patients with liver cirrhosis were found to be susceptible to NOVC bacteremia [13,14].
Other than CT, some V. cholerae strains possess various virulence factors, e.g., heat-stable toxin [15], hemolysin [16], type III secretion system [17,18], multifunctional autoprocessing repeats-in-toxin [19], accessory toxins like zonula occludens toxin [20,21], Cholix toxin [22,23]. Cholix toxin (Cholix) belongs to a mono-ADP-ribosyltransferase (mART) family, as does CT. While CT ADP-ribosylates the alpha subunit of the heterotrimeric G protein Gs, Cholix specifically targets diphthamide of eukaryotic elongation factor 2 (eEF2), a mechanism similar to that used by diphtheria toxin (DT) and Pseudomonas exotoxin A (PEA) from, respectively, Corynebacterium diphtheriae and Pseudomonas aeruginosa [22]. In addition to the three mARTs (i.e., DT, PEA, and Cholix), Fernández-Bravo et al. reported recently that some Aeromonas hydrophila strains possess exotoxin A genes [24]. The four mARTs catalyze ADP-ribosylation of the diphthamide residues using nicotinamide adenine dinucleotide (NAD+) as the ADP-ribose donor [25]. Eukaryotic elongation factor 2 catalyzes the translocation of mRNA and peptidyl-tRNA; inactivation of eEF2 by ADP-ribosylation results in inhibition of protein synthesis, followed by host cell death [26,27].
2. V. cholerae Strains Possessing Cholix Genes
The cholix genes were found in V. cholerae strains isolated in various countries, such as Bangladesh [4,23,28], Germany [29,30,31,32], India [28], Iran [33], Kenya [34], Mexico [23,29,35], and the United States [23]. While the cholix gene was present in O1 serotype strains [4,23], some researchers proposed that the gene appears to be preferentially associated with NOVC strains, with higher prevalence in NOVC than O1/O139 V. cholerae strains [28,29]. It remains unknown how the cholix gene was transferred to some V. cholerae chromosomes. The guanine-cytosine (GC) content of the cholix gene from a V. cholerae TP strain [36] (44%) is significantly lower than that of toxA (66%), which encodes Pseudomonas exotoxin A (PEA), thus strongly suggesting that the cholix gene is not the result of a recent lateral transfer from P. aeruginosa [23]. No other indicators of lateral gene transfer, such as phage-like sequences or insertion sequence elements, are seen near this region of the V. cholerae genome [23]. The IslandViewer4 web tool showed that the cholix gene is not designated to any genomic island [37]. The two genes surrounding the cholix gene were highly conserved in the chromosomes of cholix-negative V. cholerae strains. These results suggested that the probability of horizontal transfer is low.
3. Structural Insights
The crystal structure of Cholix was first reported by the Merrill group [22]. Their studies summarized structural insights into Cholix [38]. Based on the Cholix/NAD+ complex structure and site-directed mutagenesis, Fieldhouse et al. proposed a new kinetic model for NAD+ binding and mART activity by Cholix [39]. Next, Turgeon et al. tested various chemical compounds which can inhibit cytotoxicity of Cholix by competitive binding to the NAD+ binding site in Cholix [40]. In order to propose structure/function relationships of the Cholix catalytic domain, the group conducted in silico simulation of molecular dynamics [41,42]. Their findings revealed structural and enzymatic characteristics of Cholix as an eEF2-mART.
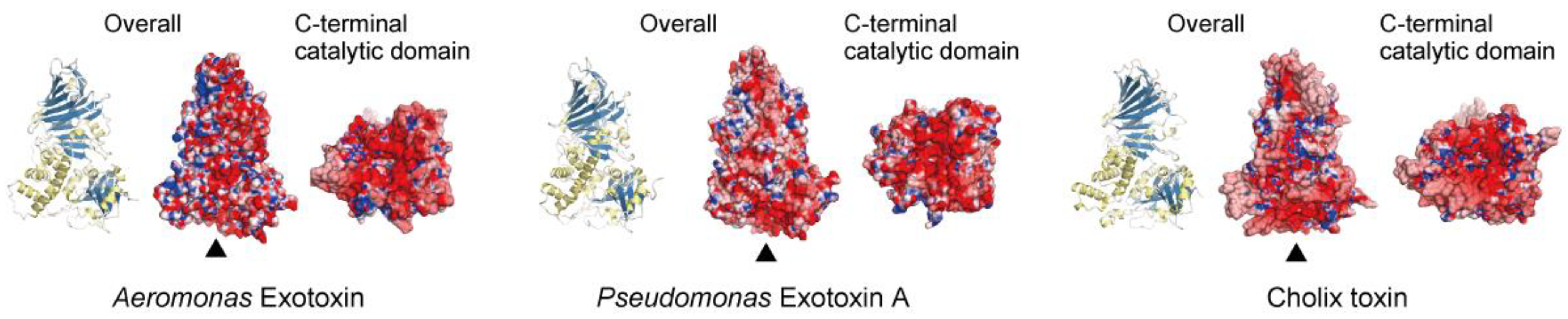
The crystal structure of a new exotoxin from Aeromonas hydrophila resembles those of PEA and Cholix [43] (Figure 1). The primary amino acid sequence of Aeromonas exotoxin A exhibited, respectively, 64% and 35% identity with PEA and Cholix, while its C-terminal mART domain had, respectively, 72% and 46% identity with amino acid sequences of PEA and Cholix [43]. As shown in Figure 1, charged amino acid residues on the surfaces of the three mART toxins are different. The structures of the NAD+-binding pockets in the C-terminal domains also vary. While Cholix possesses an EDETV sequence around the catalytic Glu581 (underlined), PEA and Aeromonas exotoxin A have an RLETI sequence [43]. Jørgensen et al. reported that the Km(NAD+) of Cholix is lower than that of PEA [22].
