Pathogenic Pore Forming Proteins of Plasmodium Triggers the Necrosis of Endothelial Cells Attributed to Malaria Severity


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
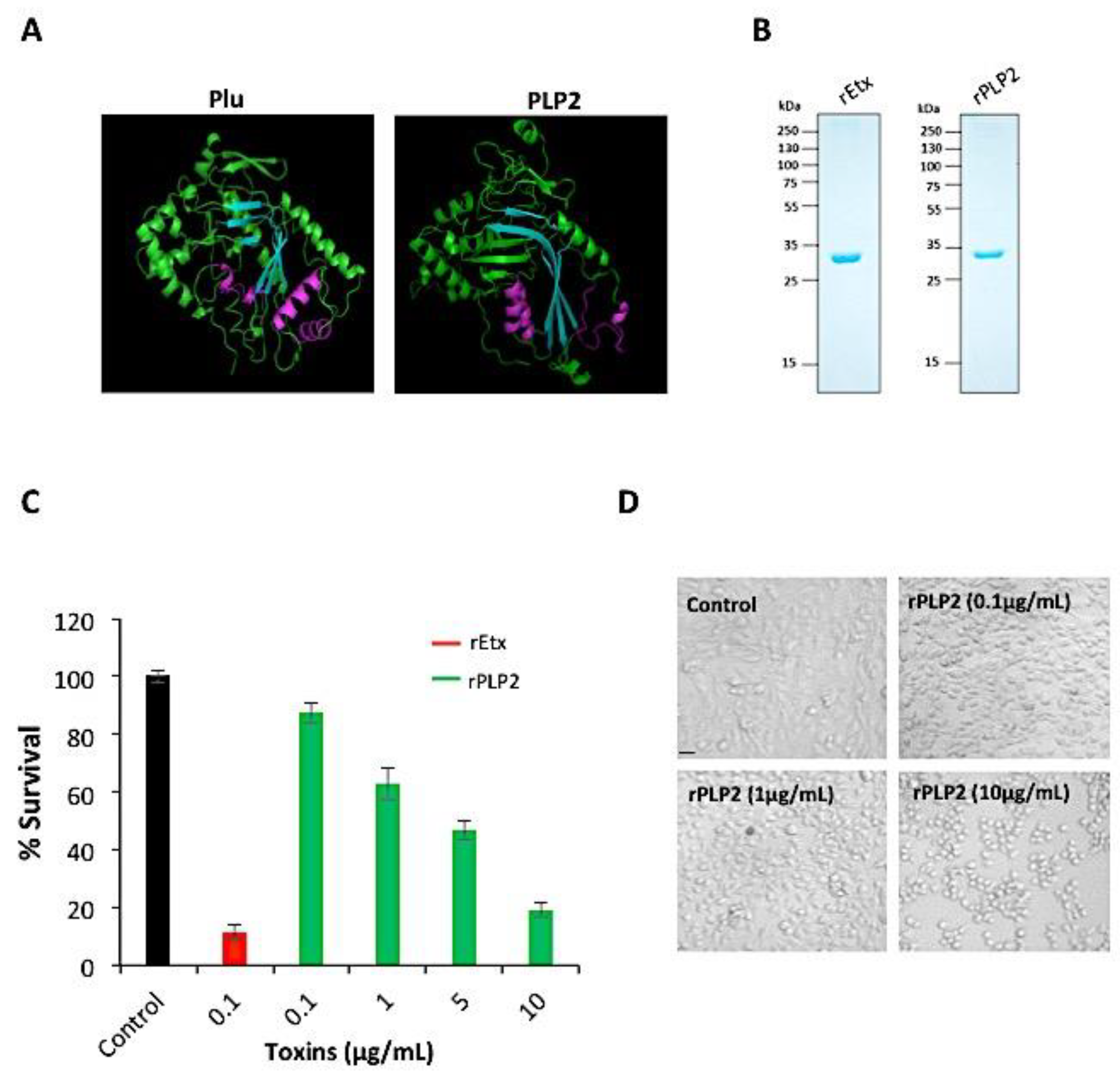
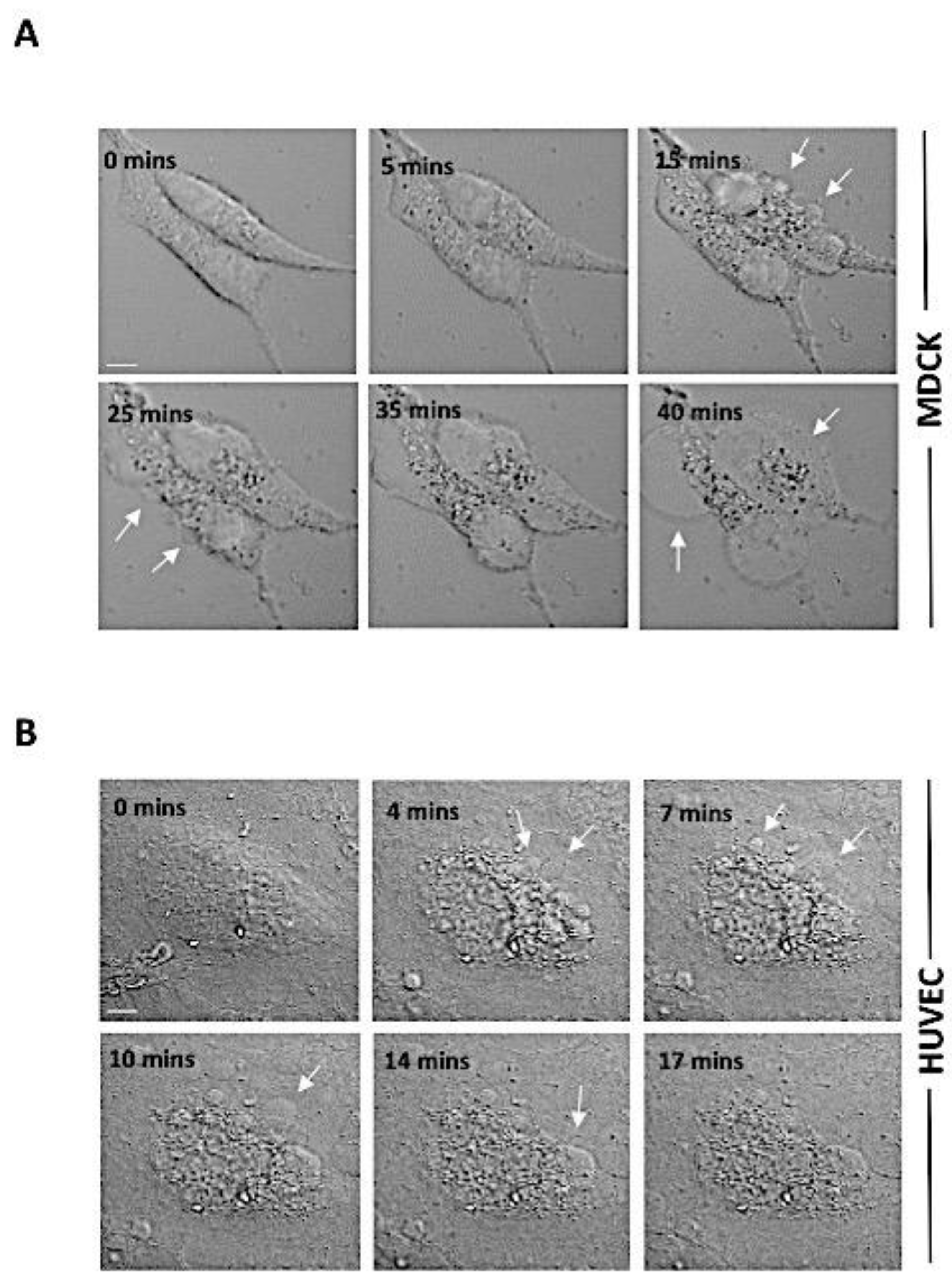
2.1. rPLP2 Induces Death of Primary Endothelial Cells In Vitro
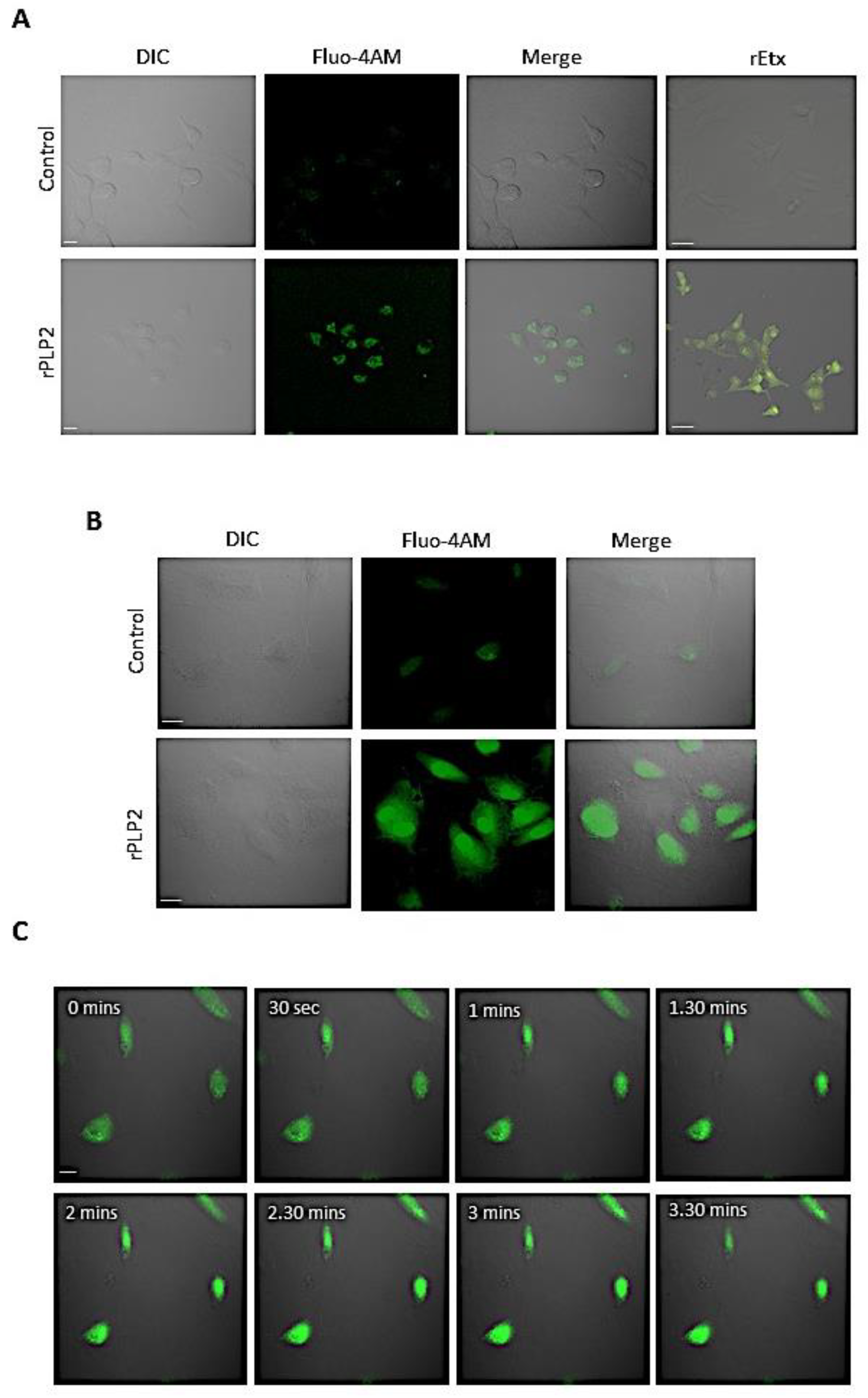
2.2. rPLP2 Mediated an Increase in the Intracellular Calcium Levels of Barrier Cells
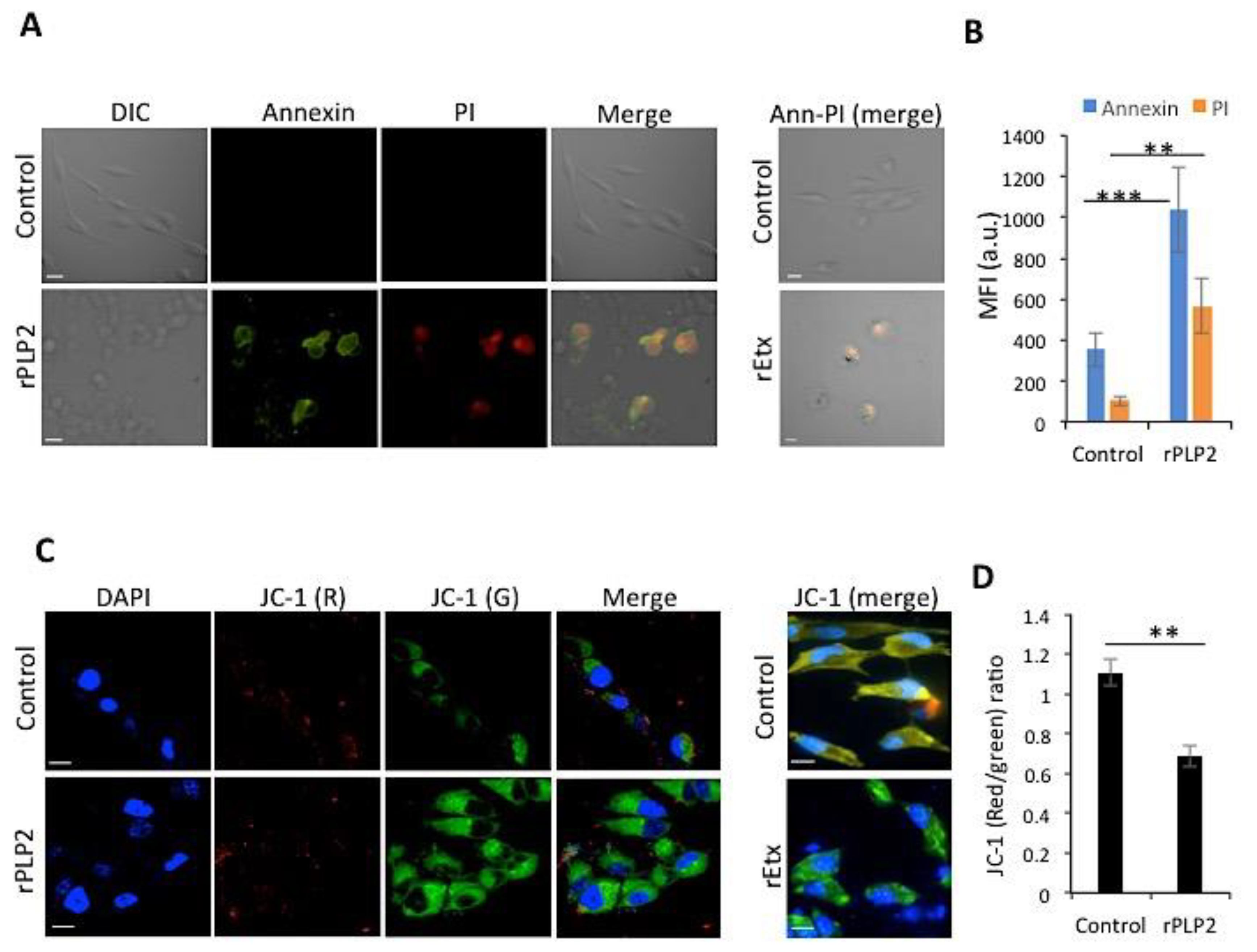
2.3. rPLP2 Leads to the Exposure of Phosphatidylserine and Propidium Iodide Positivity Resulting in Late Apoptosis/Necrosis
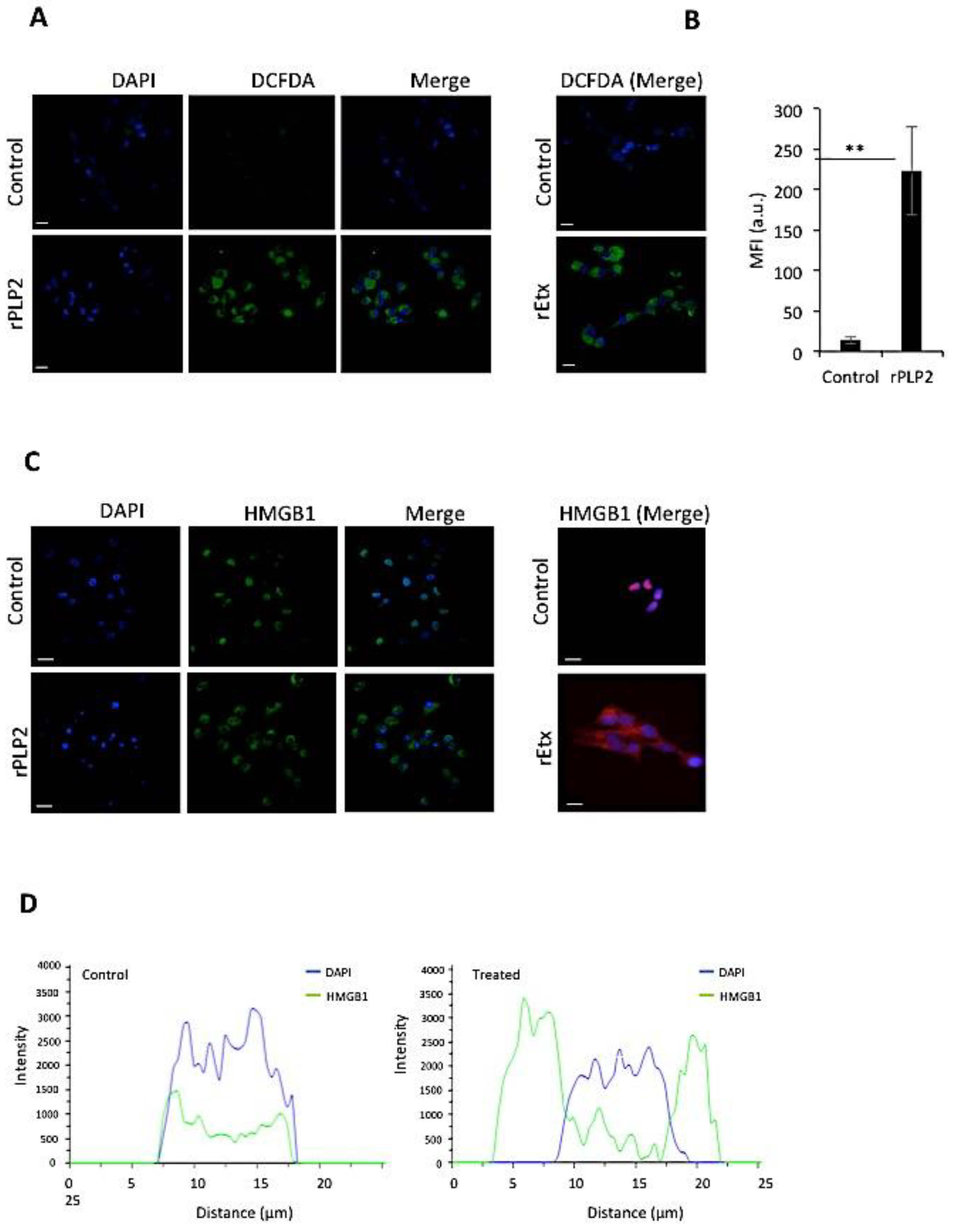
2.4. rPLP2 Increases the Levels of Intracellular Reactive Oxygen Species in Primary Cells
2.5. rPLP2 Treatment Leads to the Translocation of HMGB1 from the Nucleus to the Cytosol
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Recombinant Protein Purification
4.3. Cell Viability
4.4. Endotoxin Contamination Assay in Recombinant Protein
4.5. RBC Lysis Assay
4.6. Live-Cell Video Microscopy
4.7. Erythrocyte Calcium Assay
4.8. Annexin-PI Staining
4.9. Mitochondrial Membrane Potential and ROS Detection Assays
4.10. HMGB1 Assays
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Rocco, M.; Picco, P.; Arslanian, A.; Restagno, G.; Perfumo, F.; Buoncompagni, A.; Gattorno, M.; Borrone, C. Retinitis Pigmentosa, Hypopituitarism, Nephronophthisis, and Mild Skeletal Dysplasia (RHYNS): A New Syndrome? Am. J. Med. Gen. 1997, 73, 1–4. [Google Scholar] [CrossRef]
