Non-Transgenic CRISPR-Mediated Knockout of Entire Ergot Alkaloid Gene Clusters in Slow-Growing Asexual Polyploid Fungi


Abstract
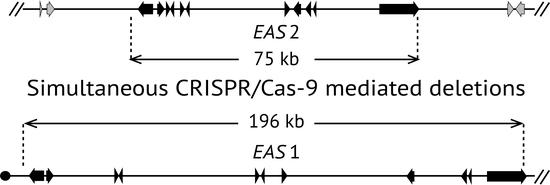
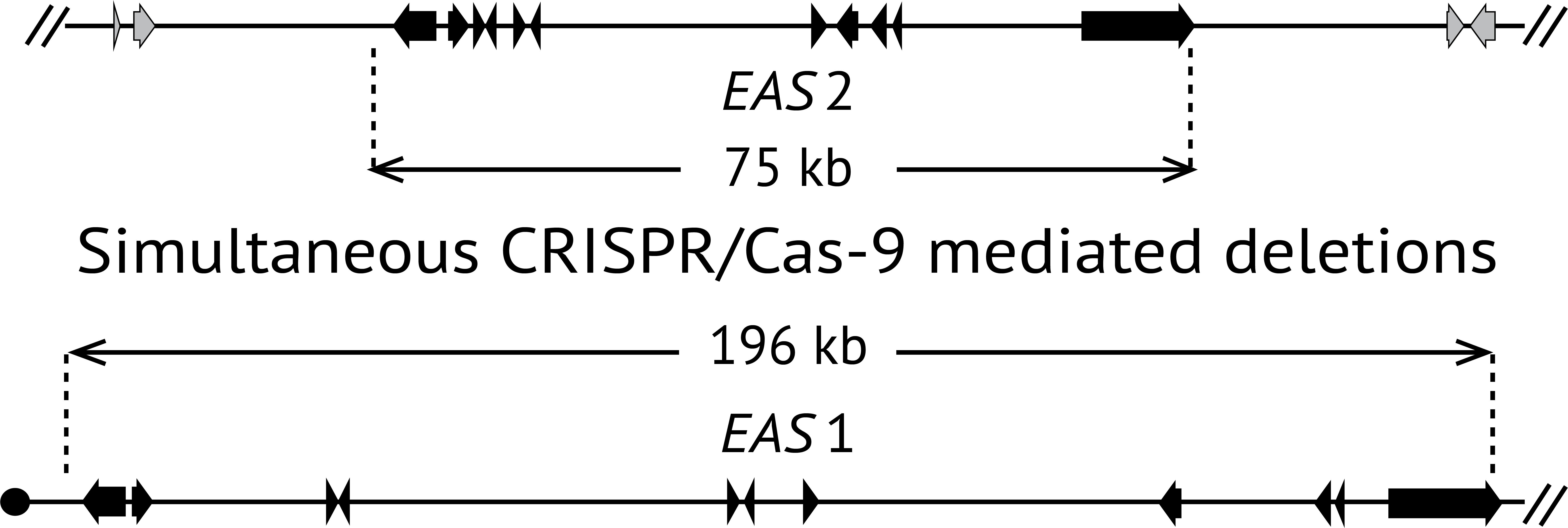
:
1. Introduction
2. Results
2.1. Assembly of E. coenophiala and E. hybrida Genome Sequences Including Nanopore Data
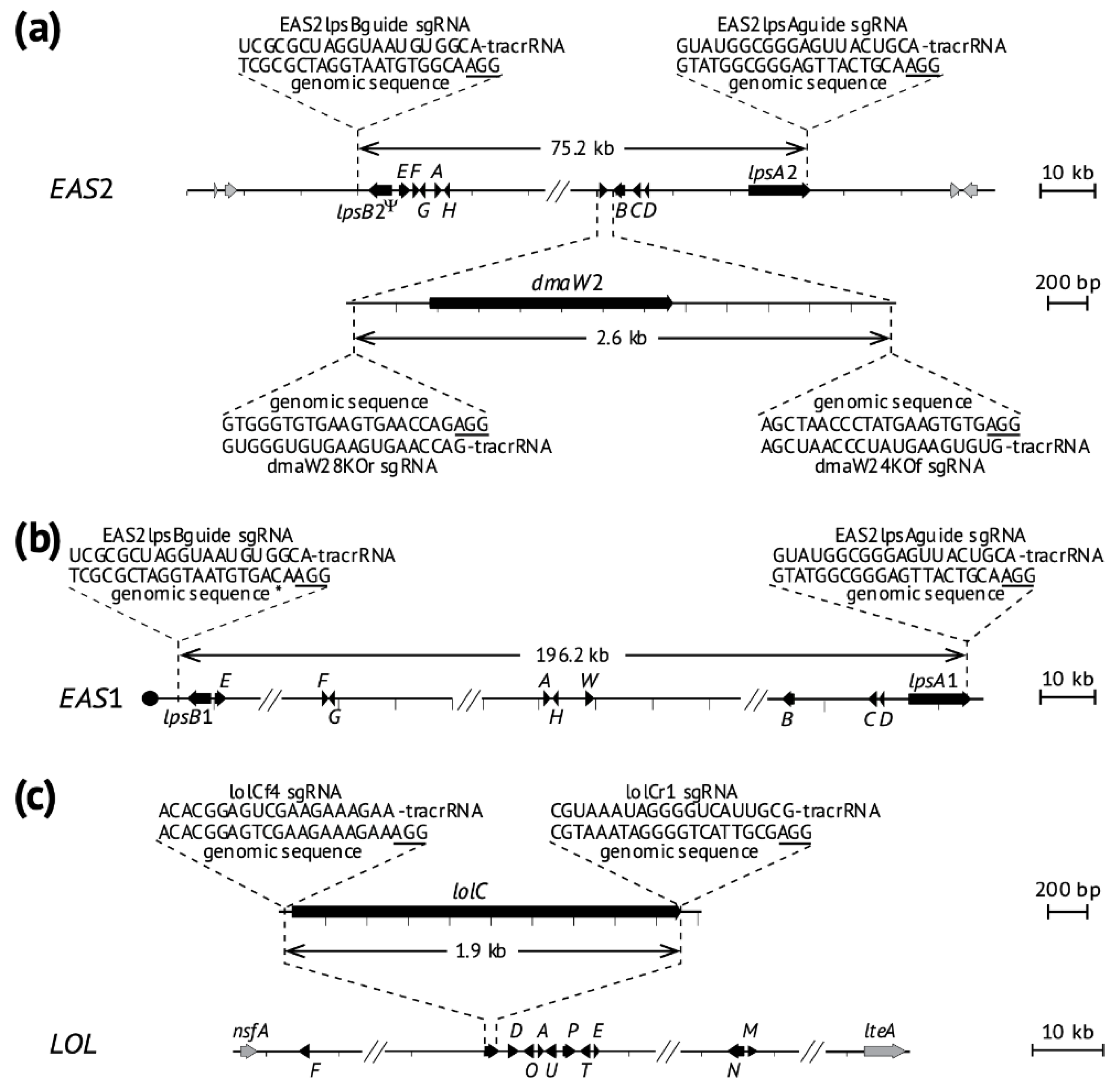
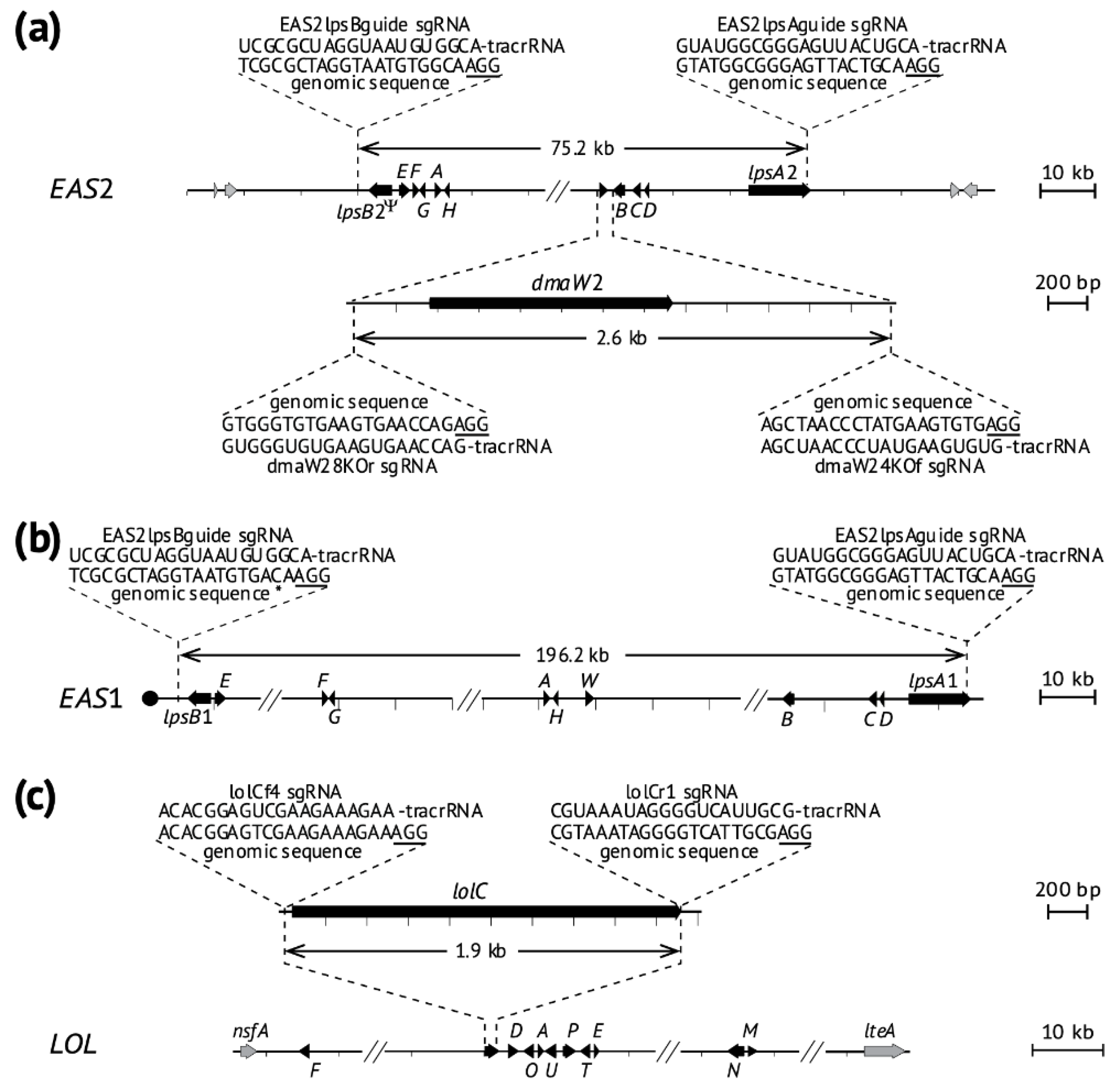
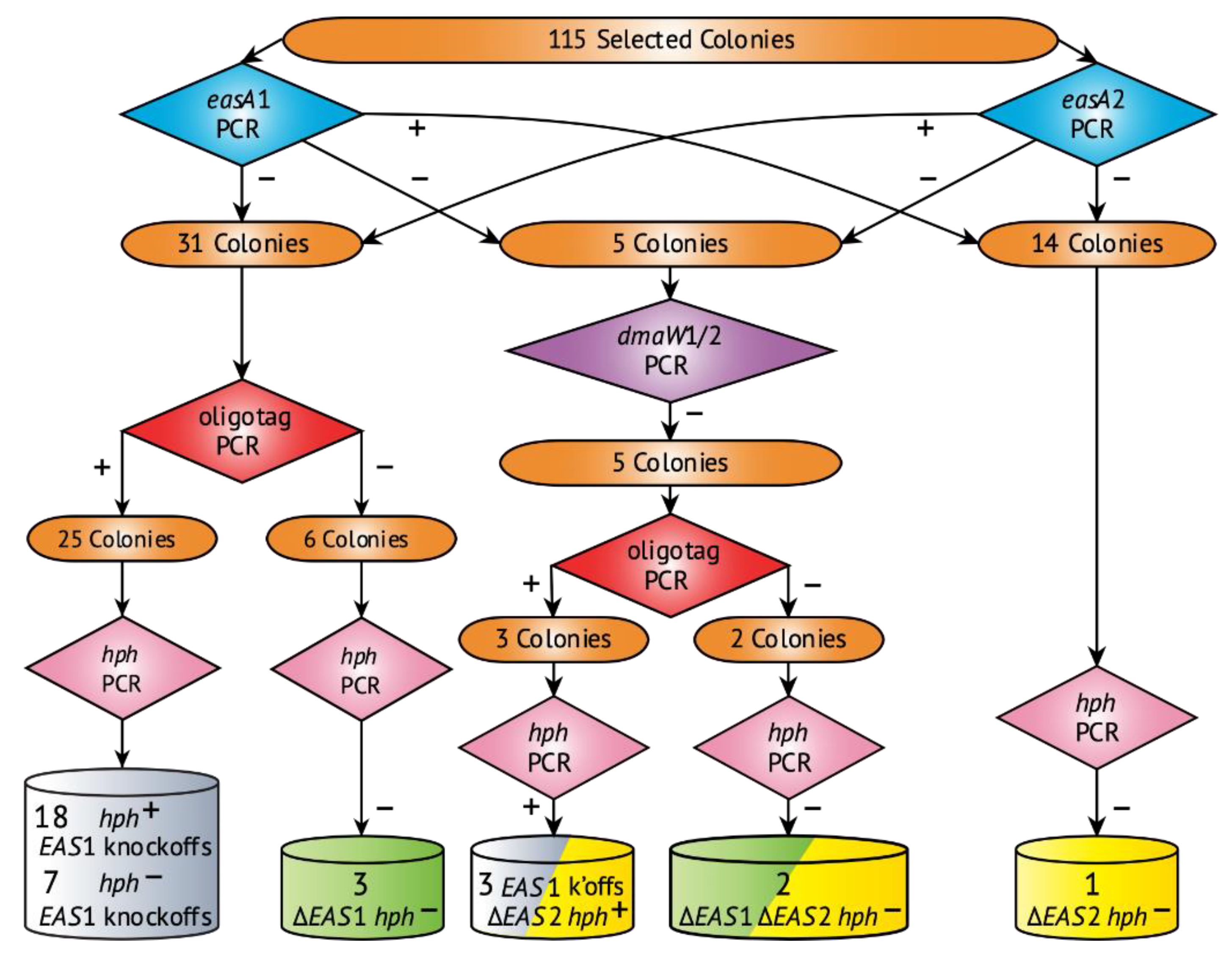
2.2. Deletion of Ergot Alkaloid Biosynthesis Gene Clusters from the E. coenophiala Genome
2.3. Deletion of dmaW2
2.4. Deletion of the EAS Cluster in E. hybrida Lp1
2.5. Deletion of lolC in E. coenophiala e19
2.6. Genome Analysis of the CRISPR-Derived Mutants
2.7. Inoculation Efficiency
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Biological Materials
5.2. Miscellaneous Molecular Methods
5.3. Nanopore Single-Molecule Sequencing of Genomic DNA
5.4. Genome Sequence Assembly
5.5. Illumina HiSeq Sequencing of Genomic DNA
5.6. Design of sgRNA Molecules and Assembly of RNP Complexes
5.7. Fungal Transformation and Selection
5.8. Screening and Analysis of Deletion Mutants
5.9. Establishment of Symbiota
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schardl, C.L.; Florea, S.; Pan, J.; Nagabhyru, P.; Bec, S.; Calie, P.J. The epichloae: Alkaloid diversity and roles in symbiosis with grasses. Curr. Opin. Plant. Biol. 2013, 16, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Schardl, C.L.; Young, C.A.; Pan, J.; Florea, S.; Takach, J.E.; Panaccione, D.G.; Farman, M.L.; Webb, J.S.; Jaromczyk, J.; Charlton, N.D.; et al. Currencies of mutualisms: Sources of alkaloid genes in vertically transmitted epichloae. Toxins 2013, 5, 1064–1088. [Google Scholar] [CrossRef]
