Transcriptome Analysis of Caco-2 Cells upon the Exposure of Mycotoxin Deoxynivalenol and Its Acetylated Derivatives


Abstract
:1. Introduction
2. Results
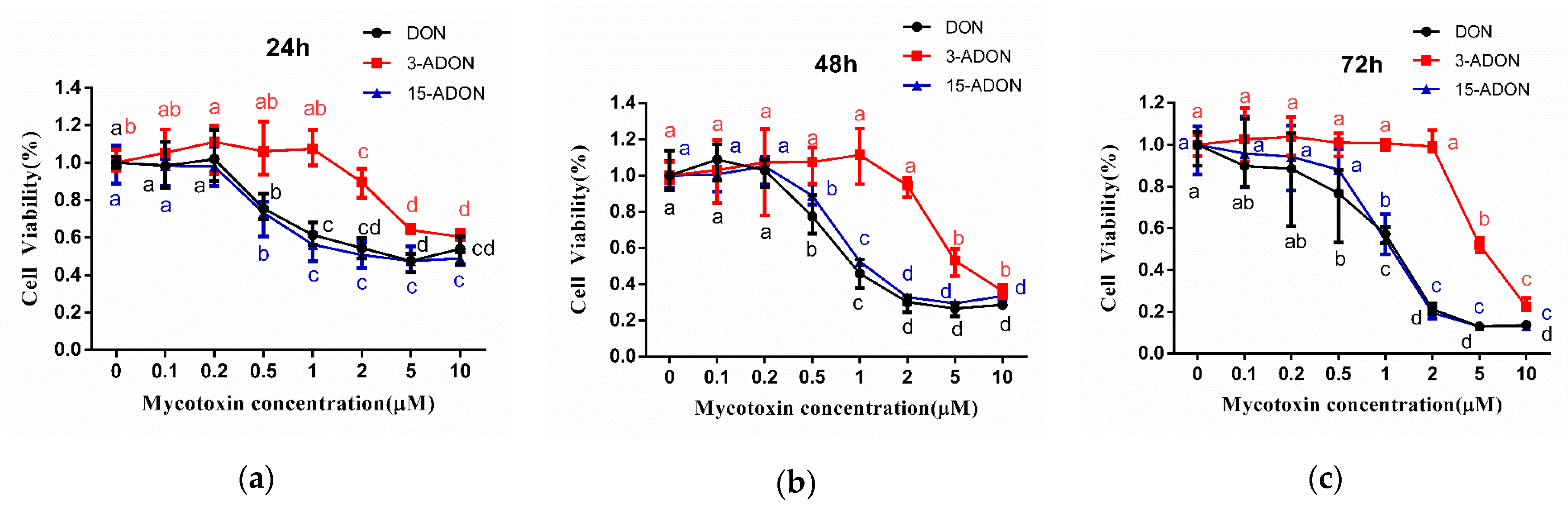
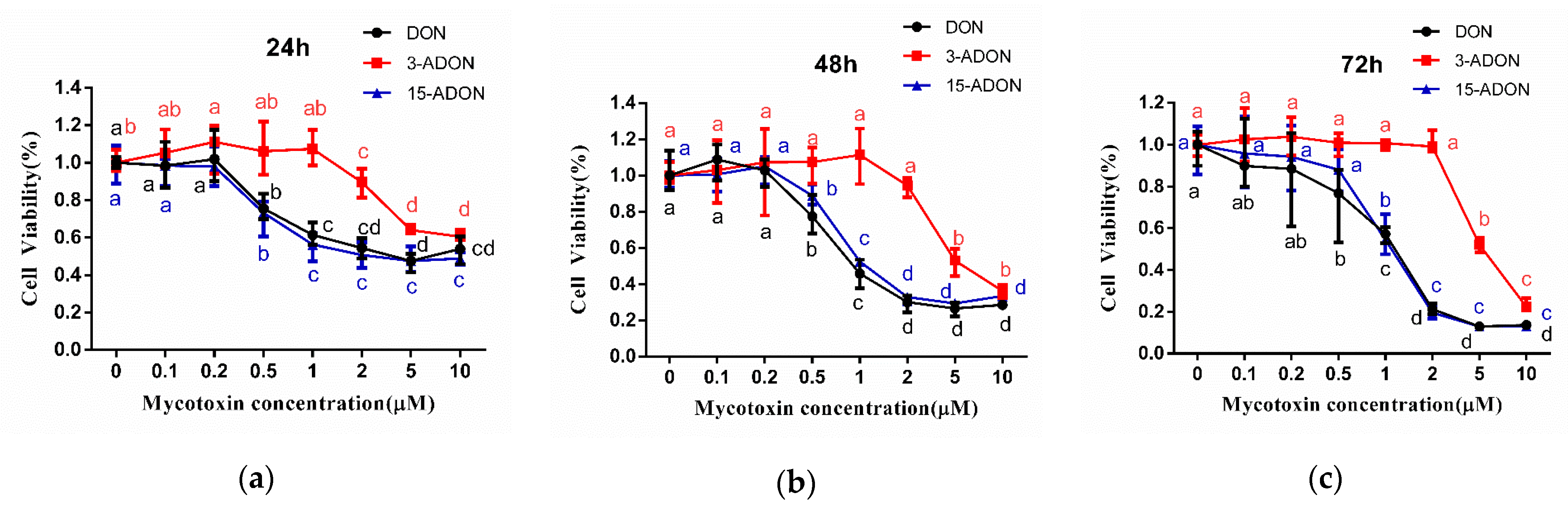
2.1. 3-ADON Exerts Less Harmful Effects Than DON and 15-ADON on Cultured Caco-2 Cells
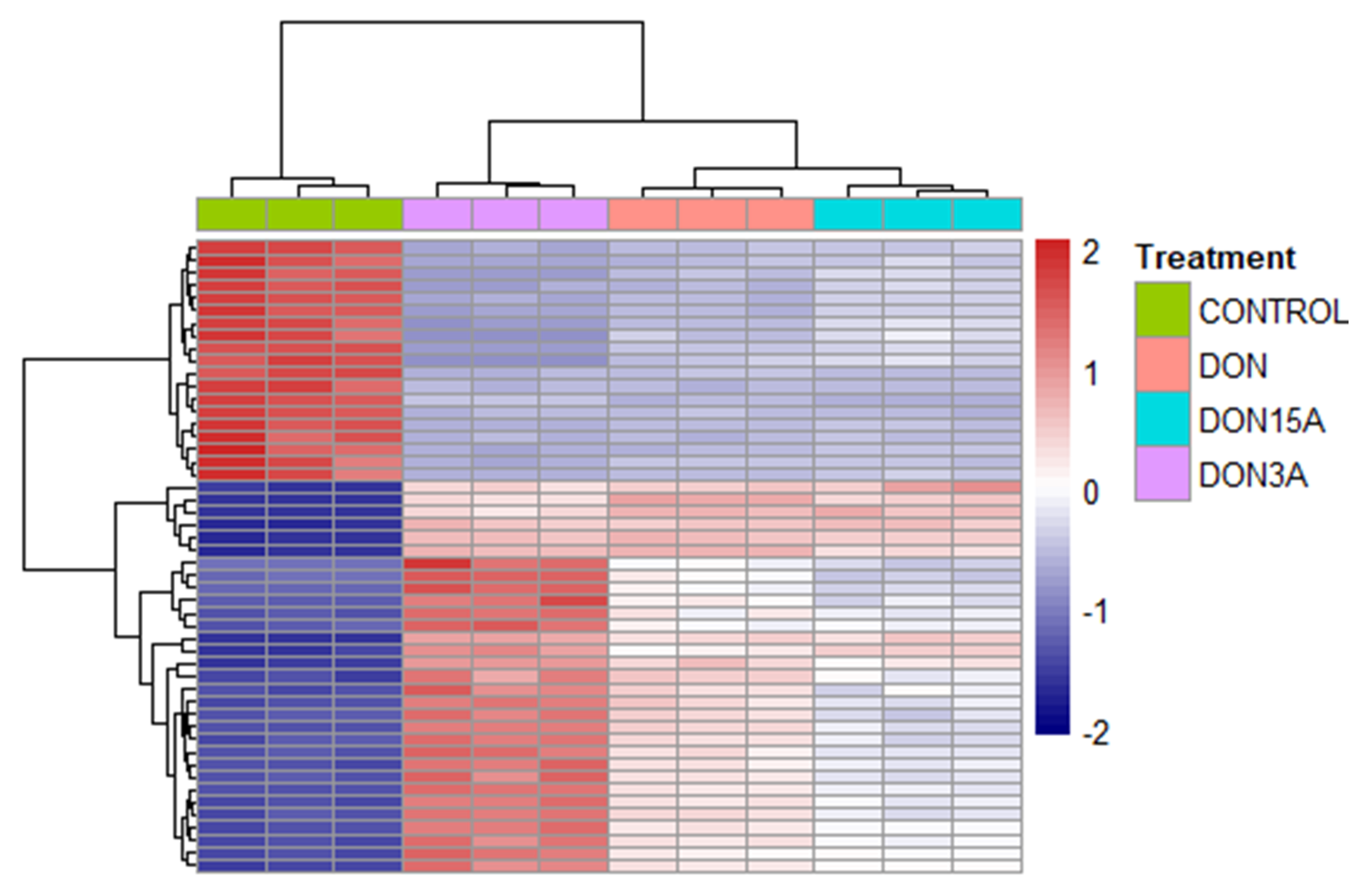
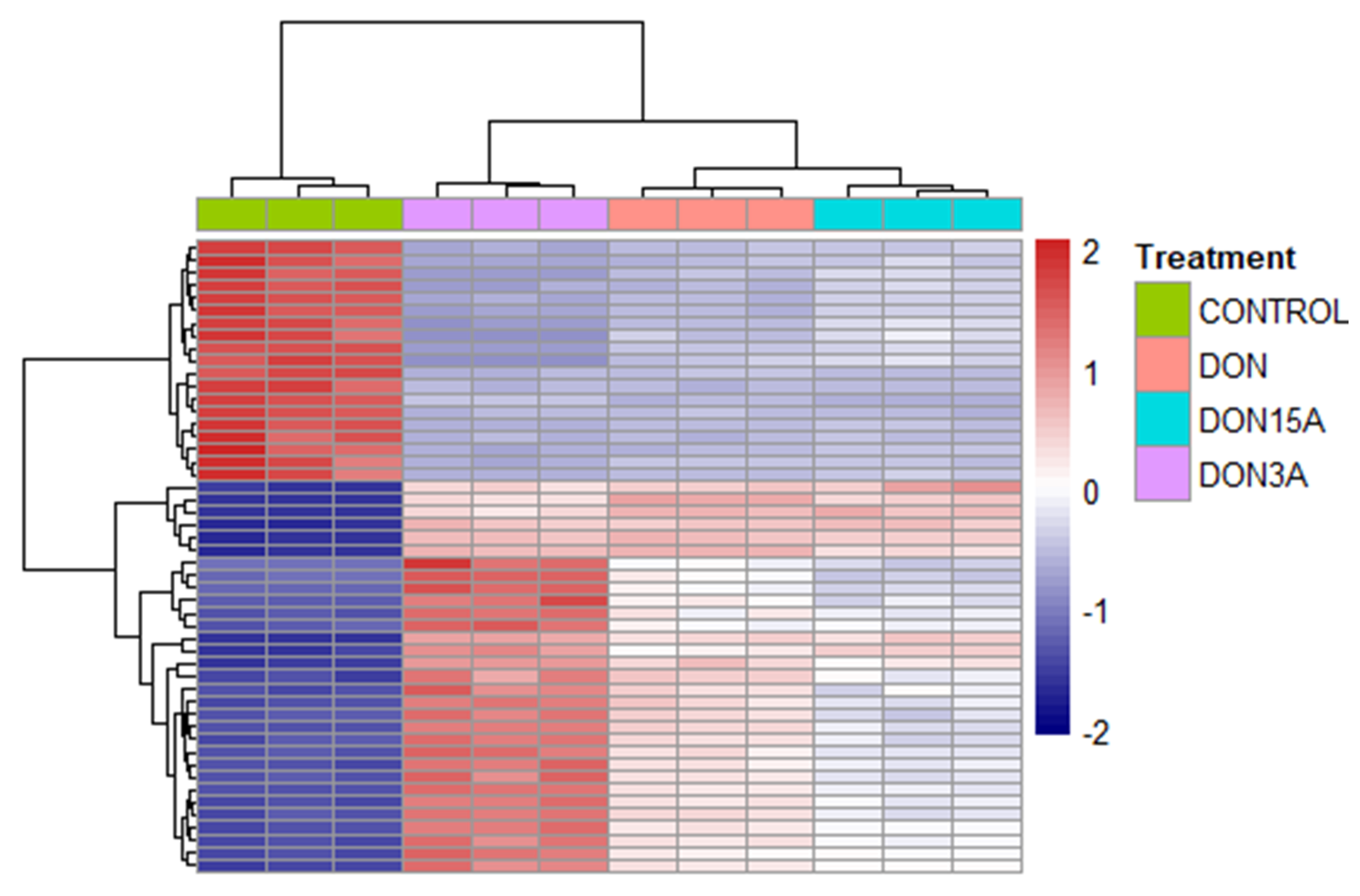
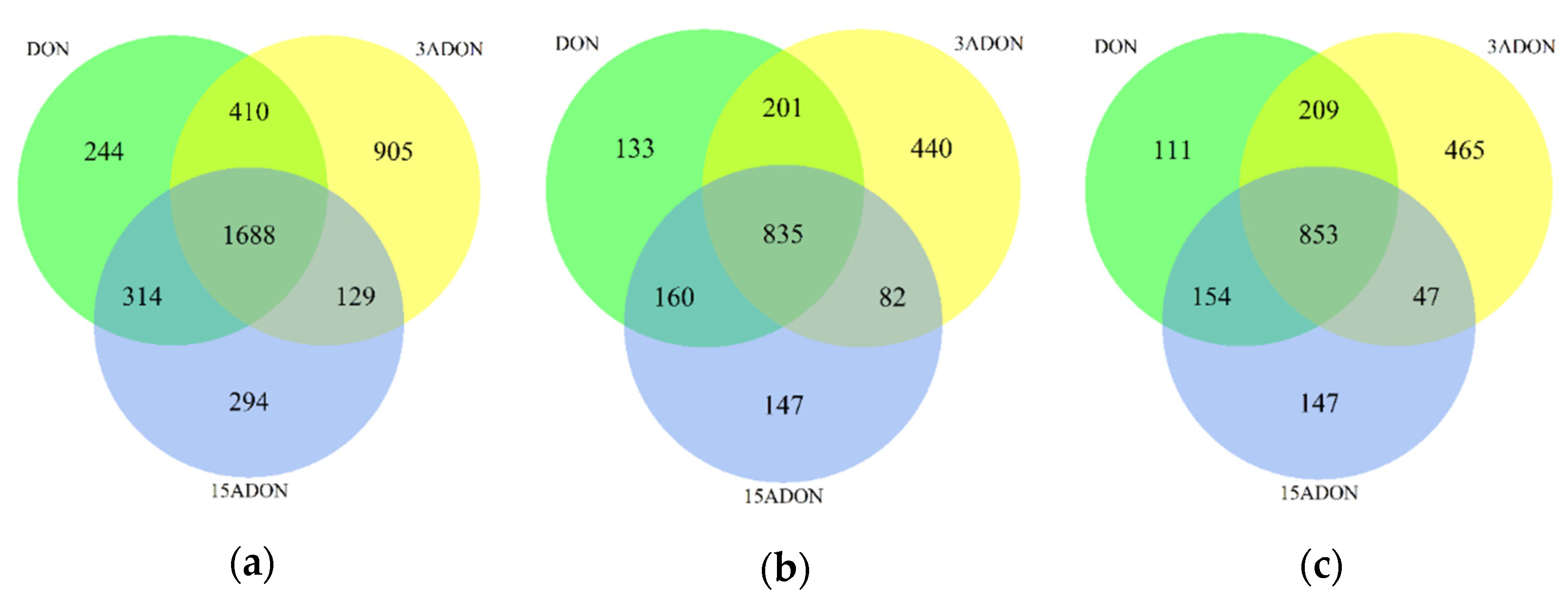
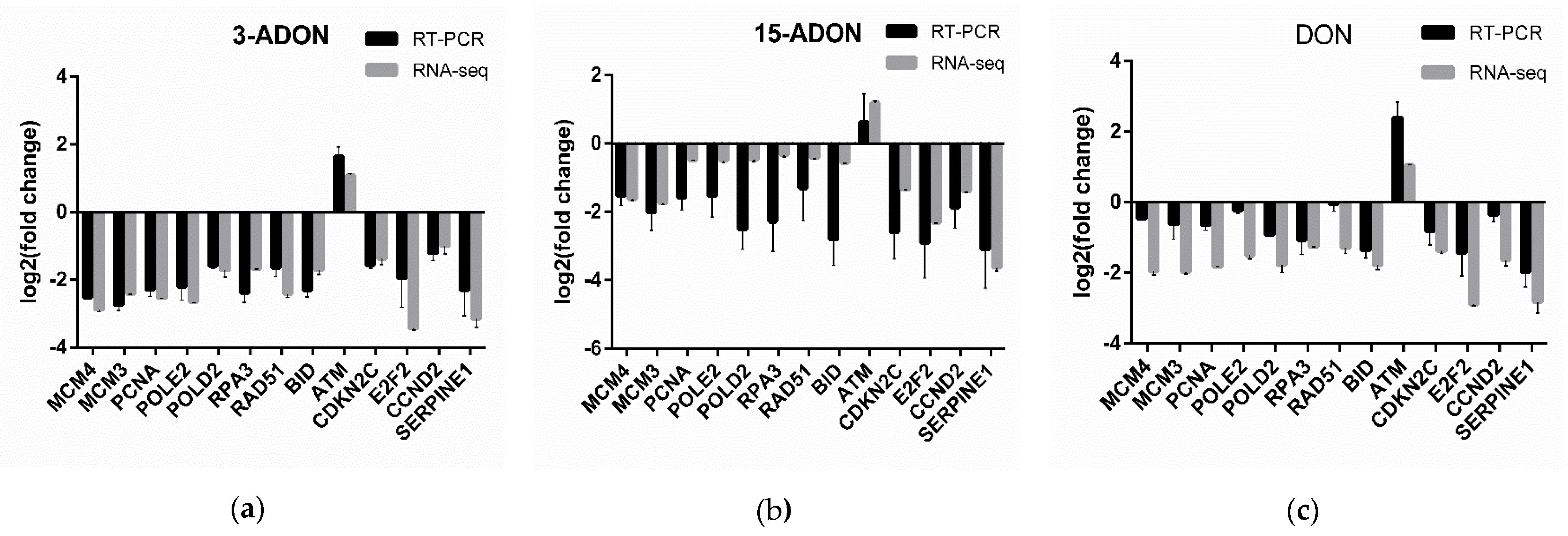
2.2. Transcriptomic Changes of Caco-2 Cells
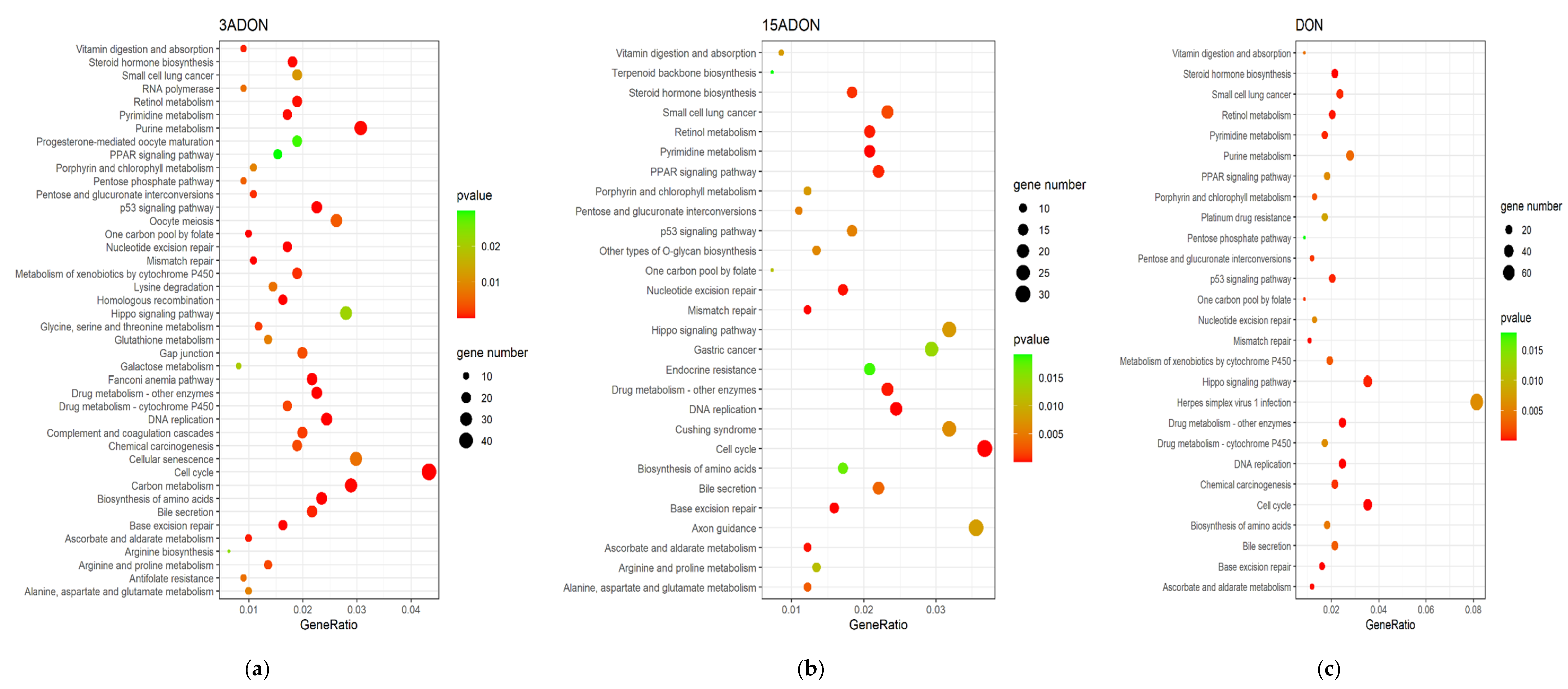
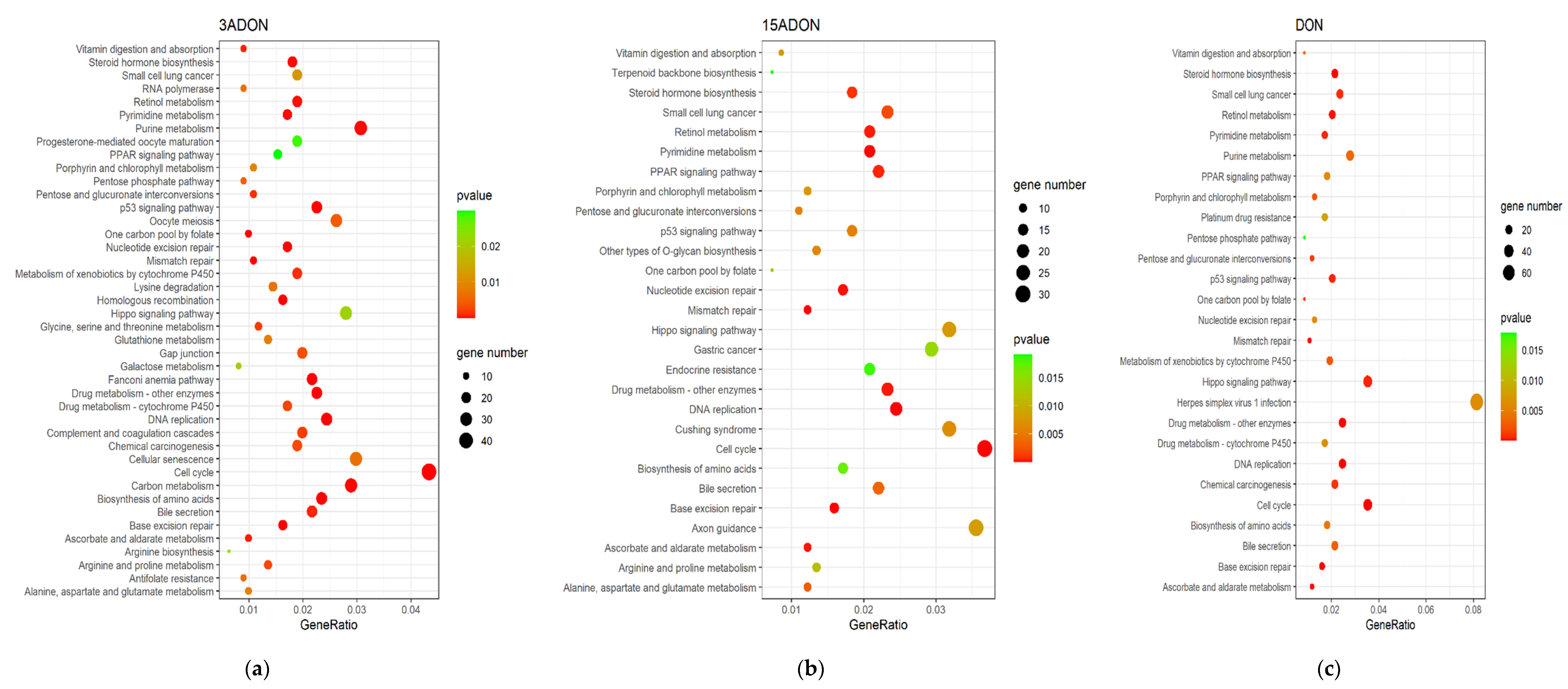
2.3. Function Enrichment Analysis of Differentially Expressed Genes (DEGs)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Cell Viability Assay
4.4. RNA Extraction
4.5. RNA-seq Analysis
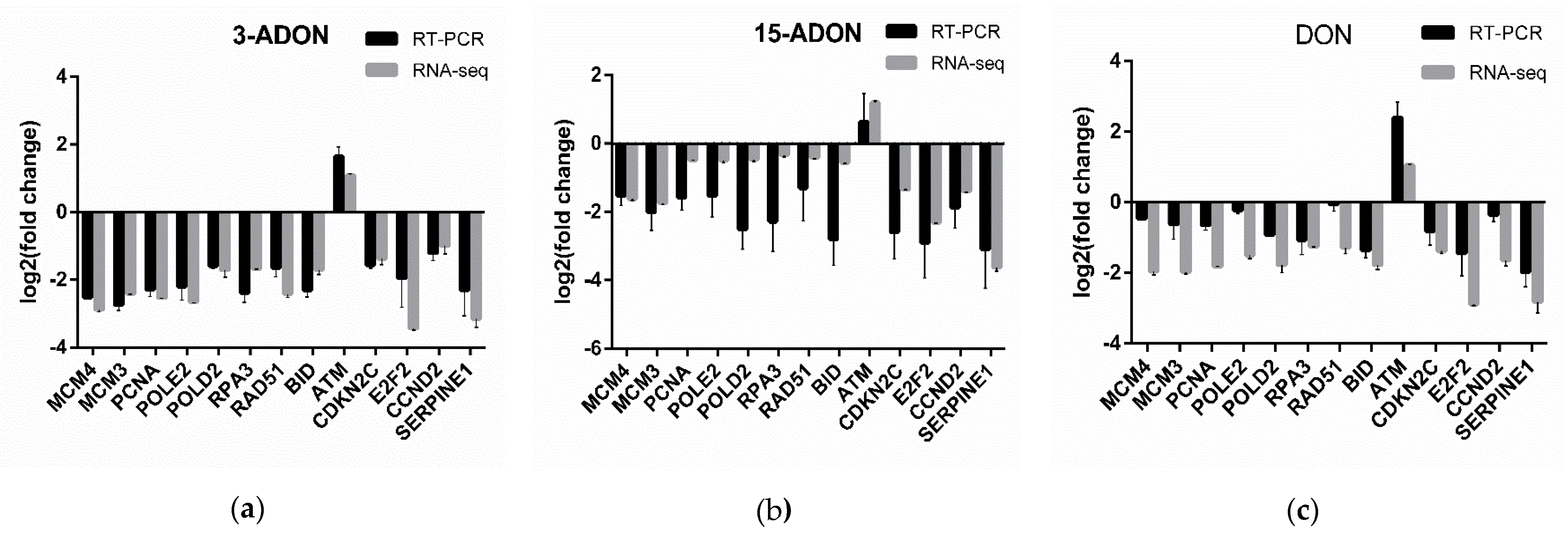
4.6. Real-Time PCR (RT-PCR) Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khaneghah, A.M.; Martins, L.M.; von Hertwig, A.M.; Bertoldo, R.; Sant’Ana, A.S. Deoxynivalenol and its masked forms: Characteristics, incidence, control and fate during wheat and wheat based products processing—A review. Trends Food Sci. Technol. 2018, 71, 13–24. [Google Scholar] [CrossRef]
- Freire, L.; Sant’Ana, A.S. Modified mycotoxins: An updated review on their formation, detection, occurrence, and toxic effects. Food Chem. Toxicol. 2018, 111, 189–205. [Google Scholar] [CrossRef]
- Ji, J.; Wang, Q.; Wu, H.; Xia, S.; Guo, H.; Blaženović, I.; Zhang, Y.; Sun, X. Insights into cellular metabolic pathways of the combined toxicity responses of Caco-2 cells exposed to deoxynivalenol, zearalenone and Aflatoxin B 1. Food Chem. Toxicol. 2019, 126, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Alassane-Kpembi, I.; Gerez, J.R.; Cossalter, A.M.; Neves, M.; Laffitte, J.; Naylies, C.; Lippi, Y.; Kolf-Clauw, M.; Bracarense, A.P.L.; Pinton, P.; et al. Intestinal toxicity of the type B trichothecene mycotoxin fusarenon-X: Whole transcriptome profiling reveals new signaling pathways. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Panel, E.; Chain, F. Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA J. 2014, 12, 3916. [Google Scholar] [CrossRef] [Green Version]
- Mousavi Khaneghah, A.; Fakhri, Y.; Raeisi, S.; Armoon, B.; Sant’Ana, A.S. Prevalence and concentration of ochratoxin A, zearalenone, deoxynivalenol and total aflatoxin in cereal-based products: A systematic review and meta-analysis. Food Chem. Toxicol. 2018, 118, 830–848. [Google Scholar] [CrossRef]