- Nishanth, G.; Schlüter, D. Blood–Brain Barrier in Cerebral Malaria: Pathogenesis and Therapeutic Intervention. Trends Parasitol. 2019, 35, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brian De Souza, J.; Hafalla, J.C.R.; Riley, E.M.; Couper, K.N. Cerebral malaria: Why experimental murine models are required to understand the pathogenesis of disease. Parasitology 2010, 137, 755–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newbold, C.; Warn, P.; Black, G.; Berendt, A.; Craig, A.; Snow, B.; Msobo, M.; Peshu, N.; Marsh, K. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. Am. J. Trop. Med. Hyg. 1997, 57, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Sullivan, D.J.; Stins, M.F. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J. Infect. Dis. 2007, 195, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Jambou, R.; Combes, V.; Jambou, M.J.; Weksler, B.B.; Couraud, P.O.; Grau, G.E. Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog. 2010, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mandala, W.L.; Msefula, C.L.; Gondwe, E.N.; Drayson, M.T.; Molyneux, M.E.; MacLennan, C.A. Cytokine profiles in Malawian children presenting with uncomplicated malaria, severe malarial anemia, and cerebral malaria. Clin. Vaccine Immunol. 2017, 24, 24. [Google Scholar] [CrossRef] [Green Version]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R.J. Cerebral malaria: Mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Schiess, N.; Villabona-Rueda, A.; Cottier, K.E.; Huether, K.; Chipeta, J.; Stins, M.F. Pathophysiology and neurologic sequelae of cerebral malaria. Malar. J. 2020, 19, 1–12. [Google Scholar] [CrossRef]
