Validation of New ELISA Technique for Detection of Aflatoxin B1 Contamination in Food Products versus HPLC and VICAM

 ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Aflatoxin B1 Detection with HPLC and VICAM
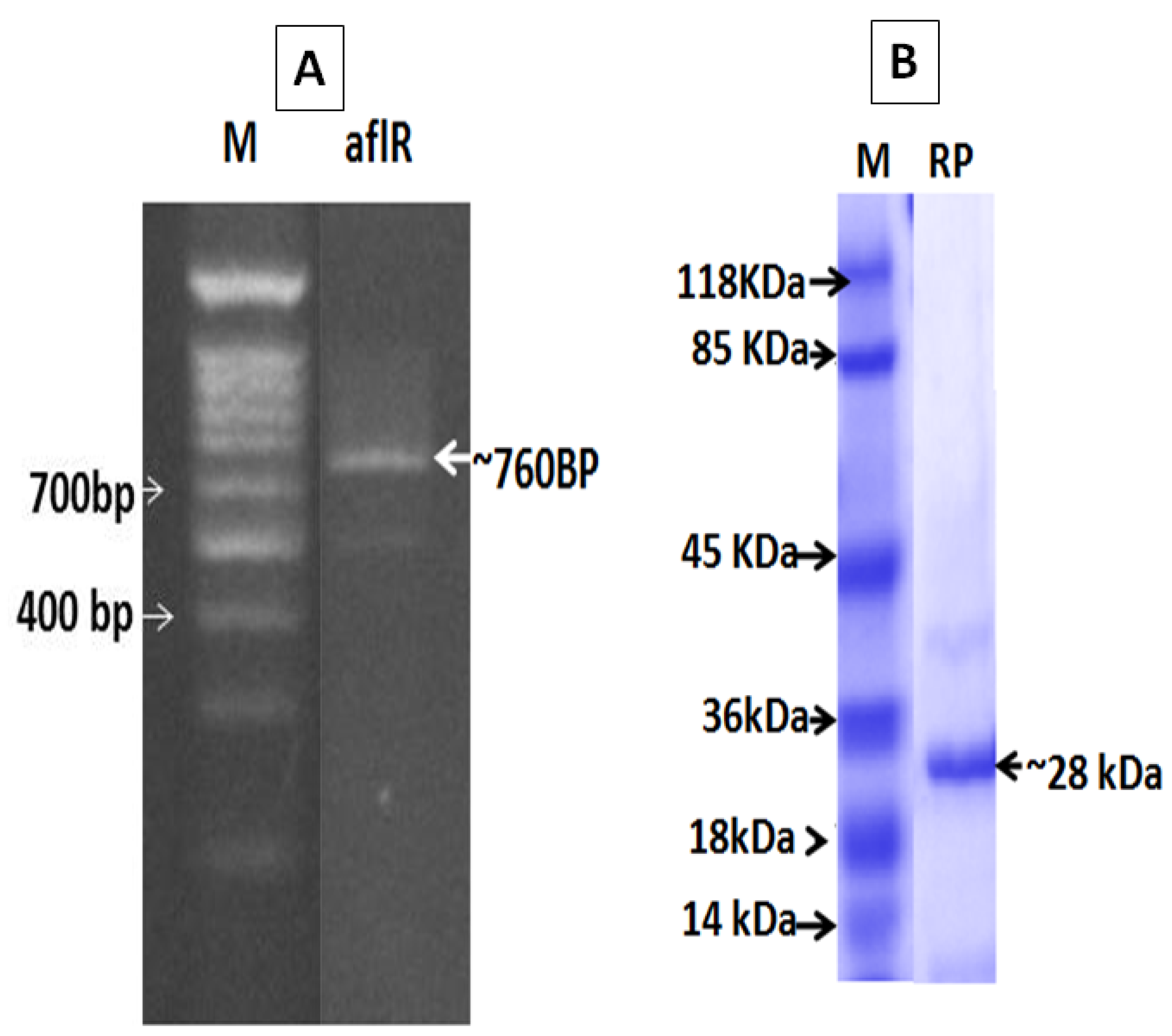
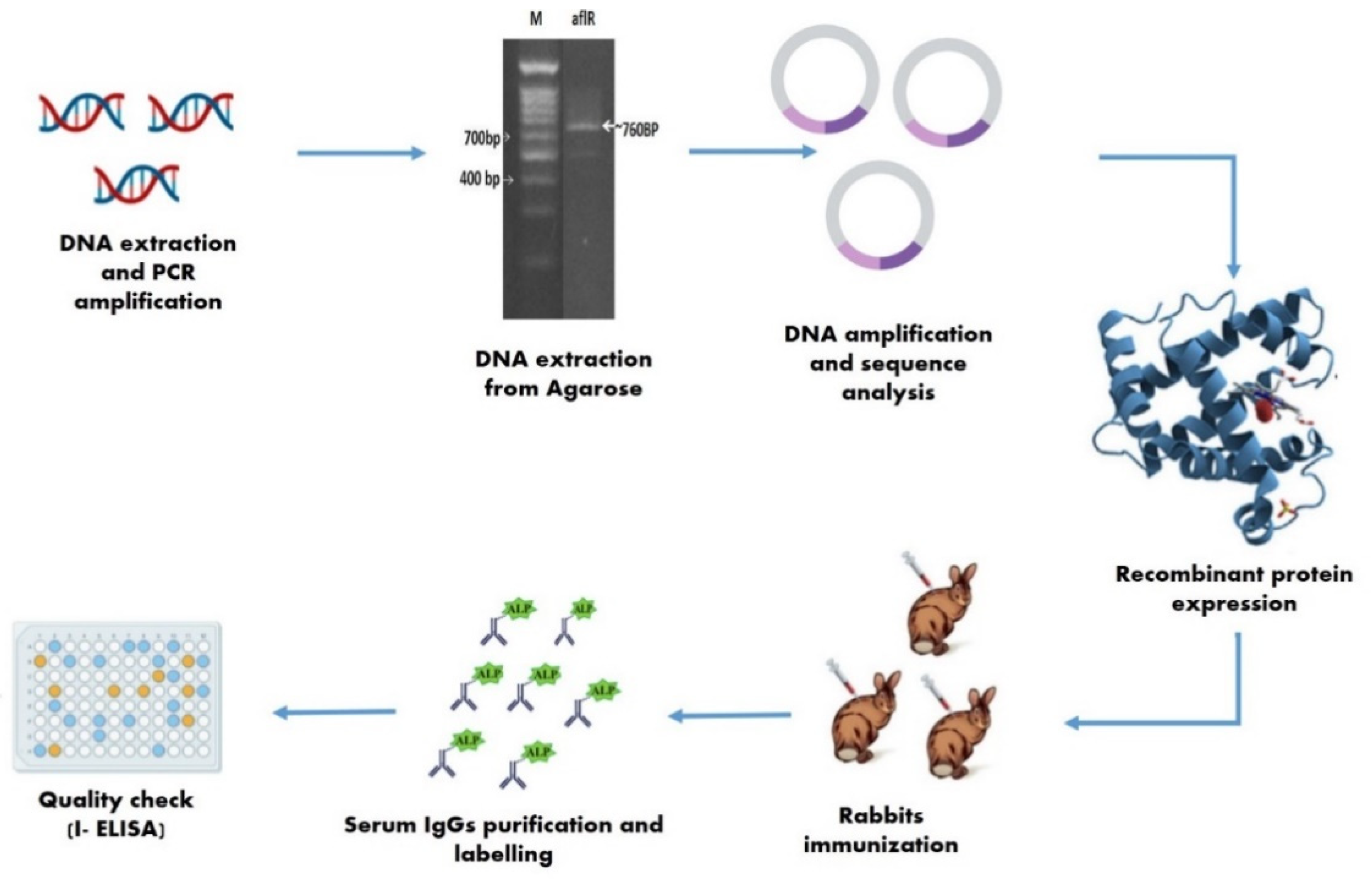
2.2. Molecular Detection and SDS-PAGE
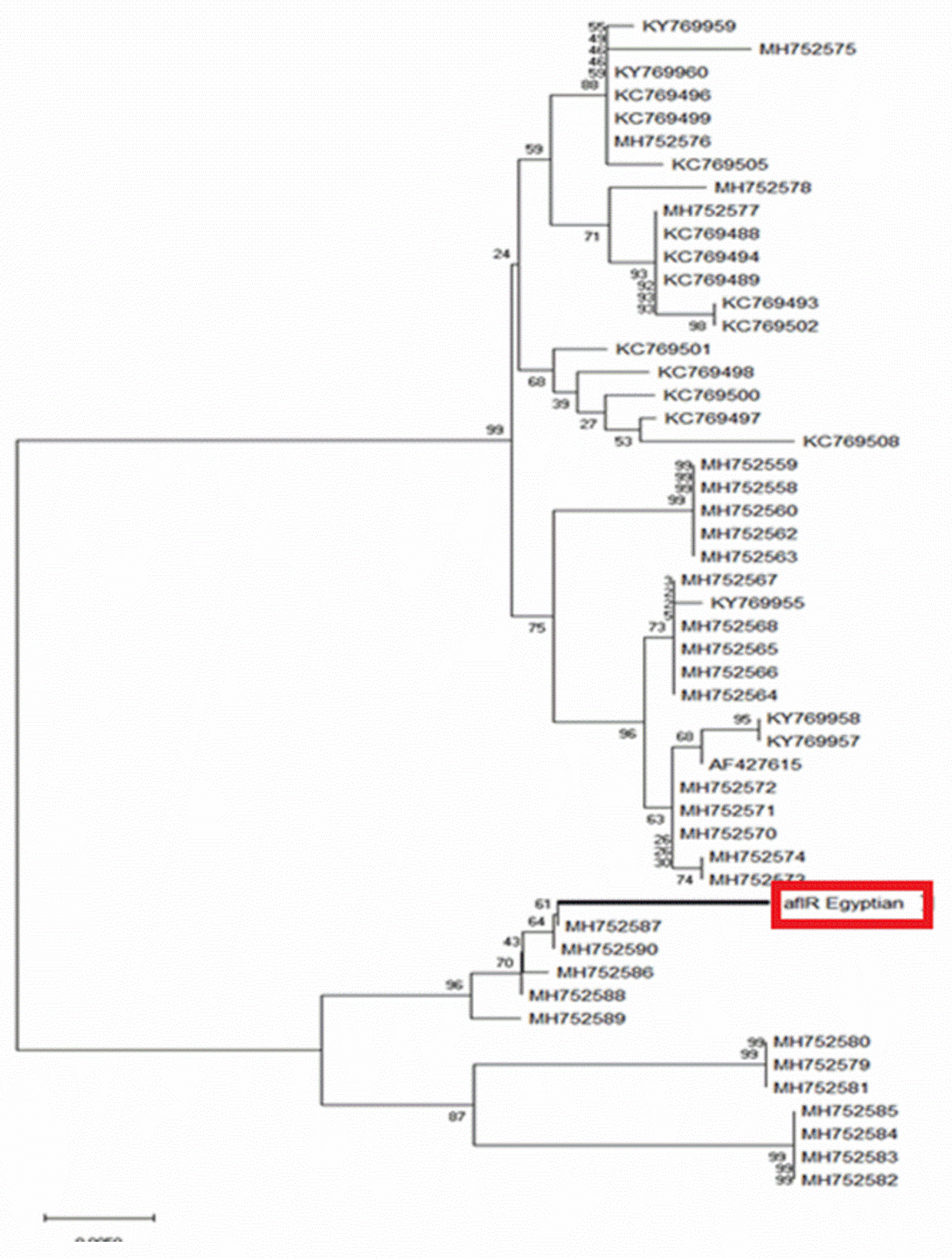
2.3. Sequence Analysis and Phylogenic Construction
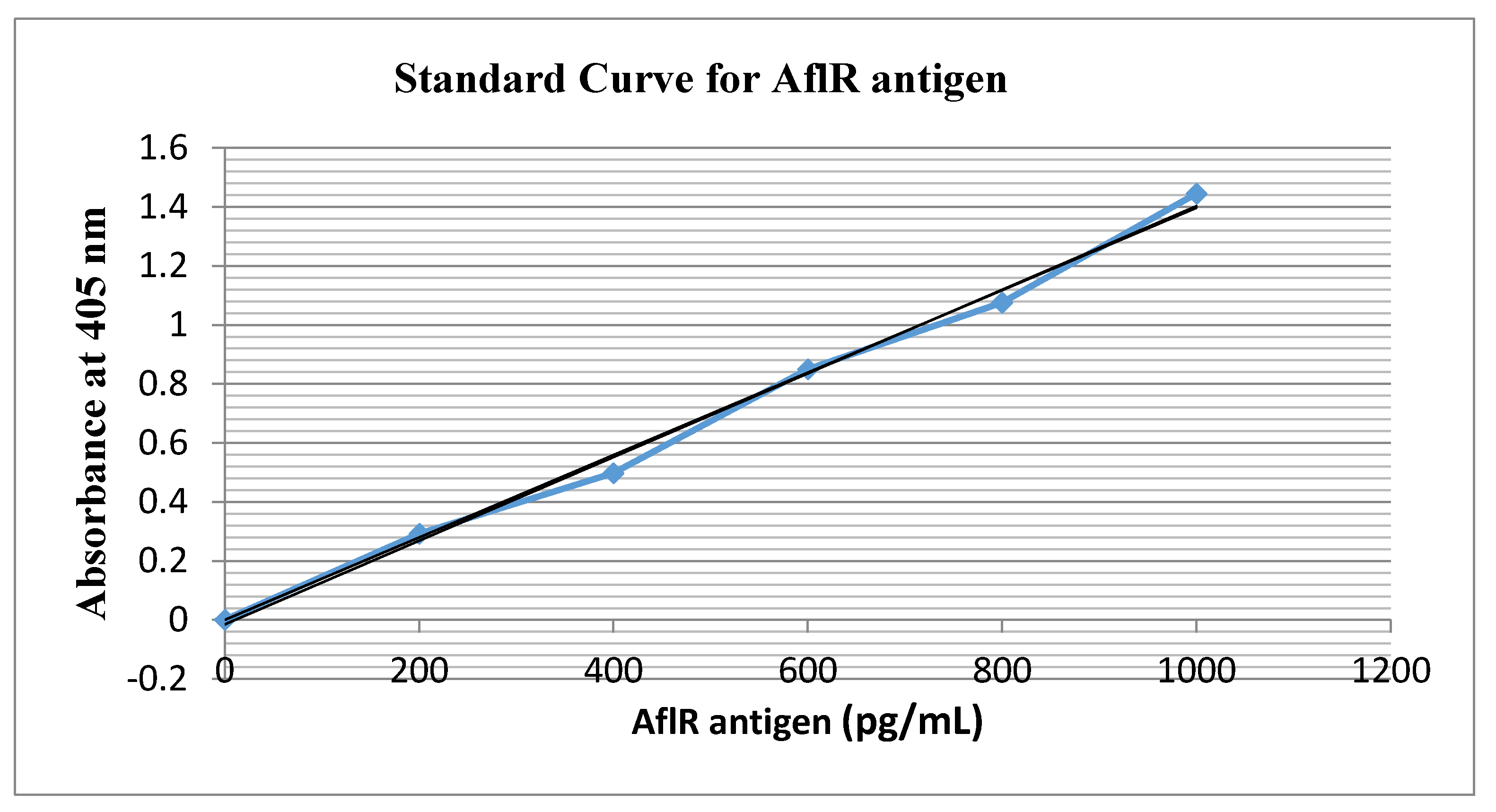
2.4. Antigenicity Test
2.5. IgG Polyclonal Antibody Labeling and Purification
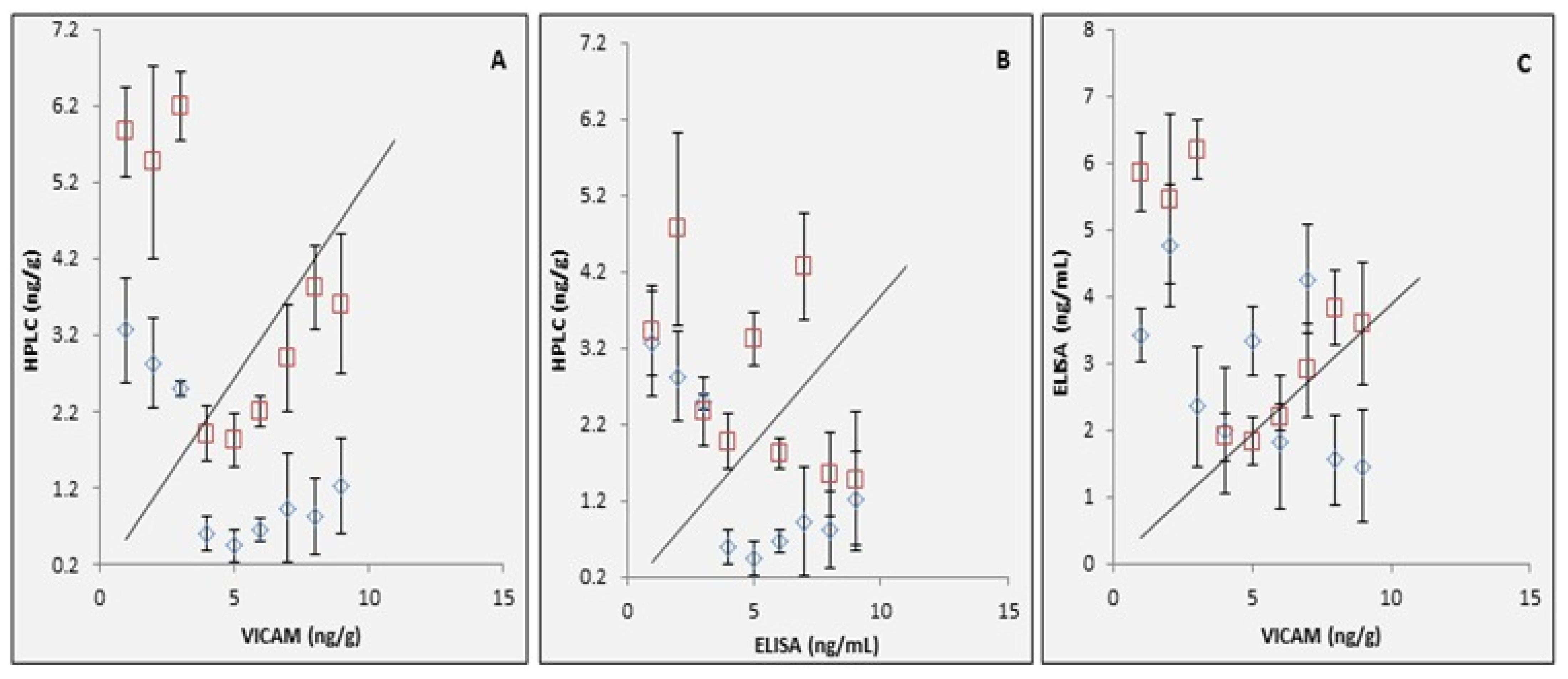
2.6. Validation of the Modified Recombinant AflR Gene Antiserum ELISA Technique with HPLC and VICAM
2.7. Limitations of the Modified Recombinant AflR Gene Antiserum ELISA Technique
3. Conclusions and Future Perspective
4. Materials and Methods
4.1. Sampling
4.2. HPLC Detection
4.3. AflA-Vt Detection
4.4. Specific PCR Detection Method
4.5. Detection via AflR Gene Analysis and Transformation
4.5.1. Cloning, Sequencing, and AflR Gene Transformation
4.5.2. Molecular Size Determination of AflR Recombinant Protein
4.5.3. Epitope Prediction and Antigenic Determination
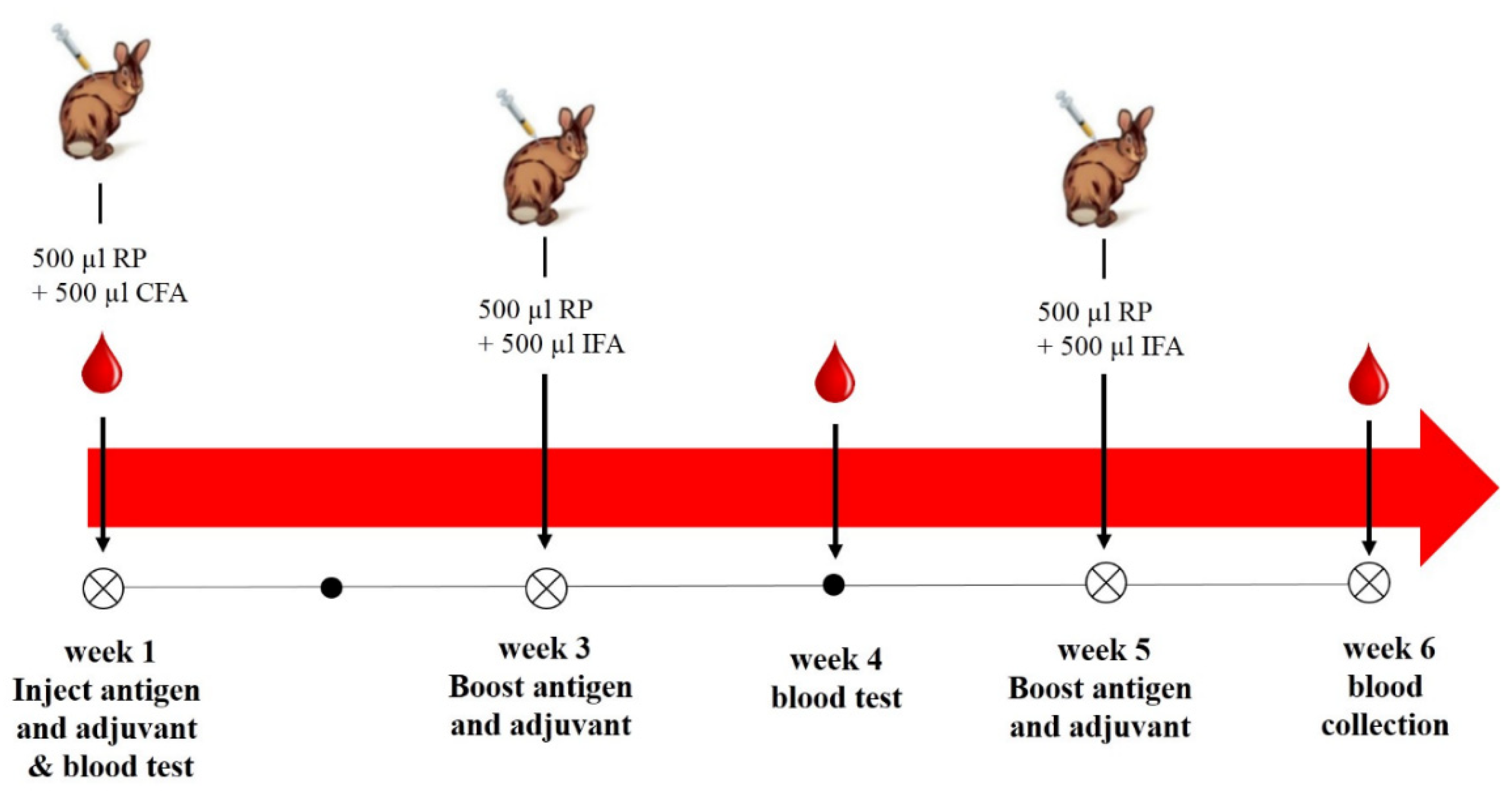
4.5.4. Immunization and Antibody Production
Rabbit Immunization with AflR Recombinant Protein