- Alizadeh, A.; Braber, S.; Akbari, P.; Kraneveld, A.; Garssen, J.; Fink-Gremmels, J. Deoxynivalenol and its modified forms: Are there major differences? Toxins 2016, 8, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, F.; Xu, J.; Liu, X.; Yin, X.; Shi, J. Natural occurrence of deoxynivalenol and zearalenone in wheat from Jiangsu province, China. Food Chem. 2014, 157, 393–397. [Google Scholar] [CrossRef]
- Singh, S.; Banerjee, S.; Chattopadhyay, P.; Borthakur, S.K.; Veer, V. Deoxynivalenol induces cytotoxicity and genotoxicity in animal primary cell culture. Toxicol. Mech. Methods 2015, 25, 184–191. [Google Scholar] [CrossRef]
- Wang, H.; Zong, Q.; Wang, S.; Zhao, C.; Wu, S.; Bao, W. Genome-Wide DNA Methylome and Transcriptome Analysis of Porcine Intestinal Epithelial Cells upon Deoxynivalenol Exposure. J. Agric. Food Chem. 2019, 67, 6423–6431. [Google Scholar] [CrossRef] [PubMed]
- Juan-García, A.; Juan, C.; Tolosa, J.; Ruiz, M.J. Effects of deoxynivalenol, 3-acetyl-deoxynivalenol and 15-acetyl-deoxynivalenol on parameters associated with oxidative stress in HepG2 cells. Mycotoxin Res. 2019, 35, 197–205. [Google Scholar] [CrossRef]
- World Health Organization. Evaluation of certain contaminants in food. In Proceedings of the World Health Organization Technical Report Series; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Juan-García, A.; Taroncher, M.; Font, G.; Ruiz, M.J. Micronucleus induction and cell cycle alterations produced by deoxynivalenol and its acetylated derivatives in individual and combined exposure on HepG2 cells. Food Chem. Toxicol. 2018, 118, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Fruhmann, P.; Wiesenberger, G.; Kluger, B.; Sarkanj, B.; Lemmens, M.; Hametner, C.; Fröhlich, J.; Adam, G.; Krska, R.; et al. Deoxynivalenol-sulfates: Identification and quantification of novel conjugated (masked) mycotoxins in wheat. Anal. Bioanal. Chem. 2015, 407, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Stadler, D.; Lambertini, F.; Woelflingseder, L.; Schwartz-Zimmermann, H.; Marko, D.; Suman, M.; Berthiller, F.; Krska, R. The influence of processing parameters on the mitigation of deoxynivalenol during industrial baking. Toxins 2019, 11, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šarkanj, B.; Warth, B.; Uhlig, S.; Abia, W.A.; Sulyok, M.; Klapec, T.; Krska, R.; Banjari, I. Urinary analysis reveals high deoxynivalenol exposure in pregnant women from Croatia. Food Chem. Toxicol. 