Non-Digestible Oligosaccharides and Short Chain Fatty Acids as Therapeutic Targets against Enterotoxin-Producing Bacteria and Their Toxins

 , , and
, , and
Abstract
:1. Introduction
2. Enterotoxin-Producing Bacteria and Related Enterotoxins
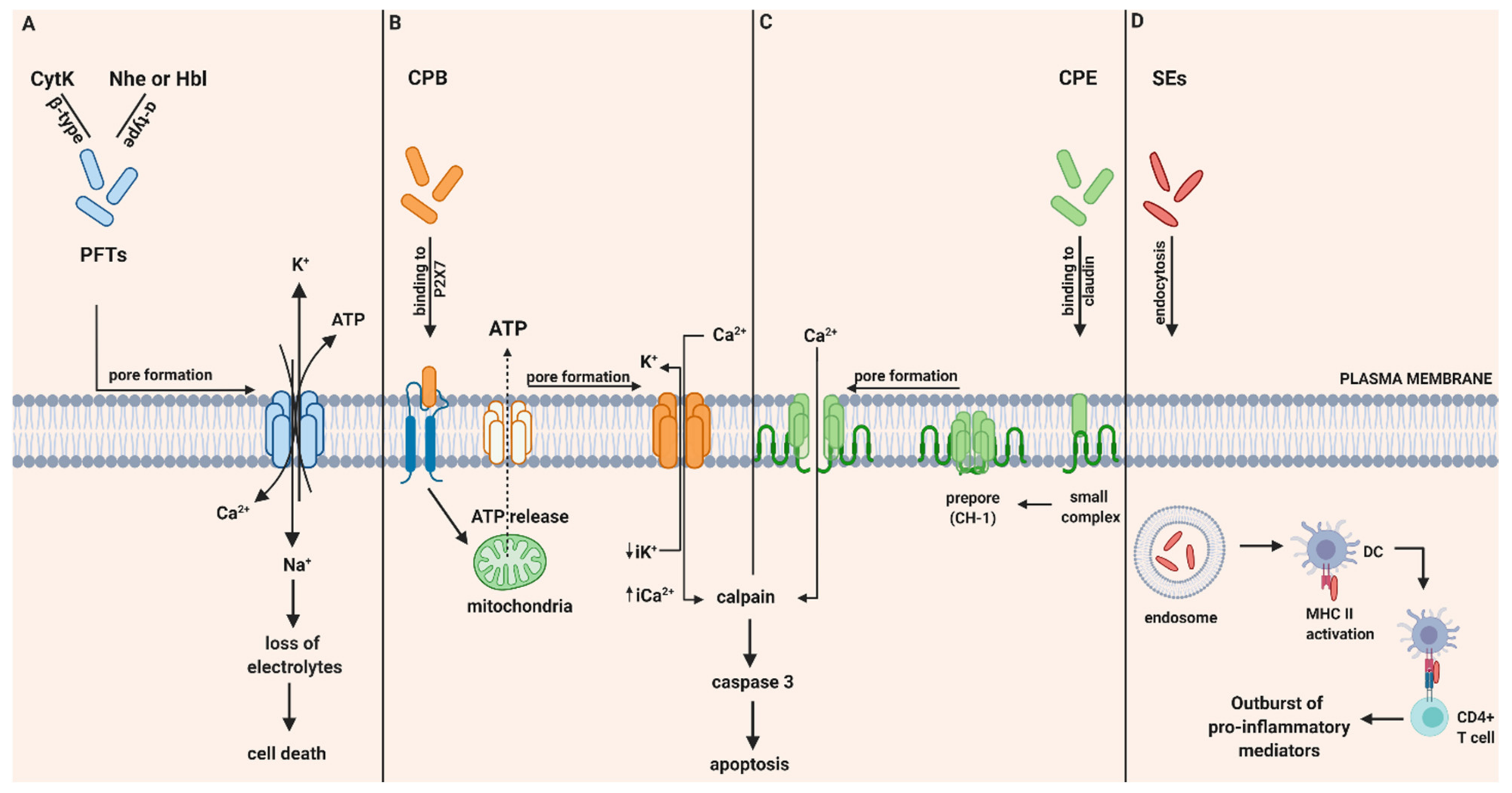
2.1. B. cereus and Related Enterotoxins
2.1.1. Epidemiology of B. cereus and Related Enterotoxins
2.1.2. Structure of Hbl, Nhe and CytK
2.1.3. Pathogenicity of Hbl, Nhe and CytK
2.2. C. perfringens and Related Enterotoxins
2.2.1. C. perfringens Beta Toxin (CPB)
Epidemiology of CPB
Structure of CPB
Mode of Action of CPB
2.2.2. C. perfringens Enterotoxin (CPE)
Epidemiology of CPE
Structure of CPE
Mode of Action of CPE
2.3. S. aureus and Staphylococcal Enterotoxins (SEs)
2.3.1. Epidemiology of S. aureus and SEs in Foodborne Poisoning Associated Diarrhea
2.3.2. Structure of SEs
2.3.3. Mechanism of Action of SEs in Gastro-Intestinal Inflammatory Injury
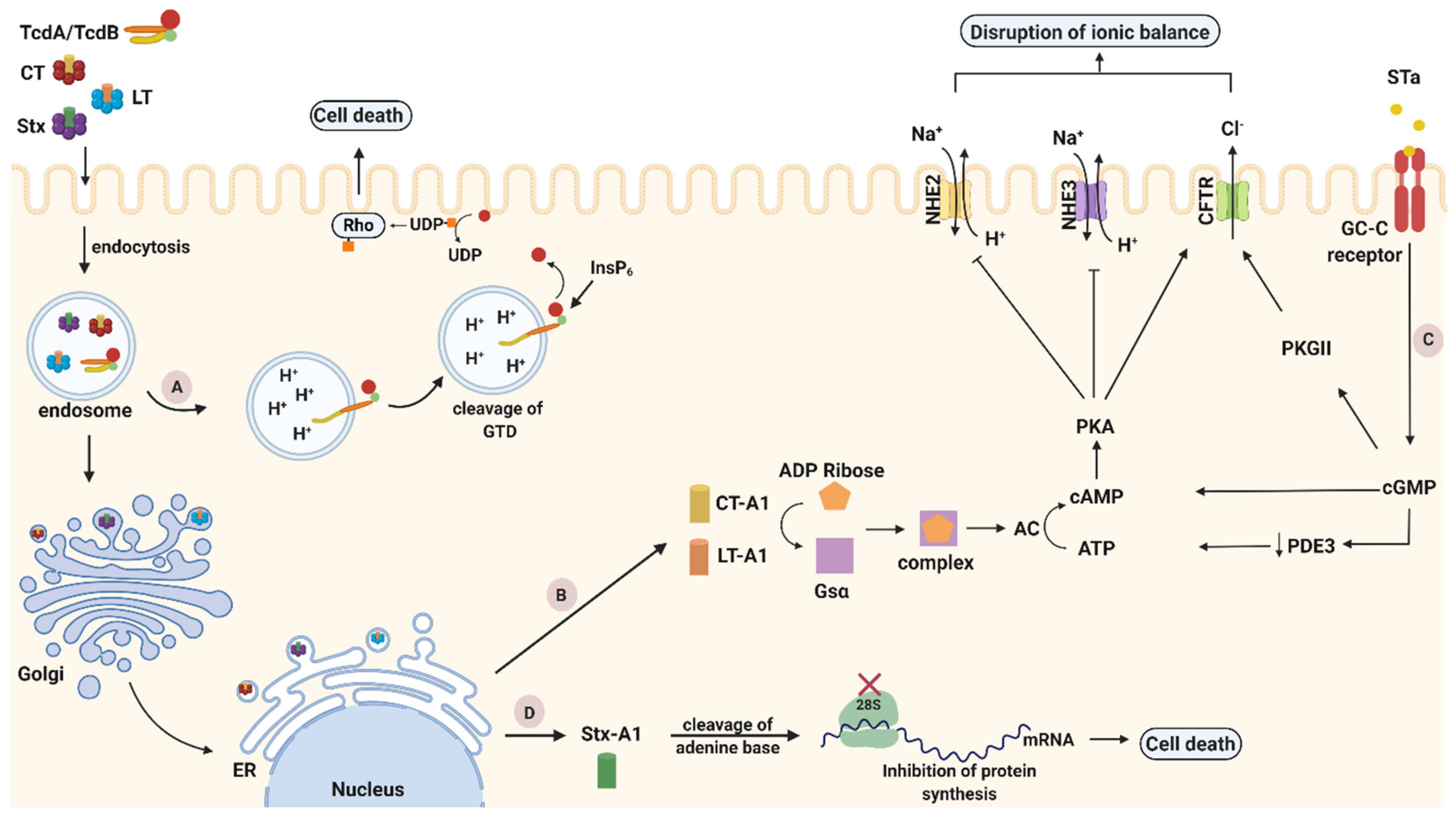
2.4. C. difficile and Related Enterotoxins (TcdA, TcdB and CDT)
2.4.1. C. difficile Toxin A (TcdA) and Toxin B (TcdB)
Epidemiology of TcdA and TcdB
Structure of TcdA and TcdB
Mechanism of Action of TcdA and TcdB
2.4.2. C. difficile Transferase (CDT)
Epidemiology of CDT
Structure of CDT
Mechanism of Action of CDT
2.5. V. cholerae and Cholera Toxin (CT)
2.5.1. Epidemiology of V. cholerae and CT
2.5.2. Structure of CT
2.5.3. Trafficking and Mechanism of Action of CT
2.6. E. coli and Related Enterotoxins
2.6.1. E. coli, LTs and STs
Epidemiology LTs and STs
Structure of LTs and STs
Trafficking and Mechanism of Action LTs
Trafficking and Mechanism of Action of STs
2.6.2. E. coli and Stxs
Epidemiology of Stxs
Structure of Stxs
Trafficking and Mechanism of Action of Stxs
3. Non-Digestible Oligosaccharides
4. Different Effects of NDOs and SCFA against Enterotoxins and Enterotoxin-Producing Bacteria
4.1. The Effects of NDOs and SCFA against Enterotoxin-Producing Bacteria
4.1.1. Direct Mechanisms of Action of NDOs against EPB
E. coli
V. cholerea
B. cereus
S. aureus
Clostridium spp.
4.1.2. Indirect Mechanisms of Action of NDOs and SCFA against EPB
Antimicrobial Activity of Beneficial Bacteria