Serum IgG Purification and Fractionation
Labelling of Antibodies
4.5.5. Quality Checks
5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Goud, K.Y.; Reddy, K.K.; Satyanarayana, M.; Kummari, S.; Gobi, K.V. A review on recent developments in optical and electrochemical aptamer-based assays for mycotoxins using advanced nanomaterials. Microchim. Acta 2019, 187, 29. [Google Scholar] [CrossRef]
- Hussain, K.K.; Malavia, D.; Johnson, E.M.; Littlechild, J.; Winlove, C.P.; Vollmer, F.; Gow, N.A.R. Biosensors and Diagnostics for Fungal Detection. J. Fungi 2020, 6, 349. [Google Scholar] [CrossRef] [PubMed]
- Bryła, M.; Waśkiewicz, A.; Podolska, G.; Szymczyk, K.; Jędrzejczak, R.; Damaziak, K.; Sułek, A. Occurrence of 26 Mycotoxins in the Grain of Cereals Cultivated in Poland. Toxins 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Ezekiel, C.; Sulyok, M.; Babalola, D.; Warth, B.; Ezekiel, V.; Krska, R. Incidence and consumer awareness of toxigenic Aspergillus section Flavi and aflatoxin B1 in peanut cake from Nigeria. Food Control 2013, 30, 596–601. [Google Scholar] [CrossRef]
- Dai, Y.; Huang, K.; Zhang, B.; Zhu, L.; Xu, W. Aflatoxin B1-induced epigenetic alterations: An overview. Food Chem. Toxicol. 2017, 109, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.A.; Widstrom, N.W. Aflatoxin in maize. Crit. Rev. Plant Sci. 1992, 10, 423–440. [Google Scholar] [CrossRef]