- Tanaka, A.; Takemoto, D.; Chujo, T.; Scott, B. Fungal endophytes of grasses. Curr. Opin. Plant. Biol. 2012, 15, 462–468. [Google Scholar] [CrossRef]
- Moon, C.D.; Craven, K.D.; Leuchtmann, A.; Clement, S.L.; Schardl, C.L. Prevalence of interspecific hybrids amongst asexual fungal endophytes of grasses. Mol. Ecol. 2004, 13, 1455–1467. [Google Scholar] [CrossRef]
- Schuster, M.; Kahmann, R. CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet. Biol. 2019, 130, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Zhai, Q.; Sun, L.; Huang, E.; Zhang, Y.; Zhu, Y.; Guo, Q.; Tian, Y.; Zhao, B.; Lu, H. CRISPR/Cas9 genome editing technology in filamentous fungi: Progress and perspective. Appl. Microbiol. Biotechnol. 2019, 103, 6919–6932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nødvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.J.; Martin-Urdiroz, M.; Yan, X.; Wright, H.S.; Soanes, D.M.; Talbot, N.J. CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Malinowski, D.P.; Belesky, D.P. Adaptations of endophyte-infected cool-season grasses to environmental stresses: Mechanisms of drought and mineral stress tolerance. Crop. Sci. 2000, 40, 923–940. [Google Scholar] [CrossRef]
- Malinowski, D.P.; Belesky, D.P. Epichloë (formerly Neotyphodium) fungal endophytes increase adaptation of cool-season perennial grasses to environmental stresses. Acta Agrobot. 2019, 72. [Google Scholar] [CrossRef]
- Clay, K.; Schardl, C. Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am. Nat. 2002, 160, S99. [Google Scholar] [CrossRef] [PubMed]
- Aiken, G.E.; Klotz, J.L.; Johnson, J.M.; Strickland, J.R.; Schrick, F.N. Postgraze assessment of toxicosis symptoms for steers grazed on toxic endophyte-infected tall fescue pasture. J. Anim. Sci. 2013, 91, 5878–5884. [Google Scholar] [CrossRef]