4. Cholix Receptor
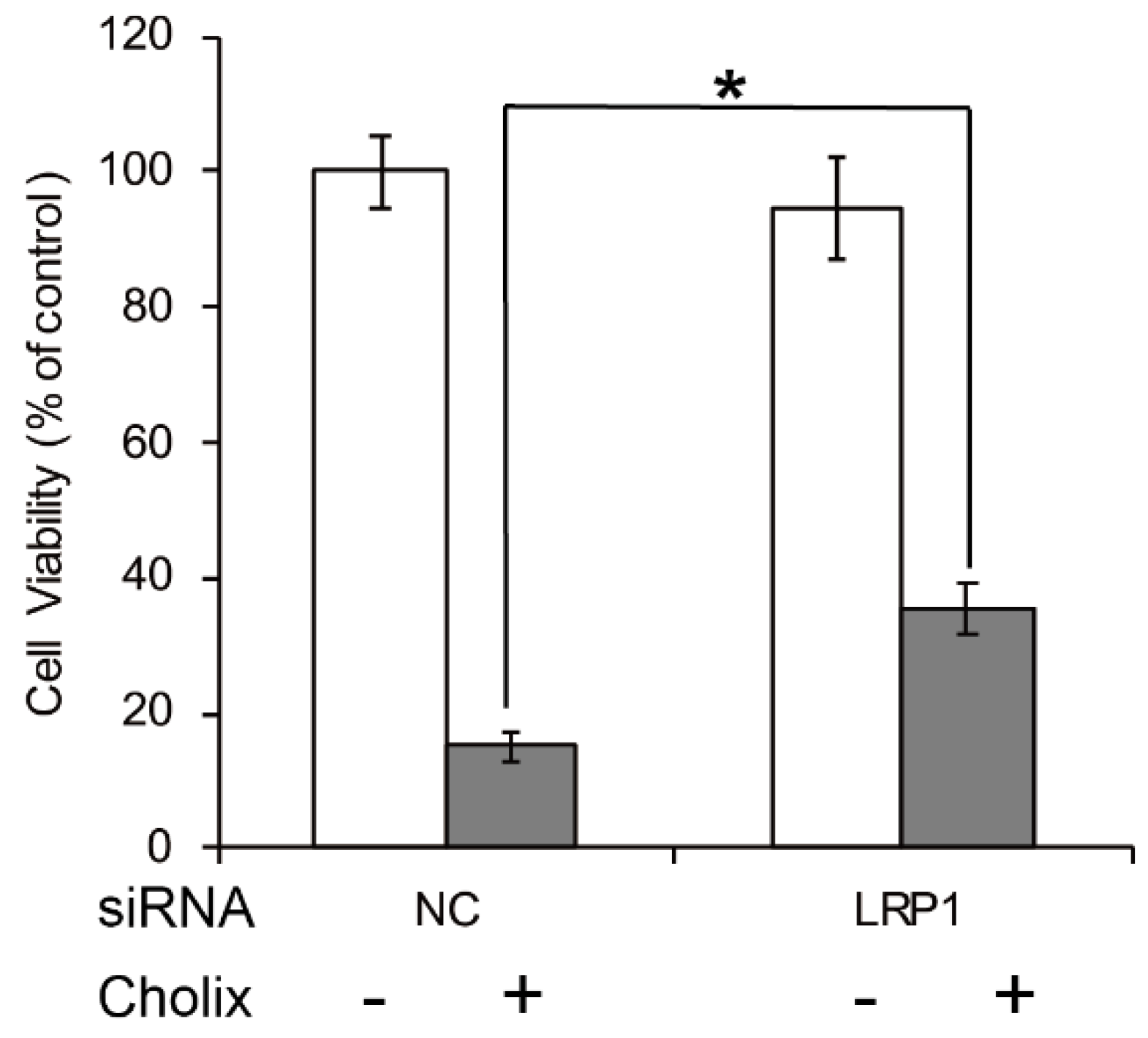
The low-density lipoprotein receptor-related protein 1 (LRP1) is a specific PEA receptor [45]. Jørgensen et al. reported that sensitivity to Cholix is significantly lower in embryonic fibroblasts derived from an LRP1-deficient (LRP−/−) mouse [22]. Consistent with that report, we also found in HeLa cells that Cholix-induced cell death was partially suppressed by LRP1 knockdown (Figure 2). Therefore, LRP1 is considered to be a receptor. However, Jørgensen et al. also reported that the LRP1-deficient cells still showed some sensitivity to Cholix [22]. The suppression of Cholix-induced cell death by LRP1 knockdown was only partial. We also reported that Cholix-induced apoptosis was not suppressed at all by LRP1 knockdown in human hepatocytes [46]. Further, Cholix induced significant cell death in human colon cell line HCT116 cells, whose LRP1 gene was mutated by frameshifts [47,48]. These reports indicate that there may be another pathway for intoxication independent of LRP1.
5. Translocation of Cholix in Host Cells
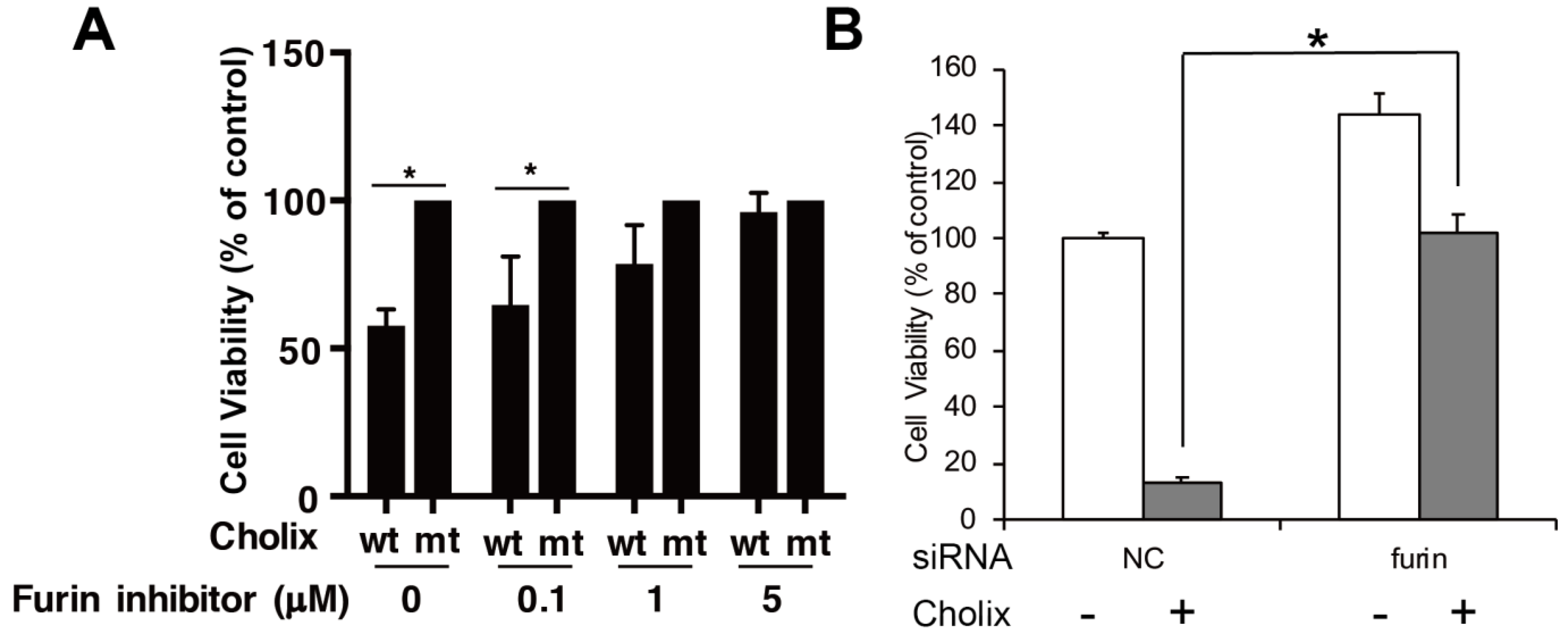
In host cells, Cholix is transported from endosomes to the endoplasmic reticulum and then reacts with eEF2 in the cytosol. PEA follows a similar intracellular path. In the acidic, early-endosomal environment, PEA is cleaved into two fragments by the protease furin, which digests a furin motif (RHRQPRG) [49,50] to generate a 28 kDa N-terminal domain and a 37 kDa C-terminal mART domain. Protein disulfide-isomerase further reduces the disulfide bond in PEA, although the disulfide bond is likely reduced in the endoplasmic reticulum (ER) [51]. Similar to PEA, Cholix has the furin cleavage site (RHKR) and the disulfide bond [22]. Cholix reduction in cell viability was suppressed in the presence of a furin inhibitor, decanoyl-Arg-Val-Lys-Arg-chloromethylketone (Figure 3). These findings suggest that cleavage of the furin site is required for activation of Cholix as well as of PEA. However, Morlon-Guyot et al. found that full-length PEA was also translocated into the cytosol in a mouse fibroblast cell line (L929 cells) [50]. It remains unclear whether Cholix, similarly to PEA, is translocated into the cytosol without processing in L929 cells.