- Shotwell, O.L.; Goulden, M.L.; Hesseltine, C.W. Aflatoxin: Distribution incontaminated corn. Cereal Chem. 1974, 51, 492–499. [Google Scholar]
- Filazi, A.; Tansel, U. Occurrence of Aflatoxins in Food. Aflatoxins Recent Adv. Future Prospect. 2013, 143–170. [Google Scholar] [CrossRef]
- Herzallah, S.M. Determination of aflatoxins in eggs, milk, meat and meat products using HPLC fluorescent and UV detectors. Food Chem. 2009, 114, 1141–1146. [Google Scholar] [CrossRef]
- Wagner, K.; Springer, B.; Pires, V.P.; Keller, P.M. Molecular detection of fungal pathogens in clinical specimens by 18S rDNA high-throughput screening in comparison to ITS PCR and culture. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Gallo, A.; Stea, G.; Battilani, P.; Logrieco, A.F.; Perrone, G. Molecular characterization of Aspergillus flavus population isolated from maize during the first outbreak of aflatoxin contamination in Italy. Phytopathol. Mediterr. 2012, 51, 198–206. [Google Scholar] [CrossRef]
- Alahi, M.E.E.; Mukhopadhyay, S.C. Detection methodologies for pathogen and toxins: A review. Sensors 2017, 17, 1885. [Google Scholar] [CrossRef]