Immunomodulation Activity
Improvement of Intestinal Barrier Function
Acidic Environment
4.2. The Effects of NDOs and SCFA against Bacterial Enterotoxins
4.2.1. B. cereus Enterotoxins
4.2.2. Cholera Toxin (CT)
NDOs against CT
SCFA against CT
4.2.3. C. difficile Enterotoxins
NDOs against C. difficile Toxin A (TcdA) and Toxin B (TcdB)
SCFA against C. difficile Toxin A (TcdA) and Toxin B (TcdB)
4.2.4. C. perfringens Enterotoxins
4.2.5. Heat-Labile (LT) and Heat-Stable (ST) Enterotoxins
NDOs against LT and ST Enterotoxins
SCFA against LT Enterotoxin
4.2.6. Shiga Toxins (Stxs)
NDOs against Stxs
SCFA against Stxs
4.2.7. Staphylococcal Enterotoxins (SEs)
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase | HMOs | Human milk oligosaccharides |
| AMPK | AMP-activated protein kinase | HMW | High-molecular weight |
| AOS | Alginate-oligosaccharides | HUS | Hemolytic uremic syndrome |
| B. breve | Bifidobacterium breve | IEC | Intestinal epithelial cells |
| B. cereus | Bacillus cereus | iK+ | intracytoplasmic K+ |
| B. fragilis | Bacteroides fragilis | IMOS | Isomalto-oligosaccharides |
| C. difficile | Clostridium difficile | InsP6 | Inositolhexakisphosphate |
| C. perfringens | Clostridium perfringens | L. rhamnosus | Lactobacillus rhamnosus |
| cAMP | cyclic AMP | LMW | Low-molecular weight |
| CDI | C. difficile infections | LNFP | Lacto-N-fucopentaose |
| CDT | C. difficile transferase | LNnh | Lacto-N-hexaose |
| CFTR | Cystic fibrosis transmembrane conductance regulatror | LSR | Lipoprotein receptor |
| cGMP | cyclic GMP | LT | Heat-labile enterotoxin |
| COS | Chito-oligosaccharides/chitosan-oligosaccharides | ManA | D-Mannuronic acid |
| CPB | C. perfringens beta toxin | MHC | Major histocompatibility complex |
| CPE | C. perfringens enterotoxin | MOS | Mannan-oligosaccharides |
| CROP | Combined repetitive peptides | MRSA | Methicillin-resistant S. Aureus |
| CSPG4 | Chondroitin sulfate proteoglycan4 | MUC2 | Mucin2 |
| CT | Cholera toxin | MW | Molecular weight |
| CypA | Cyclophilin A | NDOs | Non-digestible oligosaccharides |
| CytK | Cytotoxin K | NetB | Necrotic enteritis B-like toxin |
| DA | Degree of acetylation | Nhe | Non-hemolytic enterotoxin |