- Pehrson, C.; Mathiesen, L.; Heno, K.K.; Salanti, A.; Resende, M.; Dzikowski, R.; Damm, P.; Hansson, S.R.; King, C.L.; Schneider, H.; et al. Adhesion of Plasmodium falciparum infected erythrocytes in ex vivo perfused placental tissue: A novel model of placental malaria. Malar. J. 2016, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hunt, N.H.; Grau, G.E. Cytokines: Accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003, 24, 491–499. [Google Scholar] [CrossRef]
- Medana, I.M.; Turner, G.D.H. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 2006, 36, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Pino, P.; Vouldoukis, I.; Kolb, J.P.; Mahmoudi, N.; Desportes-Livage, I.; Bricaire, F.; Danis, M.; Dugas, B.; Mazier, D. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J. Infect. Dis. 2003, 187, 1283–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claser, C.; Malleret, B.; Gun, S.Y.; Wong, A.Y.W.; Chang, Z.W.; Teo, P.; See, P.C.E.; Howland, S.W.; Ginhoux, F.; Rénia, L. Cd8+ T cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS ONE 2011, 6, e18720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rénia, L.; Howland, S.W.; Claser, C.; Gruner, A.C.; Suwanarusk, R.; Teo, T.H.; Russell, B.; Lisa, N.P. Cerebral malaria Mysteries at the blood-brain barrier. Virulence 2012, 3, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Huggins, M.A.; Johnson, H.L.; Jin, F.; N’Songo, A.; Hanson, L.M.; LaFrance, S.J.; Butler, N.S.; Harty, J.T.; Johnson, A.J. Perforin expression by CD8 T cells is sufficient to cause fatal brain edema during experimental cerebral malaria. Infect. Immun. 