- Maggira, M.; Ioannidou, M.; Sakaridis, I.; Samouris, G. Determination of Aflatoxin M1 in Raw Milk Using an HPLC-FL Method in Comparison with Commercial ELISA Kits—Application in Raw Milk Samples from Various Regions of Greece. Vet. Sci. 2021, 8, 46. [Google Scholar] [CrossRef]
- Vosough, M.; Bayat, M.; Salemi, A. Matrix-free analysis of aflatoxins in pistachio nuts using parallel factor modeling of liquid chromatography diode-array detection data. Anal. Chim. Acta 2010, 663, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.; Ngundi, M.; Moore, M.; Lassman, M.; Shriver-Lake, L.; Taitt, C.; Ligler, F. Rapid detection of food-borne contaminants using an array biosensor. Sens. Actuators B Chem. 2006, 113, 599–607. [Google Scholar] [CrossRef]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Schmidt, A.M. Development and application of synthetic peptides as vaccines. Biotechnol. Adv. 1989, 7, 187–213. [Google Scholar] [CrossRef]
- Mahan, D.E.; Morrison, L.; Watson, L.; Haugneland, L.S. Phase change enzyme immunoassay. Anal. Biochem. 1987, 162, 163–170. [Google Scholar] [CrossRef]
- Gruzdys, V.; Cahoon, K.; Pearson, L.; Lehman, C.M. Method Verification Shows a Negative Bias between 2 Procalcitonin Methods at Medical Decision Concentrations. J. Appl. Lab. Med. 2019, 4, 69–77. [Google Scholar] [CrossRef]