After transportation through late endosomes into the trans-Golgi network [52], the C-terminal mART domain of PEA is translocated to the ER, mainly via the KDEL receptor-mediated pathway [53]. The C-terminal motif REDLK of the mART domain was cleaved at the lysine residue (REDLK) by a carboxypeptidase of the host cell surface [54]. Then, the exposed REDL motif is able to bind to a KDEL receptor, which participates in an important step of the intracellular trafficking pathway, followed by translocation to the ER in a retrograde process [55,56]. Finally, the mART domain of PEA utilizes the cellular ER-associated protein degradation (ERAD) pathway to move from the ER into the cytosol [57,58,59]. Toxins that follow the ERAD pathway to the cytosol have an arginine over lysine amino acid bias to avoid ubiquitination and degradation by the proteasome [60]. While the bias was also observed in the mART domain of Cholix (26 arginines and 12 lysines), it was lower than that of PEA (30 arginines and 3 lysines). Cholix also possesses the C-terminal KEDL motif (RKDELK). These data suggested that translocation of Cholix follows a similar pathway to that of PEA. Awasthi et al. found that a Cholix variant with HDELK in place of RKDELK did not show cytotoxicity in mice [28]. However, KDEL receptors recognize both KDEL and HDEL sequences [61,62,63,64,65]. It remains unclear why the Cholix HDELK variant lacked cytotoxicity.
In addition, by immunoblotting using an anti-Cholix antibody, we found that Cholix was detected in the mitochondrial fraction rather than in the cytosolic compartment, suggesting that Cholix can be translocated into mitochondria; however, there is no information on the translocation mechanism [46].
6. ADP-Ribosylation of eEF2 by Cholix
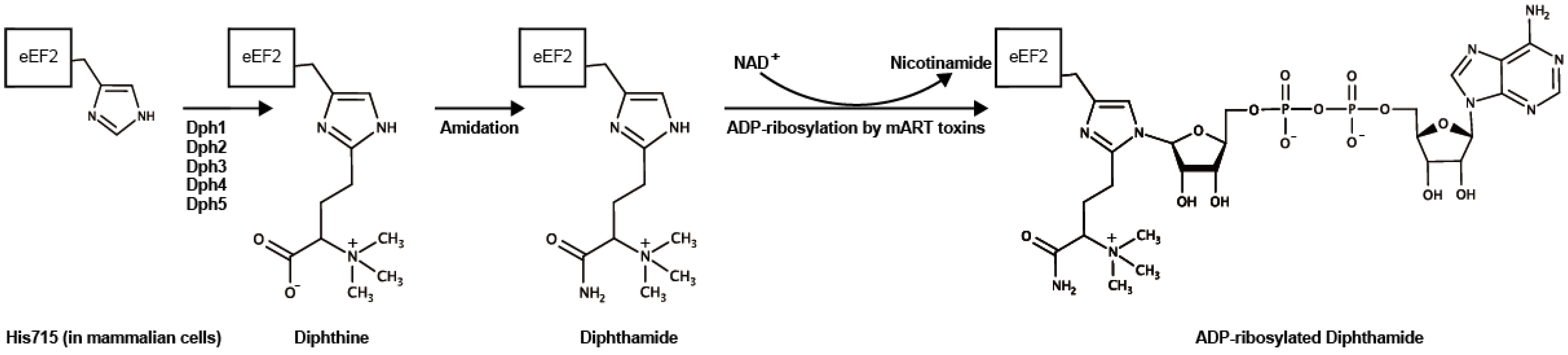
Activated Cholix specifically ADP-ribosylates the diphthamide synthesized from a histidine residue (His715 in mammalian cells) in eukaryotic elongation factor 2 (eEF2) by the diphthamide biosynthesis pathway [66,67] (Figure 4). During protein synthesis, eEF2 translocates mRNA from the ribosomal A-site to the P-site and mediates the elongation step of protein synthesis [66,68]. Cholix-induced eEF2-ADP-ribosylation inhibits ribosomal protein synthesis, resulting in the death of host cells.
7. Liver Damage Induced by Cholix
Previous studies demonstrated the effects of Cholix in vivo. Cholix did not cause fluid accumulation in the rabbit ileal loop assay [28]. Intraperitoneal administration to mice of Cholix, but not the catalytically inactivated mutant Cholix (E581A), resulted in death with severe hepatotoxicity [28,70]. Histological analysis of Cholix-injected mice showed hemorrhagic lesions with necrotic, apoptotic, and inflammatory cells, especially in the liver zone between the Glisson’s sheath and the central vein, by periodic acid-Schiff (PAS) and hematoxylin-eosin (HE) staining. Further, liver damage marker alanine transaminase (ALT) was significantly increased in Cholix-injected mice [70], consistent with the conclusion that liver is a target organ of Cholix. PEA also caused liver damage [71], which involved liver macrophage-like Kupffer cells that produced tumor necrosis factor-α (TNF-α) [72,73]. IL-18 and perforin also participated in the liver damage seen in PEA-injected mice [74]. Further, following PEA intoxication of mice, Kupffer cell-dependent, early TNF-α production required T cells [72].