2017, 85, e00985-16. [Google Scholar] [CrossRef] [Green Version]
- Tavares, J.; Amino, R.; Ménard, R. The role of MACPF proteins in the biology of malaria and other apicomplexan parasites. Subcell. Biochem. 2014, 80, 241–253. [Google Scholar]
- Alaganan, A.; Singh, P.; Chitnis, C.E. Molecular mechanisms that mediate invasion and egress of malaria parasites from red blood cells. Curr. Opin. Hematol. 2017, 24, 208–214. [Google Scholar] [CrossRef]
- Wirth, C.C.; Glushakova, S.; Scheuermayer, M.; Repnik, U.; Garg, S.; Schaack, D.; Kachman, M.M.; Weißbach, T.; Zimmerberg, J.; Dandekar, T.; et al. Perforin-like protein PPLP2 permeabilizes the red blood cell membrane during egress of Plasmodium falciparum gametocytes. Cell. Microbiol. 2014, 16, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Deligianni, E.; Morgan, R.N.; Bertuccini, L.; Wirth, C.C.; Silmon de Monerri, N.C.; Spanos, L.; Blackman, M.J.; Louis, C.; Pradel, G.; Siden-Kiamos, I. A perforin-like protein mediates disruption of the erythrocyte membrane during egress of Plasmodium berghei male gametocytes. Cell. Microbiol. 2013, 15, 1438–1455. [Google Scholar] [CrossRef]