- Harrison, B.D.; Barker, H.; Bock, K.R.; Guthrie, E.J.; Meredith, G.; Atkinson, M. Plant viruses with circular single-stranded DNA. Nat. Cell Biol. 1977, 270, 760–762. [Google Scholar] [CrossRef]
- Padidam, M.; Beachy, R.N.; Fauquet, C.M. Classification and identific-ation of geminiviruses using sequence comparison. J. Gen. Virol. 1995, 76, 249–263. [Google Scholar] [CrossRef]
- Abdel–Salam, A.M. Isolation and characterization of a whitefly-transmitted geminivirus associated with the leaf curl and mosaic symptoms on cotton in Egypt. Arab. J. Biotech. 1999, 2, 193–218. [Google Scholar]
- Whitaker, T.B. Detecting Mycotoxins in Agricultural Commodities. Mol. Biotechnol. 2003, 23, 61–72. [Google Scholar] [CrossRef]
- Lee, N.A.; Rachaputi, N.C.; Wright, G.C.; Krosch, S.; Norman, K.; Anderson, J.; Ambarwati, S.; Retnowati, I.; Dharmaputra, O.S.; Kennedy, I.R. Validation of analytical parameters of a competitive direct ELISA for aflatoxin B1in peanuts. Food Agric. Immunol. 2005, 16, 149–163. [Google Scholar] [CrossRef]
- Lee, S.D.; Yu, I.S.; Jung, K.; Kim, Y.S. Incidence and Level of Aflatoxins Contamination in Medicinal Plants in Korea. Mycobiology 2014, 42, 339–345. [Google Scholar] [CrossRef]
- Scaglioni, P.T.; Badiale-Furlong, E. Rice husk as an adsorbent: A new analytical approach to determine aflatoxins in milk. Talanta 2016, 152, 423–431. [Google Scholar] [CrossRef]