2013, 62, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Alassane-Kpembi, I.; Puel, O.; Oswald, I.P. Toxicological interactions between the mycotoxins deoxynivalenol, nivalenol and their acetylated derivatives in intestinal epithelial cells. Arch. Toxicol. 2015, 89, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, N.; Yang, L.; Deng, Y.; Wang, J.; Song, S.; Lin, S.; Wu, A.; Zhou, Z.; Hou, J. Multi-mycotoxin analysis of animal feed and animal-derived food using LC–MS/MS system with timed and highly selective reaction monitoring. Anal. Bioanal. Chem. 2015, 407. [Google Scholar] [CrossRef] [PubMed]
- Palacios, S.A.; Erazo, J.G.; Ciasca, B.; Lattanzio, V.M.T.; Reynoso, M.M.; Farnochi, M.C.; Torres, A.M. Occurrence of deoxynivalenol and deoxynivalenol-3-glucoside in durum wheat from Argentina. Food Chem. 2017, 230, 728–734. [Google Scholar] [CrossRef] [PubMed]
- De Boevre, M.; Di Mavungu, J.D.; Maene, P.; Audenaert, K. Chemistry, Analysis, Control, Exposure & Risk Assessment Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolite in different cereals and cereal-. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar]
- Mastanjević, K.; Šarkanj, B.; Krska, R.; Sulyok, M.; Warth, B.; Mastanjević, K.; Šantek, B.; Krstanović, V. From malt to wheat beer: A comprehensive multi-toxin screening, transfer assessment and its influence on basic fermentation parameters. Food Chem. 2018, 254, 115–121. [Google Scholar] [CrossRef]
- Ojuri, O.T.; Ezekiel, C.N.; Eskola, M.K.; Šarkanj, B.; Babalola, A.D.; Sulyok, M.; Hajšlová, J.; Elliott, C.T.; Krska, R. Mycotoxin co-exposures in infants and young children consuming household- and industrially-processed complementary foods in Nigeria and risk management advice. Food Control 2019, 98, 312–322. [Google Scholar] [CrossRef]
- Ajandouz, E.H.; Berdah, S.; Moutardier, V.; Bege, T.; Birnbaum, D.J.; Perrier, J.; Di Pasquale, E.; Maresca, M. Hydrolytic fate of 3/15-acetyldeoxynivalenol in humans: Specific deacetylation by the small intestine and liver revealed using in vitro and ex vivo approaches. Toxins 2016, 8, 232. [Google Scholar] [CrossRef]
- Payros, D.; Alassane-Kpembi, I.; Pierron, A.; Loiseau, N.; Pinton, P.; Oswald, I.P. Toxicology of deoxynivalenol and its acetylated and modified forms. Arch. Toxicol. 2016, 90, 2931–2957. [Google Scholar] [CrossRef]