- Thompson, F.N.; Stuedemann, J.A.; Hill, N.S. Anti-quality factors associated with alkaloids in eastern temperate pasture. J. Range Manag. 2001, 54, 474. [Google Scholar] [CrossRef]
- Klotz, J.L. Activities and effects of ergot alkaloids on livestock physiology and production. Toxins 2015, 7, 2801–2821. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Tapper, B.A.; Simpson, W.R.; Johnson, R.D.; Mace, W.J.; Ram, A.; Lukito, Y.; Dupont, P.-Y.; Johnson, L.J.; Scott, B.; et al. Epichloë hybrida, sp. nov., an emerging model system for investigating fungal allopolyploidy. Mycology 2017, 109, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anonymous. New ryegrass withdrawn after side-effects found. Manawatu Evening Standard, 16 October 1992; p. 1. [Google Scholar]
- Schardl, C.L.; Panaccione, D.G.; Tudzynski, P. Chapter 2 Ergot Alkaloids—Biology and Molecular Biology. In The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 63, pp. 45–86. [Google Scholar]
- Tsai, H.F.; Wang, H.; Gebler, J.C.; Poulter, C.D.; Schardl, C.L. The claviceps purpurea gene encoding dimethylallyltryptophan synthase, the committed step for ergot alkaloid biosynthesis. Biochem. Biophys. Res. Commun. 1995, 216, 119–125. [Google Scholar] [CrossRef]
- Wang, J.; Machado, C.; Panaccione, D.G.; Tsai, H.-F.; Schardl, C.L. The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet. Biol. 2004, 41, 189–198. [Google Scholar] [CrossRef]
- Davis, N.D.; Clark, E.M.; Schrey, K.A.; Diener, U.L. In Vitro Growth of Acremonium coenophialum, an Endophyte of Toxic Tall Fescue Grass. Appl. Environ. Microbiol. 