- Ertekin, Ö.; Pirinçci, Ş.Ş.; Öztürk, S. Monoclonal IgA Antibodies for Aflatoxin Immunoassays. Toxins 2016, 8, 148. [Google Scholar] [CrossRef]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Fishback, J.E.; Stronsky, S.M.; Green, C.A.; Bean, K.D.; Froude, J.W. Erratum: Antibody production in rabbits administered Freund’s complete adjuvant and carprofen concurrently. Lab Anim. 2016, 45, 121. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Arnaud, P. A general method for fractionation of plasma proteins. Dye-ligand affinity chromatography on immobilized Cibacron blue F3-GA. Biochem. J. 1982, 201, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Avrameas, S. Coupling of enzymes to proteins with glutaraldehyde: Use of the conjugates for the detection of antigens and antibodies. Immunochemistry 1969, 6, 43–52. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | HPLC | VICAM |
|---|---|---|
| Peanut 1 | +++ | +++ |
| Peanut 2 | +++ | ++ |
| Peanut 3 | +++ | +++ |
| Flour 1 | ++ | ++ |
| Flour 2 | +++ | ++ |
| Flour 3 | ++ | + |
| Milk-powder 1 | ++ | ++ |
| Milk-powder 2 | + | + |
| Milk-powder 3 | ++ | + |
| No. | Start | End | Peptide | Length | >aflIR d Deduced Amino Acid Sequence |