| DC | Dendritic cells | NHE3 | Na+/H+ exchanger 3 |
| DD | Degree of deacetylation | PDE3 | Phosphodiesterase 3 |
| DP | Degree of polymerization | PFT | Pore-forming toxins |
| E. coli | Escherichia coli | PKA | Protein kinase A |
| EPB | Enterotoxin-producing bacteria | PLC | Phospholipase C |
| EPEC | Enteropathogenic E. coli | POS | Pectic-oligosaccharides |
| EPS | Extracellular polymeric substance | PVRL3 | Poliovirus receptor-like protein |
| ER | Endoplasmic reticulum | S. Aureus | Staphylococcus Aureus |
| ETEC | Enterotoxigenic Escherichia coli | SAgs | Superantigens |
| FOS | Fructo-oligosaccharides | SCFA | Short chain fatty acids |
| Fuc | Fucose | Sec | Secretory |
| Gal | Galactose | SEs | Staphylococcal Enterotoxins |
| GalA | D-Galacturonic acid | SI | Sucrose-isomaltase |
| GC-C | Guanylyl cyclase C | SOS | Sialylated-oligosaccharides |
| GI | Gastrointestinal | ST | Heat-stable enterotoxin |
| Glc | Glucose | STEC | Shiga toxin-producing Escherichia coli |
| GlcN | D-glucosamine | Stx | Shiga toxin |
| GlcNAc | N-acetylglucosamine | TcdA | C. difficile toxin A |
| GOS | Galacto-oligosaccharides | TcdB | C. difficile toxin B |
| Gs | G protein | TCR | T-cell receptors |
| GTD | Glucosyltransferase domain | TJ | Tight junctions |
| GTP | Guanosine triphosphate | TSST-1 | toxic shock syndrome toxin-1 |
| GulA | D-Guluronic acid | UDP | Uridine diphosphate |
| HBL | Hemolytic hemolysin B | V. cholerea | Vibrio cholerea |
| HDAC | Histone deacetylase | VTEC | Verocytotoxin-producing Escherichia coli |
| HIF | Hypoxia-inducible factor | XOS | Xylo-oligosaccharides |
References
- Abebe, E.; Gugsa, G.; Ahmed, M. Review on Major Food-Borne Zoonotic Bacterial Pathogens. J. Trop. Med. 2020, 2020, 4674235. [Google Scholar] [CrossRef]
- Mussatto, S.I.; Mancilha, I.M. Non-digestible oligosaccharides: A review. Carbohydr. Polym. 2007, 68, 587–597. [Google Scholar] [CrossRef]