- Van De Walle, J.; During, A.; Piront, N.; Toussaint, O.; Schneider, Y.J.; Larondelle, Y. Physio-pathological parameters affect the activation of inflammatory pathways by deoxynivalenol in Caco-2 cells. Toxicol. Vitr. 2010, 24, 1890–1898. [Google Scholar] [CrossRef]
- Di, R.; Zhang, H.; Lawton, M.A. Transcriptome analysis of C. Elegans reveals novel targets for DON cytotoxicity. Toxins 2018, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springler, A.; Hessenberger, S.; Reisinger, N.; Kern, C.; Nagl, V.; Schatzmayr, G.; Mayer, E. Deoxynivalenol and its metabolite deepoxy-deoxynivalenol: Multi-parameter analysis for the evaluation of cytotoxicity and cellular effects. Mycotoxin Res. 2017, 33, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alassane-Kpembi, I.; Pinton, P.; Hupé, J.F.; Neves, M.; Lippi, Y.; Combes, S.; Castex, M.; Oswald, I.P. Saccharomyces cerevisiae boulardii reduces the deoxynivalenol-induced alteration of the intestinal transcriptome. Toxins 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, J.; Li, X.; Xu, C.; Wang, X.; Guo, X. Expression Profiles of Selenium-Related Genes in Human Chondrocytes Exposed to T-2 Toxin and Deoxynivalenol. Biol. Trace Elem. Res. 2019, 190, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Katika, M.R.; Hendriksen, P.J.M.; Shao, J.; van Loveren, H.; Peijnenburg, A. Transcriptome analysis of the human T lymphocyte cell line Jurkat and human peripheral blood mononuclear cells exposed to deoxynivalenol (DON): New mechanistic insights. Toxicol. Appl. Pharmacol. 2012, 264, 51–64. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, L.; Geng, C.; Cao, J.; Zhong, L. The role of oxidative stress in deoxynivalenol-induced DNA damage in HepG2 cells. Toxicon 2009, 54, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yang, Y.; Chen, J.; Jia, H.; Zhang, Y.; Jiang, D.; Wu, G.; Wu, Z. 3-Acetyldeoxynivalenol induces lysosomal membrane permeabilization-mediated apoptosis and inhibits autophagic flux in macrophages. Environ. Pollut. 2020, 265, 114697. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B.; Hogstrand, C.; et al. Risks to human and animal health related to the presence of deoxynivalenol and its acetylated and modified forms in food and feed. EFSA J. 2017, 15, e04718. [Google Scholar] [CrossRef]