|---|---|---|---|---|---|
| 1 | 26 | 33 | LMQVPKIY | 8 | MSHSYNTFAGWFINTPTGRTQGSLALMQVPKI |
| 2 | 66 | 76 | EHYLLFLVQFV | 11 | YLAGNKSFLGSQPAHDGLRYLEPEACMRAGQL |
| 3 | 107 | 120 | TPQLVTFVYIHLDL | 14 | AEHYLLFLVQFVNRSRSSLVTRFQPRYVNKEC |
| 4 | 136 | 143 | FTLCVPPRLA | 8 | TARQSLGQVRTPQLVTFVYIHLDLSARQRKGO |
| 5 | 170 | 179 | PGRCVPPPRLA | 10 | ATLQEKAFTLCVFFPAFNSKLYSTPSSRPPRW |
| 6 | 186 | 196 | IAVRVVPVQKC | 11 | LTIFPPGHIPGRCVPPRLAALESSGIAVRVVPVQKC |
| 7 | 205 | 215 | VLGVSNVVLPV | 11 | DAPRRNRPVLGVSNVVLPVNTWSPSGWAAT |
| 8 | 227 | 236 | RALPVPLIQL | 10 | RALPVPLIQLGDHQRVFLQPDRNRDIRRIT |
| Sample | HPLC (ng/g) | VICAM (ng/g) | Specific PCR | ELISA (ng/mL) |
|---|---|---|---|---|
| Peanut 1 | 3.26 ± 0.68 a | 5.86 ± 0.58 a | + | 3.43 ± 0.40 a,b |
| Peanut 2 | 2.83 ± 0.58 a | 5.46 ± 1.26 a | + | 4.76 ± 0.92 a |
| Peanut 3 | 2.50 ± 0.10 a | 6.20 ± 0.45 a | + | 2.36 ± 0.90 b,c |
| Flour 1 | 0.60 ± 0.23 b | 1.90 ± 0.36 c | + | 1.98 ± 0.94 b,c |
| Flour 2 | 0.44 ± 0.22 b | 1.83 ± 0.35 c | + | 3.33 ± 0.51 a,b |
| Flour 3 | 0.66 ± 0.15 b | 2.20 ± 0.20 c | + | 1.82 ± 1.01 c |
| Milk-powder 1 | 0.93 ± 0.71 b | 2.90 ± 0.70 b,c | + | 4.26 ± 0.81 a |
| Milk-powder 2 | 0.82 ± 0.50 b | 3.83 ± 0.55 b | + | 1.55 ± 0.67 c |
| Milk-powder 3 | 1.23 ± 0.62 b | 3.60 ± 0.91 b | + | 1.46 ± 0.84 c |