- Singh, P. Functional Oligosaccharides: Physico-chemical Properties, Synthesis, Structures and Biological Applications. JICS 2019, 9, 674–690. [Google Scholar]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat. Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Multifaceted interactions of bacterial toxins with the gastrointestinal mucosa. Futur. Microbiol. 2011, 6, 763–797. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, S. Antibiotic treatments and microbes in the gut. Environ. Microbiol. 2014, 16, 919–924. [Google Scholar] [CrossRef]
- Akbari, P.; Fink-Gremmels, J.; Willems, R.H.A.M.; Difilippo, E.; Schols, H.A.; Schoterman, M.H.C.; Garssen, J.; Braber, S. Characterizing microbiota-independent effects of oligosaccharides on intestinal epithelial cells: Insight into the role of structure and size: Structure–activity relationships of non-digestible oligosaccharides. Eur. J. Nutr. 2017, 56, 1919–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Goyal, A. Functional oligosaccharides: Production, properties and applications. World J. Microbiol. Biotechnol. 2011, 27, 1119–1128. [Google Scholar] [CrossRef]
- Sun, Y.; O’Riordan, M.X.D. Regulation of bacterial pathogenesis by intestinal short-chain fatty acids. In Advances in Applied Microbiology; Academic Press Inc.: Cambridge, MA, USA, 2013; Volume 85, pp. 93–118. [Google Scholar]
- Jessberger, N.; Dietrich, R.; Granum, P.; Märtlbauer, E. The Bacillus cereus Food Infection as Multifactorial Process. Toxins 2020, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Granum, P.E.; Lund, T. Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Lett. 2006, 157, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.; De Buyser, M.-L.; Granum, P.E. A new cytotoxin from Bacillus cereus that may cause necrotic enteritis. Mol. Microbiol. 2000, 38, 254–261. [Google Scholar] [CrossRef]
- Messelhäußer, U.; Ehling-Schulz, M. Bacillus cereus—A Multifaceted Opportunistic Pathogen. Curr. Clin. Microbiol. Rep. 2018, 5, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Senesi, S.; Ghelardi, E. Production, Secretion and Biological Activity of Bacillus cereus Enterotoxins. Toxins 2010, 2, 1690–1703. [Google Scholar] [CrossRef]