1986, 52, 888–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.F.; Liu, J.S.; Staben, C.; Christensen, M.J.; Latch, G.C.; Siegel, M.R.; Schardl, C.L. Evolutionary diversification of fungal endophytes of tall fescue grass by hybridization with Epichloe species. Proc. Natl. Acad. Sci. USA 1994, 91, 2542–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, S.; Andreeva, K.; Machado, C.; Mirabito, P.M.; Schardl, C.L. Elimination of marker genes from transformed filamentous fungi by unselected transient transfection with a CRE-expressing plasmid. Fungal Genet. Biol. 2009, 46, 721–730. [Google Scholar] [CrossRef]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-Symbiotic Fungi as Chemical Engineers: Multi-Genome Analysis of the Clavicipitaceae Reveals Dynamics of Alkaloid Loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [Green Version]
- Florea, S.; Phillips, T.D.; Panaccione, D.G.; Farman, M.L.; Schardl, C.L. Chromosome-End Knockoff Strategy to Reshape Alkaloid Profiles of a Fungal Endophyte. G3 (Bethesda) 2016, 6, 2601–2610. [Google Scholar] [CrossRef] [Green Version]
- Florea, S.; Panaccione, D.G.; Schardl, C.L. Ergot Alkaloids of the Family Clavicipitaceae. Phytopathology 2017, 107, 504–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiering, M.J.; Moon, C.D.; Wilkinson, H.H.; Schardl, C.L. Gene Clusters for Insecticidal Loline Alkaloids in the Grass-Endophytic Fungus Neotyphodium uncinatum. Genetics 2005, 169, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Hou, Y.; Zhang, P.; Zhang, Z.; Xu, Y.; Zhang, L.; Niu, L.; Yang, Y.; Liang, D.; Yi, F.; et al. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 2017, 7, 40638. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Feng, Y.-L.; Xiao, J.-J.; Liu, Q.; Sun, X.-N.; Xiang, J.-F.; Kong, N.; Liu, S.-C.; Chen, G.-Q.; Wang, Y.; et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018, 19, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lemos, B.R.; Kaplan, A.C.; Bae, J.E.; Ferrazzoli, A.E.; Kuo, J.; Anand, R.P.; Waterman, D.P.; Haber, J.E. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. USA 2018, 115, E2040–E2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, K.A.; Sampson, J.K.; Panaccione, D.G. Genetic reprogramming of the ergot alkaloid pathway of metarhizium brunneum. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Khan, H.; McDonald, M.C.; Williams, S.J.; Solomon, P.S. Assessing the efficacy of CRISPR/Cas9 genome editing in the wheat pathogen Parastagonspora nodorum. Fungal Biol. Biotechnol. 2020, 7, 4–8. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Siegel, M.R.; Schardl, C.L. Transformation of Acremonium coenophialum, a protective fungal symbiont of the grass Festuca arundinacea. Curr. Genet. 1992, 22, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Leuchtmann, A.; Rowan, D.D.; Tapper, B. Taxonomy of Acremonium endophytes of tall fescue (Festuca arundinacea), meadow fescue (F. pratensis) and perennial ryegrass (Lolium perenne). Mycol. Res. 1993, 97, 1083–1092. [Google Scholar] [CrossRef]
- Florea, S.; Schardl, C.L.; Hollin, W. Detection and isolation of epichloë species, fungal endophytes of grasses. Curr. Protoc. Microbiol. 2015, 38, 19A.1.1–19A.1.24. [Google Scholar] [CrossRef] [PubMed]
- Al-Samarrai, T.H.; Schmid, J. A simple method for extraction of fungal genomic DNA. Lett. Appl. Microbiol. 2000, 30, 53–56. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.-C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R.A. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Panaccione, D.G.; Johnson, R.D.; Wang, J.; Young, C.A.; Damrongkool, P.; Scott, B.; Schardl, C.L. Elimination of ergovaline from a grass-Neotyphodium endophyte symbiosis by genetic modification of the endophyte. Proc. Natl. Acad. Sci. USA 2001, 98, 12820–12825. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, L.; Sutherland, B. Sheep responses to grazing ryegrass with AR37 endophyte. Proc. N. Z. Grassl. Assoc. 2009, 71, 127–132. [Google Scholar] [CrossRef]
- Latchs, G.C.M.; Christensen, M.J. Artificial infection of grasses with endophytes. Ann. Appl. Biol. 1985, 107, 17–24. [Google Scholar] [CrossRef]
- Chung, K.-R.; Hollin, W.; Siegel, M.R.; Schardl, C.L. Genetics of Host Specificity in Epichloë typhina. Phytopathology 1997, 87, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.Q.; Siegel, M.R.; Hollin, W.; Tsai, H.F.; Schmidt, D.; Schardl, C.L. Relationships among non-Acremonium sp. fungal endophytes in five grass species. Appl. Environ. Microbiol. 1993, 59, 1540–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scheme 1. | Target Site | PAM | Modified sgRNA Sequence 1 |
|---|---|---|---|
| dmaW24KOf | dmaW2 3′-flank | AGG | A*G*C*UAACCCUAUGAAGUGUG |
| dmaW28KOr | dmaW2 5′-flank | AGG | G*U*G*GGUGUGAAGUGAACCAG |
| EAS2lpsAguide | lpsA 3′-region | AGG | G*U*A*UGGCGGGAGUUACUGCA |
| EAS2lpsBguide | lpsB 3′-flank | AGG | U*C*G*CGCUAGGUAAUGUGGCA |
| lolCf4 | lolC 5′-flank | AGG | A*C*A*CGGAGUCGAAGAAAGAA |
| lolCr1 | lolC 3′-flank | AGG | C*G*U*AAAUAGGGGUCAUUGCG |
| Primer Name | Target Gene(s) or Sites | Sequence |
|---|---|---|
| dmaW1&2f | dmaW1, dmaW2 | GCAAAGACACTCCACCAGGAAGTT |
| dmaW1&2r | dmaW1, dmaW2 | AGTTGCGGCGTTAATAGGCTCGTA |
| hph.4d | hph | GACCTGATGCAGCTCTCGGA |
| hph.3u | hph | TCGGCGAGTACTTCTACACA |
| RTeasA1f | easA1 | ACAACTTTGGGCGACTGGG |
| RteasA1r | easA1 | CCGTTGGTTGCAAGAAGATTGA |
| RteasA2 | easA2 | GCAGCTTTGGGCGACTGGA |
| RteasA2r | easA2 | ACGTTGGTTGCAAGGAGATTGG |