- Desjardins, A.E.; McCormick, S.P.; Appell, M. Structure-activity relationships of trichothecene toxins in an Arabidopsis thaliana leaf assay. J. Agric. Food Chem. 2007, 55, 6487–6492. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Wannemacher, R.W. Structure—Function relationships of 12,13-epoxytrichothecene mycotoxins in cell culture: Comparison to whole animal lethality. Toxicon 1986, 24, 985–994. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Mun Tho, L.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 108. [Google Scholar]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM–CHK2 and ATR–CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Lygerou, Z. Control of DNA replication licensing in a cell cycle. Genes Cells 2002, 7, 523–534. [Google Scholar] [CrossRef]
- Nevins, J.R.; Leone, G.; DeGregori, J.; Jakoi, L. Role of the Rb/E2F pathway in cell growth control. J. Cell. Physiol. 1997, 173, 233–236. [Google Scholar] [CrossRef]
- Elbæk, C.R.; Petrosius, V.; Sørensen, C.S. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2020, 819–820, 111694. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ye, Y.; Lin, S.; Deng, L.; Fan, X.; Zhang, Y.; Deng, X.; Li, Y.; Yan, H.; Ma, Y. Evaluation of deoxynivalenol-induced toxic effects on DF-1 cells in vitro: Cell-cycle arrest, oxidative stress, and apoptosis. Environ. Toxicol. Pharmacol. 2014, 37, 141–149. [Google Scholar] [CrossRef]
- Danuta, P.; Sliwinska, E. Trichothecene fusarial toxins perturb the cell cycle in meristem-atic cells of Secale cereale L., Triticum aestivum L. and Vicia faba L. Caryologia 2005, 58, 86–93. [Google Scholar] [CrossRef]
- Payros, D.; Dobrindt, U.; Martin, P.; Secher, T.; Bracarense, A.P.F.L.; Boury, M.; Laffitte, J.; Pinton, P.; Oswald, E.; Oswald, I.P. The food contaminant deoxynivalenol exacerbates the genotoxicity of gut microbiota. MBio 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Lim, W.; Park, S.; Kim, J.; You, S.; Song, G. Deoxynivalenol induces apoptosis and disrupts cellular homeostasis through MAPK signaling pathways in bovine mammary epithelial cells. Environ. Pollut. 2019, 252, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef]
- Rafia, S.; Saran, S. Sestrin-like protein from Dictyostelium discoideum is involved in autophagy under starvation stress. Microbiol. Res. 2019, 220, 61–71. [Google Scholar] [CrossRef]