- Garg, S.; Agarwal, S.; Kumar, S.; Shams Yazdani, S.; Chitnis, C.E.; Singh, S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat. Commun. 2013, 4, 1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadota, K.; Ishino, T.; Matsuyama, T.; Chinzei, Y.; Yuda, M. Essential role of membrane-attack protein in malarial transmission to mosquito host. Proc. Natl. Acad. Sci. USA 2004, 101, 16310–16315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishino, T.; Chinzei, Y.; Yuda, M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell. Microbiol. 2005, 7, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Ecker, A.; Pinto, S.B.; Baker, K.W.; Kafatos, F.C.; Sinden, R.E. Plasmodium berghei: Plasmodium perforin-like protein 5 is required for mosquito midgut invasion in Anopheles stephensi. Exp. Parasitol. 2007, 116, 504–508. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.S.P.; O’Neill, M.T.; Jennison, C.; Lopaticki, S.; Allison, C.C.; Armistead, J.S.; Erickson, S.M.; Rogers, K.L.; Ellisdon, A.M.; Whisstock, J.C.; et al. Cell Traversal Activity Is Important for Plasmodium falciparum Liver Infection in Humanized Mice. Cell Rep. 2017, 18, 3105–3116. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.J.C.; Mikelj, M.; Dalla Serra, M.; Froelich, C.J.; Anderluh, G. Effects of MACPF/CDC proteins on lipid membranes. Cell. Mol. Life Sci. 2013, 70, 2083–2098. [Google Scholar] [CrossRef]
- Garg, S.; Shivappagowdar, A.; Hada, R.S.; Ayana, R.; Bathula, C.; Sen, S.; Kalia, I.; Pati, S.; Singh, A.P.; Singh, S. Plasmodium Perforin-Like Protein Pores on the Host Cell Membrane Contribute in Its Multistage Growth and