| Parameters | HPLC | VICAM | I-ELISA |
|---|---|---|---|
| Limit of detection | <0.008 ng/mL | <2 ng/mL | <1 ng/mL |
| Time of analysis | 120 min | 90 min | 30 min |
| Cost of analysis | High | Moderate | Moderate |
| Use of organic solvents | Yes | Yes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafez, E.; Abd El-Aziz, N.M.; Darwish, A.M.G.; Shehata, M.G.; Ibrahim, A.A.; Elframawy, A.M.; Badr, A.N. Validation of New ELISA Technique for Detection of Aflatoxin B1 Contamination in Food Products versus HPLC and VICAM. Toxins 2021, 13, 747. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13110747
Hafez E, Abd El-Aziz NM, Darwish AMG, Shehata MG, Ibrahim AA, Elframawy AM, Badr AN. Validation of New ELISA Technique for Detection of Aflatoxin B1 Contamination in Food Products versus HPLC and VICAM. Toxins. 2021; 13(11):747. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13110747
Chicago/Turabian StyleHafez, Elsayed, Nourhan M. Abd El-Aziz, Amira M. G. Darwish, Mohamed G. Shehata, Amira A. Ibrahim, Asmaa M. Elframawy, and Ahmed N. Badr. 2021. "Validation of New ELISA Technique for Detection of Aflatoxin B1 Contamination in Food Products versus HPLC and VICAM" Toxins 13, no. 11: 747. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13110747