- Yadav, A.; Sharma, V.; Pal, J.; Gulati, P.; Goel, M.; Chandra, U.; Bansal, N.; Saha, S. DNA replication protein Cdc45 directly interacts with PCNA via its PIP box in Leishmania donovani and the Cdc45 PIP box is essential for cell survival. PLoS Pathog. 2020, 16, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tye, B.K. MCM proteins in DNA replication. Annu. Rev. Biochem. 1999, 68, 649–686. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.; Chou, D.M.; You, Z.; Hunter, T.; Walter, J.C.; Walter, G. Protein Phosphatase 2A Antagonizes ATM and ATR in a Cdk2- and Cdc7-Independent DNA Damage Checkpoint. Mol. Cell. Biol. 2006, 26, 1997–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celis, J.E.; Madsen, P.; Celis, A.; Nielsen, H.V.; Gesser, B. Cyclin (PCNA, auxiliary protein of DNA polymerase δ) is a central component of the pathway(s) leading to DNA replication and cell division. FEBS Lett. 1987, 220, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sameer, A.S.; Nissar, S.; Fatima, K. Mismatch repair pathway: Molecules, functions, and role in colorectal carcinogenesis. Eur. J. Cancer Prev. 2014, 23, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, Z.; Chatzinikolaou, G.; Stratigi, K.; Garinis, G.A. Nucleotide Excision Repair and Transcription-Associated Genome Instability. BioEssays 2019, 41, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Gassman, N.R. Transcriptional dysregulation of base excision repair proteins in breast cancer. DNA Repair (Amst) 2020, 93, 102922. [Google Scholar] [CrossRef]
- Ceylan, D.; Tuna, G.; Kirkali, G.; Tunca, Z.; Can, G.; Arat, H.E.; Kant, M.; Dizdaroglu, M.; Özerdem, A. Oxidatively-induced DNA damage and base excision repair in euthymic patients with bipolar disorder. DNA Repair (Amst) 2018, 65, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-replicative repair of oxidized bases maintains fidelity in mammalian genomes: The cowcatcher role of NEIL1 DNA glycosylase. Genes 2017, 8, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemann, U.; Viergutz, T.; Jonas, L.; Schneider, F. Influence of the mycotoxins α- and β-zearalenol and deoxynivalenol on the cell cycle of cultured porcine endometrial cells. Reprod. Toxicol. 2003, 17, 209–218. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure Time (h) | IC50 (µM) ± SD | ||
|---|---|---|---|
| DON | 3-ADON | 15-ADON | |
| 24 | 6.17 ± 0.93 | 13.19 ± 0.71 | 3.86 ± 0.31 |
| 48 | 3.86 ± 2.21 | 10.81 ± 3.84 | 2.33 ± 0.36 |
| 72 | 1.46 ± 0.42 | 7.84 ± 2.01 | 1.44 ± 0.67 |
| Toxin | Top 5 Pathways | Downregulated Genes | Upregulated Genes | p-Value | Count |
|---|---|---|---|---|---|