Previously, we analyzed Cholix-induced cell death of human epithelial hepatoblastoma-derived (HepG2) liver cells and immortalized human hepatocytes [70]. HepG2 cells showed slightly decreased cell viability in the presence of Cholix. Addition of TNF-α with Cholix to HepG2 cells significantly enhanced activation of caspases, resulting in poly (ADP-ribose) polymerase (PARP) cleavage followed by increased cytotoxicity. Inhibition of TNF-α/Cholix-activated JNK or ERK by a specific inhibitor (SB20350 or U0126) suppressed PARP cleavage. Cholix-induced PARP cleavage was enhanced in the presence of PKC activator phorbol 12-myristate 13-acetate (PMA) as well as by TNF-α and suppressed by the PKC inhibitor even in the presence of TNF-α. Further, ROS inhibitor N-acetyl cysteine suppressed JNK activation, which partially inhibited the TNF-α/Cholix apoptosis-signaling pathway. These findings suggest that TNF-α is an enhancer of Cholix-induced apoptosis, which was involved in ROS generation, PKC activation, and MAPK activation. On the other hand, immortalized human hepatocytes were more sensitive to Cholix compared to HepG2 cells. In hepatocytes, Cholix-induced apoptosis was mediated through severe mitochondrial damage that was not promoted by addition of TNF-α. Neither the JNK nor the ERK inhibitor (SB20350 or U0126) suppressed Cholix-induced apoptosis.
There are a few reports regarding NOVC-related liver disease [75,76]. However, it is not clear if these NOVC strains have Cholix. In addition, the pathway used by Cholix to translocate from the gastrointestinal tract to the liver remains unknown. Interestingly, Taverner et al. recently reported that catalytically inactive mutant Cholix (E581A) and its N-terminal domain can be transported across human intestinal epithelia in vitro (confluent monolayers of human small intestinal tissues) and rat jejunum in vivo by apical to basal transcytosis [77]. This apical-to-basal transcytosis pathway utilizes a vesicular trafficking mechanism, independent of intoxication by mART.
8. Other Cell Death Mechanisms
In addition to hepatocytes, cytotoxicity of Cholix was also tested with other cells, such as mouse fibroblasts [22], intestinal cell lines (i.e., Caco-2, HCT116, and RKO) [47], and HeLa cells [28,47]. Although Cholix did not cause fluid accumulation in rabbit ileal loop assays [28], this toxin exhibited significant cytotoxicity to the intestinal cell lines [47]. Intestinal cell death by Cholix was not affected by the presence of a general caspase inhibitor (Z-VAD-FMK), indicating that intestinal cell death was not a result of apoptosis.
HeLa cells are a commonly used epithelial cell line derived from human cervical cancer. Cholix-treated HeLa cells showed decreased viability, which was significantly repressed by Z-VAD-FMK [47]. In HeLa cells, Cholix stimulated an apoptotic pathway dependent on activation of inflammatory caspases (caspase-1, -4, and -5), followed by mitochondrial outer membrane permeabilization that occurred through rapid degradation of the anti-apoptotic Bcl-2 family protein Mcl-1 and conformational changes of pro-apoptotic Bcl-2 family members, Bak and Bax.
Mcl-1 is a protein with a short half-life, which is rapidly lost in the presence of the protein synthesis inhibitor, cycloheximide [78]. In addition, the loss of Mcl-1 induced by Cholix was suppressed in the presence of proteasome inhibitor MG132. Thus, Cholix decreased Mcl-1 content by inhibition of protein synthesis combined with rapid proteasomal degradation and promoted conformational changes of pro-apoptotic Bcl-2 family members, Bak and Bax [47].
9. Prohibitin Binding and Mitochondrial Dysfunction
Recently, we found by immunoprecipitation that Cholix interacts with prohibitin (PHB) 1 and 2 on the cell surface [46]. PHBs are ubiquitously expressed in various cells and mainly localized in mitochondria, but also expressed in nuclei and cell membranes [79]. PHBs contribute to mitochondrial biology [79] and homeostasis [80,81] by regulating ROS formation, protein degradation, mitochondrial morphology, and the oxidative phosphorylation (OXPHOS) complex [82]. The expression of PHB1 was decreased by Cholix in hepatocytes. PHB deficiency impairs respiratory supercomplex formation [83] and causes overproduction of reactive oxygen species (ROS) [84]. In agreement with previous studies, Cholix-induced PARP cleavage, ROS generation, and apoptotic chromatin assembly were significantly increased in PHB knockdown cells compared with control cells. In PHB-overexpressing cells, Cholix-induced apoptotic signaling was inhibited [46]. These findings suggest that PHB is a Cholix interaction protein, not its receptor. Cholix-dependent reduction in PHB1 expression triggers PHB dysfunction, which might lead to altered mitochondrial respiratory supercomplexes, followed by promotion of ROS production.
Rho-associated, coiled coil-containing protein kinase protein 1 (ROCK1) is a crucial factor in mitochondrial fission and ROS generation [85] and is a direct cleavage substrate of activated caspase-3, which is involved in apoptosis [86]. ROCK1 knockdown and ROCK inhibitor Y27632 suppressed Cholix-induced cleavage of PARP and ROCK1 and ROS generation [46]. In PHB knockdown cells, Cholix increased ROCK1 cleavage, followed by enhanced apoptosis, plasma membrane blebbing, and ROS generation, suggesting that ROCK1 participates in a Cholix-induced apoptotic pathway [46].
10. Conclusions
Common and different characteristics between PEA and Cholix are summarized in Table 1. Although PEA utilizes LRP1 as a receptor, Cholix binds to LRP1 and/or unknown receptor(s). Translocation of PEA and Cholix is commonly mediated by furin cleavage and KDEL pathways. Intraperitoneal administration of PEA and Cholix to mice results in liver damage. In human hepatocytes, Cholix, but not PEA, induces a decrease in PHB1 followed by PHB dysfunction.
The proposed cell death mechanism is shown in Figure 5. Cholix produced by NOVC binds to host cell receptor LRP1 and/or unknown cell surface receptor(s) and then its catalytic domain is translocated to the cytosol through furin cleavage and KDEL pathways. Cholix-induced cell death pathways varied among cell types. In HepG2 cells, TNF-α promoted Cholix-induced cell death, which includes PKC/MAPK activation, followed by induction of caspase-dependent apoptosis and a caspase-independent, yet to be defined, cell death signaling pathway. Human hepatocytes showed more sensitivity to Cholix than HepG2 cells. In Cholix-treated hepatocytes, mitochondrial dysfunction stimulates a caspase-dependent apoptotic signal pathway, which involves ROCK-1 cleavage and ROS generation. In HeLa cells, Cholix induced mitochondrial-dependent and -independent caspase activation. It remains unknown why ADP-ribosylation of eEF2 by Cholix resulted in activation of different signaling pathways among the various cell types. Further studies are required to define the mechanisms underlying Cholix cytotoxicity in different cell types.