Erythrocyte Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Rosado, C.J.; Buckle, A.M.; Law, R.H.P.; Butcher, R.E.; Kan, W.T.; Bird, C.H.; Ung, K.; Browne, K.A.; Baran, K.; Bashtannyk-Puhalovich, T.A.; et al. A common fold mediates vertebrate defense and bacterial attack. Science 2007, 317, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Mathur, D.D.; Deshmukh, S.; Kaushik, H.; Garg, L.C. Functional and structural characterization of soluble recombinant epsilon toxin of Clostridium perfringens D, causative agent of enterotoxaemia. Appl. Microbiol. Biotechnol. 2010, 88, 877–884. [Google Scholar] [CrossRef]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Lee, S.E.; Kang, I.-C.; Nam, K.I.; Choy, H.E.; Rhee, J.H. A bacterial RTX toxin causes programmed necrotic cell death through calcium-mediated mitochondrial dysfunction. J. Infect. Dis. 2013, 207, 1406–1415. [Google Scholar] [CrossRef]
- Barros, L.F.; Kanaseki, T.; Sabirov, R.; Morishima, S.; Castro, J.; Bittner, C.X.; Maeno, E.; Ando-Akatsuka, Y.; Okada, Y. Apoptotic and necrotic blebs in epethelial cells display similar neck diameters but different kinase dependency. Cell Death Differ. 2003, 10, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Ghibelli, L.; Cerella, C.; Diederich, M. The dual role of calcium as messenger and stressor in cell damage, death, and survival. Int. J. Cell Biol. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Shivappagowdar, A.; Pati, S.; Narayana, C.; Ayana, R.; Kaushik, H.; Sah, R.; Garg, S.; Khanna, A.; Kumari, J.; Garg, L.; et al. A small bioactive glycoside inhibits epsilon toxin and prevents cell death. DMM Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Zeh, H.J.; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef] [Green Version]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4-dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [Green Version]
- Tunon-Ortiz, A.; Lamb, T.J. Blood brain barrier disruption in cerebral malaria: Beyond endothelial cell activation. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensson, K.; Masocha, W.; Bentivoglio, M. Mechanisms of CNS invasion and damage by parasites. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 114, pp. 11–22. [Google Scholar]
- Kumar Sah, R.; Garg, S.; Dangi, P.; Ponnusamy, K.; Singh, S. Phosphatidic acid homeostasis regulated by a type-2 phosphatidic acid phosphatase represents a novel druggable target in malaria intervention. Cell Death Discov. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Cabanes, D.; Sarmento Mesquita, F.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; De Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Ren. Physiol. 2007, 293. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.L.; Smith, D.J.; Lyras, D.; Chakravorty, A.; Rood, J.I. Programmed cellular necrosis mediated by the pore-forming α-toxin from Clostridium septicum. PLoS Pathog. 2009, 5, e1000516. [Google Scholar] [CrossRef]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell. Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef]