- Stenfors Arnesen, L.P.; Fagerlund, A.; Granum, P.E. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 2008, 32, 579–606. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Märtlbauer, E.; Dietrich, R.; Luo, H.; Ding, S.; Zhu, K. Multifaceted toxin profile, an approach toward a better understanding of probioticBacillus cereus. Crit. Rev. Toxicol. 2019, 49, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Sastalla, I.; Fattah, R.; Coppage, N.; Nandy, P.; Crown, D.; Pomerantsev, A.P.; Leppla, S.H. The Bacillus cereus Hbl and Nhe Tripartite Enterotoxin Components Assemble Sequentially on the Surface of Target Cells and Are Not Interchangeable. PLoS ONE 2013, 8, e76955. [Google Scholar] [CrossRef]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; Mcclane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins 2018, 10, 212. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, M.; Ochi, S.; Oda, M.; Miyamoto, K.; Takehara, M.; Kobayashi, K. Recent Insights into Clostridium perfringens Beta-Toxin. Toxins 2015, 7, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Miyakawa, M.E.; Redondo, L.M. Role of Clostridium perfringens α, α, ξ, and Iota Toxins in Enterotoxemia of Monogastrics and Ruminants. In Microbial Toxins; Springer: Dordrecht. The Netherlands, 2018. [Google Scholar]
- Theoret, J.R.; Uzal, F.A.; McClane, B.A. Identification and Characterization of Clostridium perfringens Beta Toxin Variants with Differing Trypsin Sensitivity andIn VitroCytotoxicity Activity. Infect. Immun. 2015, 83, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States-Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kiu, R.; Hall, L.J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens Enterotoxin: Action, Genetics, and Translational Applications. Toxins 2016, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.L.; Smedley, J.G.; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 2007, 9, 2734–2755. [Google Scholar] [CrossRef]
- Chen, J.; Theoret, J.R.; Shrestha, A.; Smedley, J.G.; McClane, B.A. Cysteine-Scanning Mutagenesis Supports the Importance of Clostridium perfringens Enterotoxin Amino Acids 80 to 106 for Membrane Insertion and Pore Formation. Infect. Immun. 2012, 80, 4078–4088. [Google Scholar] [CrossRef] [Green Version]
- NCBI Bookshelf. StatPearls. In Staphylococcus Aureus. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK441868/ (accessed on 18 November 2020).