Funding
Our studies were supported by Japan Society for the Promotion of Science KAKENHI (22790401, 25460526, and 19K07534), Takeda Science Foundation, and the Research Program on Emerging and Re-emerging Infectious Diseases from the Japan Agency for Medical Research and Development, AMED (20fkfk0108065h0402). J.M. was supported by the Division of Intramural Research, National Institutes of Health, National Heart, Lung, and Blood Institute.
Data Availability Statement
Data are available upon request, please contact the contributing authors.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| eEF2 | Eukaryotic elongation factor 2 |
| Cholix | Cholix toxin |
| PEA | Pseudomonas exotoxin A |
| CT | Cholera toxin |
| mART | Mono-ADP-ribosyltransferase |
| TNF-α | Tumor necrosis factor-α |
| TNFR | Tumor necrosis factor receptor |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| ROCK | Rho-associated, coiled coil-containing protein kinase |
| PHB | Prohibitin |
| cPARP | Cleaved poly (ADP-ribose) polymerase |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
References
- Morris, J.G. Cholera and other types of vibriosis: A story of human pandemics and oysters on the half shell. Clin. Infect. Dis. 2003, 37, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Clemens, J.D.; Nair, G.B.; Ahmed, T.; Qadri, F.; Holmgren, J. Cholera. Lancet 2017, 390, 1539–1549. [Google Scholar] [CrossRef]
- Colwell, R.R. Global climate and infectious disease: The cholera paradigm. Science 1996, 274, 2025–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, Z.Z.; Farhana, I.; Tulsiani, S.M.; Begum, A.; Jensen, P.K.M. Transmission and Toxigenic Potential of Vibrio cholerae in Hilsha Fish (Tenualosa ilisha) for Human Consumption in Bangladesh. Front. Microbiol. 2018, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Halpern, M.; Izhaki, I. Fish as Hosts of Vibrio cholerae. Front. Microbiol. 2017, 8, 282. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, F.R.; Nur, Z.; Hassan, N.; Seidlein, L.; Dunachie, S. Pandemics, pathogenicity and changing molecular epidemiology of cholera in the era of global warming. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Vezzulli, L.; Baker-Austin, C.; Kirschner, A.; Pruzzo, C.; Martinez-Urtaza, J. Global emergence of environmental non-O1/O139 Vibrio cholerae infections linked with climate change: A neglected research field? Environ. Microbiol. 2020, 4342–4355. [Google Scholar] [CrossRef]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1913. [Google Scholar] [CrossRef] [Green Version]
- Dalsgaard, A.; Serichantalergs, O.; Forslund, A.; Lin, W.; Mekalanos, J.; Mintz, E.; Shimada, T.; Wells, J.G. Clinical and environmental isolates of vibrio cholerae serogroup O141 carry the CTX phage and the genes encoding the toxin-coregulated pili. J. Clin. Microbiol. 2001, 39, 4086–4092. [Google Scholar] [CrossRef] [Green Version]
- Aydanian, A.; Tang, L.; Morris, J.G.; Johnson, J.A.; Stine, O.C. Genetic diversity of O-antigen biosynthesis regions in Vibrio cholerae. Appl. Environ. Microbiol. 2011, 77, 2247–2253. [Google Scholar] [CrossRef] [Green Version]
- Tobin-D’Angelo, M.; Smith, A.R.; Bulens, S.N.; Thomas, S.; Hodel, M.; Izumiya, H.; Arakawa, E.; Morita, M.; Watanabe, H.; Marin, C.; et al. Severe diarrhea caused by cholera toxin-producing Vibrio cholerae serogroup O75 infections acquired in the southeastern United States. Clin. Infect. Dis. 2008, 47, 1035–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Reytor, D.; Jaña, V.; Pavez, L.; Navarrete, P.; García, K. Accessory toxins of vibriopathogens and their role in epithelial disruption during infection. Front. Microbiol. 2018, 9, 2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanley, J.; Kanj, A.; El Zein, S.; Tabaja, H.; Trzcinski, B.; Horman, J.; Salimnia, H.; Fairfax, M.; Singh, M. Non-O1, non-O139 Vibrio cholerae bacteremia in an urban academic medical center in the United States. IDCases 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, Y.; Qian, H.; Liu, G.; Mei, Y.; Jin, F.; Xia, W.; Ni, F. Non-O1, Non-O139 Vibrio cholerae (NOVC) Bacteremia: Case Report and Literature Review, 2015–2019. Infect. Drug Resist. 2020, 13, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, A.; Takeda, T.; Ogawa, A. The Gene Encoding the Heat-Stable Enterotoxin of Vibrio cholerae is Flanked by 123-Base Pair Direct Repeats. Microbiol. Immunol. 1993, 37, 774–777. [Google Scholar] [CrossRef]
- Cinar, H.N.; Kothary, M.; Datta, A.R.; Tall, B.D.; Sprando, R.; Bilecen, K.; Yildiz, F.; McCardell, B. Vibrio cholerae Hemolysin Is Required for Lethality, Developmental Delay, and Intestinal Vacuolation in Caenorhabditis elegans. PLoS ONE 2010, 5, e11558. [Google Scholar] [CrossRef] [Green Version]
- Dziejman, M.; Serruto, D.; Tam, V.C.; Sturtevant, D.; Diraphat, P.; Faruque, S.M.; Rahman, M.H.; Heidelberg, J.F.; Decker, J.; Li, L.; et al. Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc. Natl. Acad. Sci. USA 2005, 102, 3465–3470. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.C.; Serruto, D.; Dziejman, M.; Brieher, W.; Mekalanos, J.J. A Type III Secretion System in Vibrio cholerae Translocates a Formin/Spire Hybrid-like Actin Nucleator to Promote Intestinal Colonization. Cell Host Microbe 2007, 1, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Satchell, K.J.F. Multifunctional-autoprocessing repeats-in-toxin (MARTX) Toxins of Vibrios. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA 1991, 88, 5242–5246. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, M.; Lu, R.; Uzzau, S.; Wang, W.; Margaretten, K.; Pazzani, C.; Maimone, F.; Fasano, A. Zonula occludens toxin structure-function analysis: Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J. Biol. Chem. 2001, 276, 19160–19165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, R.; Purdy, A.E.; Fieldhouse, R.J.; Kimber, M.S.; Bartlett, D.H.; Merrill, A.R. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdy, A.E.; Balch, D.; Lizárraga-Partida, M.L.; Islam, M.S.; Martinez-Urtaza, J.; Huq, A.; Colwell, R.R.; Bartlett, D.H. Diversity and distribution of cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. Environ. Microbiol. Rep. 2010, 2, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bravo, A.; Kilgore, P.B.; Andersson, J.A.; Blears, E.; Figueras, M.J.; Hasan, N.A.; Colwell, R.R.; Sha, J.; Chopra, A.K. T6SS and ExoA of flesh-eating Aeromonas hydrophila in peritonitis and necrotizing fasciitis during mono- And polymicrobial infections. Proc. Natl. Acad. Sci. USA 2019, 116, 24084–24092. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Honjo, T.; Nishizuka, Y.; Hayaishi, O. Adenosine diphosphoribosylation of aminoacyl transferase II by diphtheria toxin. Cold Spring Harb. Symp. Quant. Biol. 1969, 34, 603–608. [Google Scholar] [CrossRef]