- Krause, K.H.; Fivaz, M.; Monod, A.; Van Gisou Der Goot, F. Aerolysin induces G-protein activation and Ca2+ release from intracellular stores in human granulocytes. J. Biol. Chem. 1998, 273, 18122–18129. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Varshney, S.K.; Khan, H.M.; Malik, M.A.; Mehdi, A.A.; Shukla, I. Superoxide dismutase activity in patients of cerebral malaria. Asian Pac. J. Trop. Dis. 2015, 5, S51–S53. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Ditsworth, D.; Zong, W.X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shivappagowdar, A.; Garg, S.; Srivastava, A.; Hada, R.S.; Kalia, I.; Singh, A.P.; Garg, L.C.; Pati, S.; Singh, S. Pathogenic Pore Forming Proteins of Plasmodium Triggers the Necrosis of Endothelial Cells Attributed to Malaria Severity. Toxins 2021, 13, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010062
Shivappagowdar A, Garg S, Srivastava A, Hada RS, Kalia I, Singh AP, Garg LC, Pati S, Singh S. Pathogenic Pore Forming Proteins of Plasmodium Triggers the Necrosis of Endothelial Cells Attributed to Malaria Severity. Toxins. 2021; 13(1):62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010062
Chicago/Turabian StyleShivappagowdar, Abhishek, Swati Garg, Akriti Srivastava, Rahul S. Hada, Inderjeet Kalia, Agam P. Singh, Lalit C. Garg, Soumya Pati, and Shailja Singh. 2021. "Pathogenic Pore Forming Proteins of Plasmodium Triggers the Necrosis of Endothelial Cells Attributed to Malaria Severity" Toxins 13, no. 1: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13010062