| lolC-3a | lolC | GGTCTAGTATTACGTTGCCAGGG |
| lolC-5b | lolC | TCTAAACTTGACGCAGTTCGGC |
| oligoscreen(f) | Oligotag | GATGGCCTTTAAAGTCTACGTACTC |
| lpsAoligoR | lpsA1, lpsA2 | ATATCATGGCAACATTCAGCGCAC |
| Mutant | Parental Strain | Genotype |
|---|---|---|
| e7799 | e7480 | EAS1-knockoff ∆dmaW2 |
| e7800 | e7480 | EAS1-knockoff ∆dmaW2 |
| e7801 | e19 | ∆EAS1 ∆EAS2 |
| e7802 | e19 | EAS1-knockoff ∆EAS2 |
| e7803 | e19 | ∆EAS2 |
| e7804 | e19 | ∆lolC |
| e7805 | e19 | ∆lolC |
| e7806 | Lp1 | ∆EAS |
| Genotype Inoculated | Parental Strain | Plant Species | Positive/Total | Infection Rate % |
|---|---|---|---|---|
| ∆EAS1 ∆EAS2 | Epichloë coenophiala e19 | Lolium arundinaceum | 65/143 | 45 |
| ∆EAS2 | E. coenophiala e19 | L. arundinaceum | 22/48 | 46 |
| ∆EAS | Epichloë hybrida Lp1 | Lolium perenne | 32/46 | 70 |
| ∆dmaW2 | E. coenophiala e7480 | L. arundinaceum | 125/251 | 50 |
| ∆lolC | E. coenophiala e19 | L. arundinaceum | 25/81 | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florea, S.; Jaromczyk, J.; Schardl, C.L. Non-Transgenic CRISPR-Mediated Knockout of Entire Ergot Alkaloid Gene Clusters in Slow-Growing Asexual Polyploid Fungi. Toxins 2021, 13, 153. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020153
Florea S, Jaromczyk J, Schardl CL. Non-Transgenic CRISPR-Mediated Knockout of Entire Ergot Alkaloid Gene Clusters in Slow-Growing Asexual Polyploid Fungi. Toxins. 2021; 13(2):153. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020153
Chicago/Turabian StyleFlorea, Simona, Jolanta Jaromczyk, and Christopher L. Schardl. 2021. "Non-Transgenic CRISPR-Mediated Knockout of Entire Ergot Alkaloid Gene Clusters in Slow-Growing Asexual Polyploid Fungi" Toxins 13, no. 2: 153. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020153