- Jørgensen, R.; Merrill, A.R.; Andersen, G.R. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 2006, 34, 1. [Google Scholar] [CrossRef]
- Awasthi, S.P.; Asakura, M.; Chowdhury, N.; Neogi, S.B.; Hinenoya, A.; Golbar, H.M.; Yamate, J.; Arakawa, E.; Tada, T.; Ramamurthy, T.; et al. Novel cholix toxin variants, ADP-ribosylating toxins in Vibrio cholerae non-O1/non-O139 strains, and their pathogenicity. Infect. Immun. 2013, 81, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Schirmeister, F.; Dieckmann, R.; Bechlars, S.; Bier, N.; Faruque, S.M.; Strauch, E. Genetic and phenotypic analysis of Vibrio cholerae non-O1, non-O139 isolated from German and Austrian patients. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, K.; Hammerl, J.A.; Göllner, C.; Strauch, E. Environmental and Clinical Strains of Vibrio cholerae Non-O1, Non-O139 from Germany Possess Similar Virulence Gene Profiles. Front. Microbiol. 2019, 10, 733. [Google Scholar] [CrossRef]
- Hackbusch, S.; Wichels, A.; Gimenez, L.; Döpke, H.; Gerdts, G. Potentially human pathogenic Vibrio spp. in a coastal transect: Occurrence and multiple virulence factors. Sci. Total Environ. 2020, 707, 136113. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, N.; Kappe, E.; Gangl, A.; Schwartz, K.; Mayer-Scholl, A.; Hammerl, J.A.; Strauch, E. Phenotypic and genotypic properties of vibrio cholerae non-o1, non-o139 isolates recovered from domestic ducks in germany. Microorganisms 2020, 8, 1104. [Google Scholar] [CrossRef] [PubMed]
- Tangestani, M.G.; Alinezhad, J.; Khajeian, A.; Gharibi, S.; Haghighi, M.A. Identification of cholix toxin gene in vibrio cholerae non-O1/non-O139 isolated from diarrhea patients in Bushehr, Iran. Iran. J. Microbiol. 2020, 12, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.M.; Chowdhury, N.; Awasthi, S.P.; Asakura, M.; Hinenoya, A.; Iijima, Y.; Yamasaki, S. Prevalence of Vibrio cholerae O1 El Tor variant in a cholera-endemic zone of Kenya. J. Med. Microbiol. 2014, 63, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Villicaña, C.; Amarillas, L.; Soto-Castro, L.; Gómez-Gil, B.; Lizárraga-Partida, M.L.; León-Félix, J. Occurrence and abundance of pathogenic Vibrio species in raw oysters at retail seafood markets in northwestern Mexico. J. Food Prot. 2019, 82, 2094–2099. [Google Scholar] [CrossRef]
- Purdy, A.; Rohwer, F.; Edwards, R.; Azam, F.; Bartlett, D.H. A glimpse into the expanded genome content of Vibrio cholerae through identification of genes present in environmental strains. J. Bacteriol. 2005, 187, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Lugo, M.R.; Merrill, A.R. The Father, Son and Cholix Toxin: The Third Member of the DT Group Mono-ADP-Ribosyltransferase Toxin Family. Toxins 2015, 7, 2757–2772. [Google Scholar] [CrossRef] [Green Version]
- Fieldhouse, R.J.; Jørgensen, R.; Lugo, M.R.; Merrill, A.R. The 1.8 Å cholix toxin crystal structure in complex with NAD+ and evidence for a new kinetic model. J. Biol. Chem. 2012, 287, 21176–21188. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, Z.; Jørgensen, R.; Visschedyk, D.; Edwards, P.R.; Legree, S.; McGregor, C.; Fieldhouse, R.J.; Mangroo, D.; Schapira, M.; Merrill, A.R. Newly discovered and characterized antivirulence compounds inhibit bacterial mono-ADP-ribosyltransferase toxins. Antimicrob. Agents Chemother. 2011, 55, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Lugo, M.R.; Merrill, A.R. Pocket analysis of the full-length cholix toxin. An assessment of the structure-dynamics of the apo catalytic domain. J. Biomol. Struct. Dyn. 2015, 33, 2452–2468. [Google Scholar] [CrossRef] [PubMed]
- Lugo, M.R.; Merrill, A.R. A comparative structure-function analysis of active-site inhibitors of Vibrio cholerae cholix toxin. J. Mol. Recognit. 2015, 28, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Masuyer, G. Crystal structure of exotoxin a from aeromonas pathogenic species. Toxins 2020, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Kounnas, M.Z.; Morris, R.E.; Thompson, M.R.; FitzGerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar] [PubMed]
- Yahiro, K.; Ogura, K.; Terasaki, Y.; Satoh, M.; Miyagi, S.; Terasaki, M.; Yamasaki, E.; Moss, J. Cholix toxin, an eukaryotic elongation factor 2 ADP-ribosyltransferase, interacts with Prohibitins and induces apoptosis with mitochondrial dysfunction in human hepatocytes. Cell. Microbiol. 2019, 21, e13033. [Google Scholar] [CrossRef]
- Ogura, K.; Yahiro, K.; Tsutsuki, H.; Nagasawa, S.; Yamasaki, S.; Moss, J.; Noda, M. Characterization of cholix toxin-induced apoptosis in HeLa cells. J. Biol. Chem. 2011, 286. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Ogata, M.; Chaudhary, V.K.; Pastan, I.; FitzGerald, D.J. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J. Biol. Chem. 1990, 265, 20678–20685. [Google Scholar]
- Morlon-Guyot, J.; Méré, J.; Bonhoure, A.; Beaumelle, B. Processing of Pseudomonas aeruginosa exotoxin A is dispensable for cell intoxication. Infect. Immun. 2009, 77, 3090–3099. [Google Scholar] [CrossRef] [Green Version]
- Mckee, M.L.; Fitzgerald, D.J. Reduction of Furin-Nicked Pseudomonas Exotoxin A: An Unfolding Story. Biochemistry 1999, 38, 16507–16513. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Spooner, R.A.; Watson, P.D.; Murray, J.L.; Hodge, T.W.; Amessou, M.; Johannes, L.; Lord, J.M.; Roberts, L.M. Internalized pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic 2006, 7, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessler, J.L.; Kreitman, R.J. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry 1997, 36, 14577–14582. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112 Pt 4, 467–475. [Google Scholar]