| 3-ADON | DNA replication | PCNA, MCM4, MCM3, MCM5, MCM6, MCM7, MCM2, POLD1, RFC3, FEN1, POLE2, RFC4, RPA3, RNASEH2A, POLE3, LIG1, POLD3, PRIM1, POLA2, POLD2, RPA1, RFC5, POLE, RNASEH1, RFC2, POLA1, RNASEH2C | − | 1.06 × 10−16 | 27 |
| Cell cycle | PCNA, MCM4, E2F1, MCM3, MCM5, CDC6, MCM6, MAD2L1, MCM7, MCM2, CDK1, CCNB1, PTTG1, E2F2, ORC6, CHEK2, CCNA2, CCND1, YWHAB, CDC45, PKMYT1, E2F3, ORC1, CDC25A, CCNE1, CDK2, CDKN2C, ESPL1, TFDP1, CHEK1, BUB1, DBF4, PLK1, RBX1, BUB1B, CDKN2A, CDC25C, MAD2L2, CDC7, CCNE2, SMC1B, CCND3, CDKN2D, CCND2 | WEE2, STAG1, STAG2, ATM | 2.74 × 10−12 | 48 | |
| Base excision repair | PCNA, PARP1, POLD1, FEN1, POLE2, POLE3, UNG, LIG1, POLD3, NEIL3, NTHL1, POLE, PARP2, NEIL2, HMGB1, POLD2 | NEIL1, PARP4 | 3.53 × 10−8 | 18 | |
| Fanconi anemia pathway | RMI2, UBE2T, RAD51, CENPX, RPA3, EME1, RPA1, TELO2, FANCI, FANCA, FANCG, FANCD2, BRCA1, FANCM, BLM, FANCB, FAAP24, RAD51C, CENPS, CENPS-CORT, ATRIP | EME2, POLK, POLI | 3.58 × 10−8 | 24 | |
| Homologous recombination | POLD1, RAD51, RPA3, RAD54L, EME1, POLD3, RPA1, POLD2, XRCC2, BRCA1, RAD54B, XRCC3, RBBP8, BLM, BARD1, RAD51C | ATM, ABRAXAS1 | 2.31 × 10−6 | 18 | |
| DON | DNA replication | PCNA, MCM4, MCM3, MCM5, MCM6, MCM2, MCM7, POLD1, POLD2, RFC3, POLE2, RFC4, FEN1, RPA3, RNASEH1, POLE3, LIG1, POLD3, RNASEH2A, PRIM1, POLA2, RFC2, RNASEH2C | − | 1.17 × 10−13 | 23 |
| Base excision repair | PCNA, PARP1, POLD1, UNG, POLD2, POLE2, FEN1, POLE3, LIG1, POLD3, NEIL2, HMGB1, NTHL1 | PARP4, NEIL1 | 1.10 × 10−6 | 15 | |
| Cell cycle | PCNA, E2F1, MCM4, MCM3, MCM5, E2F2, MCM6, MCM2, MCM7, CCND1, CDK2, MAD2L1, CDC6, ORC6, CCNE1, CDKN2C, TFDP1, CHEK2, CDC45, E2F3, ESPL1, PKMYT1, CDC25A, ORC1, CCND3, CDKN2A, MAD2L2, CDKN2B, CCND2 | WEE2, STAG1, STAG2, ATM | 2.69 × 10−6 | 33 | |
| Steroid hormone biosynthesis | CYP1A1, UGT1A6, HSD17B8, HSD11B2, UGT1A4, UGT1A1 | UGT2A3, UGT2B15, UGT2B17, AKR1C2, UGT2B7, UGT2B11, STS, AKR1C1, AKR1C3, CYP17A1, CYP3A5, SULT1E1, UGT2B10, AKR1D1 | 9.00 × 10−6 | 20 | |
| Drug metabolism—other enzymes | TK1, NME1, NME4, DUT, NME2, HPRT1, IMPDH1, UCK2, UGT1A6, UPP1, TK2, MGST1, UGT1A4, CDA, UGT1A1 | UGT2A3, GSTA2, UGT2B15, UGT2B17, UGT2B7, UGT2B11, GSTA1, UGT2B10 | 1.81 × 10−5 | 23 | |
| 15-ADON | DNA replication | PCNA, MCM3, MCM4, MCM5, POLD1, MCM6, MCM2, MCM7, POLE2, POLD2, RFC4, RFC3, RPA3, LIG1, POLE3, RNASEH1, PRIM1, POLD3, POLA2, RFC2 | − | 1.54 × 10−11 | 20 |