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal Enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-Related Illness and Death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, D.; Corti, S.; Muehlherr, J.E.; Zweifel, C.; Stephan, R. Phenotypic and genotypic characteristics of Staphylococcus aureus isolates from raw bulk-tank milk samples of goats and sheep. Vet. Microbiol. 2004, 101, 101–107. [Google Scholar] [CrossRef]
- Benkerroum, N. Staphylococcal enterotoxins and enterotoxin-like toxins with special reference to dairy products: An overview. Crit. Rev. Food Sci. Nutr. 2018, 58, 1943–1970. [Google Scholar] [CrossRef]
- Krakauer, T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins 2019, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.S.H.; Monaghan, T.M.; Wilcox, M.H. Clostridium difficile infection: Epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.A.; Longshaw, C.M.; Davis, G.L.; Bouza, E.; Barbut, F.; Barna, Z.; Delmée, M.; Fitzpatrick, F.; Ivanova, K.; Kuijper, E.; et al. Underdiagnosis of Clostridium difficile across Europe: The European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect. Dis. 2014, 14, 1208–1219. [Google Scholar] [CrossRef]
- Center of Disease Control. Healthcare-associated Infections. In Clostridioides Difficile Infection. Available online: https://www.cdc.gov/hai/organisms/cdiff/cdiff_infect.html (accessed on 28 May 2020).
- Voth, D.E.; Ballard, J.D. Clostridium difficile Toxins: Mechanism of Action and Role in Disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficileToxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C.; Galili, U.; Castagliuolo, I.; Kelly, C.P.; Nikulasson, S.; Dudeja, P.K.; Brasitus, T.A.; Lamont, J.T. A human antibody binds to α-galactose receptors and mimics the effects of Clostridium difficile toxin A in rat colon. Gastroenterology 1996, 110, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Na, X.; Kim, H.; Moyer, M.P.; Pothoulakis, C.; Lamont, J.T. gp96 Is a Human Colonocyte Plasma Membrane Binding Protein for Clostridium difficile Toxin A. Infect. Immun. 2008, 76, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Zhang, H.; Cai, C.; Zhu, S.; Zhou, Y.; Yang, X.; He, R.; Li, C.; Guo, S.; Li, S.; et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 2015, 25, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFrance, M.E.; Farrow, M.A.; Chandrasekaran, R.; Sheng, J.; Rubin, D.H.; Lacy, D.B. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 7073–7078. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, R.; Kenworthy, A.K.; Lacy, D.B. Clostridium difficile Toxin A Undergoes Clathrin-Independent, PACSIN2-Dependent Endocytosis. PLoS Pathog. 2016, 12, e1006070. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Zamboglou, C.; Genisyuerek, S.; Guttenberg, G.; Aktories, K. Clostridial Glucosylating Toxins Enter Cells via Clathrin-Mediated Endocytosis. PLoS ONE 2010, 5, e10673. [Google Scholar] [CrossRef] [Green Version]