- Ogata, M.; Fryling, C.M.; Pastan, I.; FitzGerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- Theuer, C.P.; Buchner, J.; FitzGerald, D.; Pastan, I. The N-terminal region of the 37-kDa translocated fragment of Pseudomonas exotoxin A aborts translocation by promoting its own export after microsomal membrane insertion. Proc. Natl. Acad. Sci. USA 1993, 90, 7774–7778. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska-Gołacka, J.; Sominka, H.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. Toxins utilize the endoplasmic reticulum-associated protein degradation pathway in their intoxication process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef] [Green Version]
- London, E.; Luongo, C.L. Domain-specific bias in arginine/lysine usage by protein toxins. Biochem. Biophys. Res. Commun. 1989, 160, 333–339. [Google Scholar] [CrossRef]
- Semenza, J.C.; Hardwick, K.G.; Dean, N.; Pelham, H.R.B. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 1990, 61, 1349–1357. [Google Scholar] [CrossRef]
- Lewis, M.J.; Sweet, D.J.; Pelham, H.R.B. The ERD2 gene determines the specificity of the luminal ER protein retention system. Cell 1990, 61, 1359–1363. [Google Scholar] [CrossRef] [Green Version]
- Vitale, A.; Ceriotti, A.; Denecke, J. The role of the endoplasmic reticulum in protein synthesis, modification and intracellular transport. J. Exp. Bot. 1993, 44, 1417–1444. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R.B. A human homologue of the yeast HDEL receptor. Nature 1990, 348, 162–163. [Google Scholar] [CrossRef]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Van, D.N.; Latva-Ranta, M.; Ruddock, L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, S.; Lajoie, G.; Merrill, A.R. The role of the diphthamide-containing loop within eukaryotic elongation factor 2 in ADP-ribosylation by Pseudomonas aeruginosa exotoxin A. Biochem. J. 2008, 413, 163–174. [Google Scholar] [CrossRef]
- Su, X.; Lin, Z.; Lin, H. The biosynthesis and biological function of diphthamide. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Proud, C.G. Peptide-chain elongation in eukaryotes. Mol. Biol. Rep. 1994, 19, 161–170. [Google Scholar] [CrossRef]
- Uthman, S.; Bär, C.; Scheidt, V.; Liu, S.; ten Have, S.; Giorgini, F.; Stark, M.J.R.; Schaffrath, R. The Amidation Step of Diphthamide Biosynthesis in Yeast Requires DPH6, a Gene Identified through Mining the DPH1-DPH5 Interaction Network. PLoS Genet. 2013, 9, e1003334. [Google Scholar] [CrossRef] [Green Version]
- Ogura, K.; Terasaki, Y.; Miyoshi-Akiyama, T.; Terasaki, M.; Moss, J.; Noda, M.; Yahiro, K. Vibrio cholerae Cholix toxin-induced HepG2 cell death is enhanced by tumor necrosis factor-alpha through ROS and intracellular signal-regulated kinases. Toxicol. Sci. 2017, 156. [Google Scholar] [CrossRef] [Green Version]
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin Aiadenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schümann, J.; Angermüller, S.; Bang, R.; Lohoff, M.; Tiegs, G. Acute hepatotoxicity of Pseudomonas aeruginosa exotoxin A in mice depends on T cells and TNF. J. Immunol. 1998, 161, 5745–5754. [Google Scholar] [PubMed]
- Schümann, J.; Mühlen, K.; Kiemer, A.K.; Vollmar, A.M.; Tiegs, G. Parenchymal, but not leukocyte, TNF receptor 2 mediates T cell-dependent hepatitis in mice. J. Immunol. 2003, 170, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlen, K.A.; Schümann, J.; Wittke, F.; Stenger, S.; Van Rooijen, N.; Van Kaer, L.; Tiegs, G. NK cells, but not NKT cells, are involved in Pseudomonas aeruginosa exotoxin A-induced hepatotoxicity in mice. J. Immunol. 2004, 172, 3034–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, R.; Ghafoor, M.A.; Nasralah, A.Y. Unusual non-serogroup O1 Vibrio cholerae bacteremia associated with liver disease. J. Clin. Microbiol. 1989, 27, 2853–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petsaris, O.; Nousbaum, J.B.; Quilici, M.L.; Le Coadou, G.; Payan, C.; Abalain, M.L. Non-O1, non-O139 Vibrio cholerae bacteraemia in a cirrhotic patient. J. Med. Microbiol. 2010, 59, 1260–1262. [Google Scholar] [CrossRef] [Green Version]
- Taverner, A.; MacKay, J.; Laurent, F.; Hunter, T.; Liu, K.; Mangat, K.; Song, L.; Seto, E.; Postlethwaite, S.; Alam, A.; et al. Cholix protein domain I functions as a carrier element for efficient apical to basal epithelial transcytosis. Tissue Barriers 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Adams, K.W.; Cooper, G.M. Rapid turnover of Mcl-1 couples translation to cell survival and apoptosis. J. Biol. Chem. 2007, 282, 6192–6200. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.T.; Chen, P.; Ouyang, R.Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.J.; Kahl, A.; Qian, L.; Stepanova, A.; Starkov, A.; Manfredi, G.; Iadecola, C.; Zhou, P. Prohibitin is a positive modulator of mitochondrial function in PC12 cells under oxidative stress. J. Neurochem. 2018, 146, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Wang, M.; Mao, C.; Yu, F.; Wang, Y.; Xiao, R.; Jiang, C.; Zheng, L.; Xu, Q.; Zheng, M.; et al. COMP-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 2009, 20, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, C.; Xu, F.; Hou, T.; Sun, T.; Li, J.; Cheng, H.; Wang, X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J. Cell Sci. 2017, 130, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.J.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Xie, M.; Shah, V.R.; Schneider, M.D.; Entman, M.L.; Wei, L.; Schwartz, R.J. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14495–14500. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Crystal structures of three eEF2 mART cytotoxins. The overall structures (ribbon models and electrostatic surface models) and C-terminal catalytic domains (observed from the bottom, as indicated by triangles) of Aeromonas exotoxin (Protein Data Bank (PDB) ID 6Z5H), Pseudomonas exotoxin A (PDB ID 1IKQ), and Cholix toxin (PDB ID 3Q9O). The electrostatic potentials of molecular surfaces were calculated using Adaptive Poisson-Boltzmann Solver (APBS) [44]. The models were drawn by CueMol: Molecular Visualization Framework (http://www.cuemol.org/).