| Cell cycle | PCNA, MCM3, E2F1, MCM4, MCM5, E2F2, CDK2, TFDP1, MCM6, CCND1, MAD2L1, MCM2, MCM7, ANAPC5, CCNE1, CDKN2C, CHEK2, E2F3, ORC6, CDC45, ORC1, PKMYT1, CDC25A, MAD2L2, CCND3, CCND2 | WEE2, STAG1, ATM, CDKN2B | 4.03 × 10−6 | 30 | |
| Base excision repair | PCNA, PARP1, UNG, POLD1, POLE2, POLD2, LIG1, POLE3, POLD3, NTHL1, NEIL2 | NEIL1, OGG1 | 8.87 × 10−6 | 13 | |
| Pyrimidine metabolism | TYMS, NME1, TK1, NME4, DUT, DTYMK, UCK2, CAD, NME2, DCK, UPP1, TK2, DCTPP1, DHODH, CDA | ENTPD8, AK9 | 2.92 × 10−5 | 17 | |
| Mismatch repair | PCNA, POLD1, POLD2, RFC4, RFC3, EXO1, RPA3, LIG1, POLD3, RFC2 | − | 3.57 × 10−5 | 10 |
| Gene | Length (bp) | Primer Sequence |
|---|---|---|
| β-actin | 19 | F: CCTTCCTGGGCATGGAGTC |
| 21 | R: TGATCTTCATTGTGCTGGGTG | |
| MCM3 | 19 | F: AAGCAGATGAGCAAGGATG |
| 19 | R: CAAGAGCAAGCAGAGGATT | |
| MCM4 | 17 | F: CACCTGGTCGCACTGTA |
| 17 | R: GGCTGGCTTCCTCACTT | |
| PCNA | 22 | F: ACACTAAGGGCCGAAGATAACG |
| 22 | R: ACAGCATCTCCAATATGGCTGA | |
| POLE2 | 17 | F: TGGTGGAAGCAGCAGTC |
| 21 | R: GGTTGGTCATTAACAGAGGAA | |
| POLD2 | 17 | F: CTGGTGGATGTGGTGAC |
| 17 | R: CTGTGGCTGAGGAGGTT | |
| RPA3 | 21 | F: AGCTCAATTCATCGACAAGCC |
| 22 | R: TCTTCATCAAGGGGTTCCATCA | |
| RAD51 | 22 | F: CAACCCATTTCACGGTTAGAGC |
| 21 | R: TTCTTTGGCGCATAGGCAACA | |
| BID | 17 | F: CGTCCTTGCTCCGTGAT |
| 18 | R: TGTCCGTTCAGTCCATCC | |
| ATM | 19 | F: ACTACTGCTCCAGACCAAT |
| 20 | R: TCACGACGATACAAAGAACA | |
| CDKN2C | 15 | F: CTGAGCGGCATTAGC |
| 15 | R: CGAACGGGAGTAGCA | |
| E2F2 | 18 | F: GCACTGGCATCATTCTCT |
| 18 | R: AGTCACCTCTGTCCTTGG | |
| CCND2 | 19 | F: ACCTTCCGCAGTGCTCCTA |
| 19 | R: CCCAGCCAAGAAACGGTCC | |
| SERPINE1 | 17 | F: GCTGGTGCTGGTGAATG |
| 17 | R: AGTGCTGCCGTCTGATT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Yin, X.; Dong, J.; Yang, Q.; Wu, Y.; Gong, Z. Transcriptome Analysis of Caco-2 Cells upon the Exposure of Mycotoxin Deoxynivalenol and Its Acetylated Derivatives. Toxins 2021, 13, 167. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020167
He Y, Yin X, Dong J, Yang Q, Wu Y, Gong Z. Transcriptome Analysis of Caco-2 Cells upon the Exposure of Mycotoxin Deoxynivalenol and Its Acetylated Derivatives. Toxins. 2021; 13(2):167. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020167
Chicago/Turabian StyleHe, Yuyun, Xiaoyao Yin, Jingjing Dong, Qing Yang, Yongning Wu, and Zhiyong Gong. 2021. "Transcriptome Analysis of Caco-2 Cells upon the Exposure of Mycotoxin Deoxynivalenol and Its Acetylated Derivatives" Toxins 13, no. 2: 167. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13020167