Figure 1.
Crystal structures of three eEF2 mART cytotoxins. The overall structures (ribbon models and electrostatic surface models) and C-terminal catalytic domains (observed from the bottom, as indicated by triangles) of Aeromonas exotoxin (Protein Data Bank (PDB) ID 6Z5H), Pseudomonas exotoxin A (PDB ID 1IKQ), and Cholix toxin (PDB ID 3Q9O). The electrostatic potentials of molecular surfaces were calculated using Adaptive Poisson-Boltzmann Solver (APBS) [44]. The models were drawn by CueMol: Molecular Visualization Framework (http://www.cuemol.org/).

Figure 2.
Effects of LRP1 knockdown in Cholix-treated HeLa cells. HeLa cells were treated with non-targeting control (NC) or LRP1 siRNA for 48 h and incubated with PBS (−) or Cholix (+) for 24 h. Cell viability was measured with a Cell Counting Kit (Dojindo). Asterisks indicate p < 0.05 with the Student’s t-test.
Figure 2.
Effects of LRP1 knockdown in Cholix-treated HeLa cells. HeLa cells were treated with non-targeting control (NC) or LRP1 siRNA for 48 h and incubated with PBS (−) or Cholix (+) for 24 h. Cell viability was measured with a Cell Counting Kit (Dojindo). Asterisks indicate p < 0.05 with the Student’s t-test.

Figure 3.
Effects of the furin inhibitor and its gene knockdown in Cholix-treated HeLa cells. (A) HeLa cells were incubated with catalytically inactive mutant Cholix (E581A) (mt) or wild-type Cholix (wt) in the presence of the indicated concentration of the furin inhibitor (decanoyl-RVKR-CMK). (B) HeLa cells were treated with non-targeting control (NC) or furin siRNA for 48 h and incubated with PBS (−) or Cholix (+) for 24 h. Cell viability was measured using a Cell Counting Kit. Asterisks indicate p < 0.05 with the Student’s t-test.
Figure 3.
Effects of the furin inhibitor and its gene knockdown in Cholix-treated HeLa cells. (A) HeLa cells were incubated with catalytically inactive mutant Cholix (E581A) (mt) or wild-type Cholix (wt) in the presence of the indicated concentration of the furin inhibitor (decanoyl-RVKR-CMK). (B) HeLa cells were treated with non-targeting control (NC) or furin siRNA for 48 h and incubated with PBS (−) or Cholix (+) for 24 h. Cell viability was measured using a Cell Counting Kit. Asterisks indicate p < 0.05 with the Student’s t-test.

Figure 4.
Diphthamide synthesis and ADP-ribosylation. His715 in mammalian (His699 in Saccharomyces cerevisiae) eEF2 is modified to diphthine by Dph family proteins (Dph1–5) followed by an amidation step using ATP and ammonia, presumably by Dph6 (in Saccharomyces cerevisiae) [69]. Diphtheria toxin, Pseudomonas exotoxin, Aeromonas exotoxin, and Cholix toxin ADP-ribosylate the diphthamide using NAD+ as an ADP-ribose donor. Marvin ChemAxon was used for drawing the formulas (https://www.chemaxon.com).
Figure 4.
Diphthamide synthesis and ADP-ribosylation. His715 in mammalian (His699 in Saccharomyces cerevisiae) eEF2 is modified to diphthine by Dph family proteins (Dph1–5) followed by an amidation step using ATP and ammonia, presumably by Dph6 (in Saccharomyces cerevisiae) [69]. Diphtheria toxin, Pseudomonas exotoxin, Aeromonas exotoxin, and Cholix toxin ADP-ribosylate the diphthamide using NAD+ as an ADP-ribose donor. Marvin ChemAxon was used for drawing the formulas (https://www.chemaxon.com).

Figure 5.
Proposed model of Cholix-induced cell death pathways. The cell death signal pathways in HepG2 cells [70], hepatocytes [46,70], and HeLa cells [47] are shown. This figure is described in the “Conclusion”. Marvin ChemAxon was used for drawing ADP-ribosylated diphthamide (https://www.chemaxon.com).
Figure 5.
Proposed model of Cholix-induced cell death pathways. The cell death signal pathways in HepG2 cells [70], hepatocytes [46,70], and HeLa cells [47] are shown. This figure is described in the “Conclusion”. Marvin ChemAxon was used for drawing ADP-ribosylated diphthamide (https://www.chemaxon.com).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Common and different characteristics between Pseudomonas exotoxin A and Cholix toxin [22,28,45,46,49,50,53].
| Characteristics | Pseudomonas Exotoxin A | Cholix Toxin |
|---|---|---|
| Target | Diphthamide on eEF2 | Diphthamide on eEF2 |
| Enzymatic activity Km(NAD+) (μm)/kcat (s−1) | Mono-ADP-ribosylation 121 ± 21/13 ± 2 | Mono-ADP-ribosylation 45 ± 3/10 ± 3 |
| Host receptor | LRP1 | LRP1 and/or unknown cell surface receptor(s) |
| Translocation | Furin protease-dependent KDEL motif (REDLK)-dependent | Furin protease-dependent KDEL motif (RKDELK)-dependent |
| Administration into mice | Liver damage | Liver damage |
| PHB1 expression | No change | Decrease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ogura, K.; Yahiro, K.; Moss, J. Cell Death Signaling Pathway Induced by Cholix Toxin, a Cytotoxin and eEF2 ADP-Ribosyltransferase Produced by Vibrio cholerae. Toxins 2021, 13, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010012
AMA Style
Ogura K, Yahiro K, Moss J. Cell Death Signaling Pathway Induced by Cholix Toxin, a Cytotoxin and eEF2 ADP-Ribosyltransferase Produced by Vibrio cholerae. Toxins. 2021; 13(1):12. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010012
Chicago/Turabian StyleOgura, Kohei, Kinnosuke Yahiro, and Joel Moss. 2021. "Cell Death Signaling Pathway Induced by Cholix Toxin, a Cytotoxin and eEF2 ADP-Ribosyltransferase Produced by Vibrio cholerae" Toxins 13, no. 1: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010012
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.