- Genisyuerek, S.; Papatheodorou, P.; Guttenberg, G.; Schubert, R.; Benz, R.; Aktories, K. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 2011, 79, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.F.; Schell, M.J. Back in the water: The return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium difficile binary toxin CDT: Mechanism, epidemiology, and potential clinical importance. Gut Microbes 2014, 5, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Papatheodoroua, P.; Carette, J.E.; Bell, G.W.; Schwan, C.; Guttenberg, G.; Brummelkamp, T.R.; Aktories, K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. USA 2011, 108, 16422–16427. [Google Scholar] [CrossRef] [Green Version]
- Mesli, S.; Javorschi, S.; Bérard, A.M.; Landry, M.; Priddle, H.; Kivlichan, D.; Smith, A.J.H.; Yen, F.T.; Bihain, B.E.; Darmon, M. Distribution of the lipolysis stimulated receptor in adult and embryonic murine tissues and lethality of LSR-/- embryos at 12.5 to 14.5 days of gestation. Eur. J. Biochem. 2004, 271, 3103–3114. [Google Scholar] [CrossRef]
- Ernst, K.; Schmid, J.; Beck, M.; Hägele, M.; Hohwieler, M.; Hauff, P.; Ückert, A.K.; Anastasia, A.; Fauler, M.; Jank, T.; et al. Hsp70 facilitates trans-membrane transport of bacterial ADP-ribosylating toxins into the cytosol of mammalian cells. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schwan, C.; Stecher, B.; Tzivelekidis, T.; Van Ham, M.; Rohde, M.; Hardt, W.-D.; Wehland, J.; Aktories, K. Clostridium difficile Toxin CDT Induces Formation of Microtubule-Based Protrusions and Increases Adherence of Bacteria. PLoS Pathog. 2009, 5, e1000626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-W.; Sails, A.; Poxton, I.; Liu, D.; Schwartzman, J.D. Molecular Medical Microbiology; Academic Press: Cambridge, MA, USA, 2014; Available online: http://0-ebookcentral-proquest-com.brum.beds.ac.uk/lib/uunl/detail.action?docID=1798299 (accessed on 22 May 2020).
- Niyi Awofeso, K.A. Cholera, Migration, and Global Health—A Critical Review. Int. J. Travel Med. Glob. Health 2018, 6, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Kuna, A.; Gajewski, M. Cholera—The new strike of an old foe. Int. Marit. Health 2017, 68, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, T.; Mutreja, A.; Weill, F.-X.; Das, B.; Ghosh, A.; Nair, G.B. Revisiting the Global Epidemiology of Cholera in Conjunction With the Genomics of Vibrio cholerae. Front. Public Health 2019, 7, 203. [Google Scholar] [CrossRef]
- WHO. Cholera. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cholera (accessed on 26 May 2020).
- Keya, C.; Chatterjee, S.N. Cholera Toxins; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Sánchez, J.; Holmgren, J. Cholera toxin—A foe & a friend. Indian J. Med. Res. 2011, 133, 153–163. [Google Scholar]
- Aureli, M.; Mauri, L.; Ciampa, M.G.; Prinetti, A.; Toffano, G.; Secchieri, C.; Sonnino, S. GM1 Ganglioside: Past Studies and Future Potential. Mol. Neurobiol. 2015, 53, 1824–1842. [Google Scholar] [CrossRef]
- Merritt, E.A.; Sarfaty, S.; Van Den Akker, F.; L’hoir, C.; Martial, J.A.; Hol, W.G.J. Crystal Structure of Cholera Toxin B-Pentamer Bound to Receptor GMl Pentasaccharide; Cambridge University Press: Cambridge, UK, 1994; Volume 3. [Google Scholar]
- Chinnapen, D.J.-F.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Orlandi, P.A.; Fishman, P.H. Filipin-dependent Inhibition of Cholera Toxin: Evidence for Toxin Internalization and Activation through Caveolae-like Domains. J. Cell Biol. 1998, 141, 905–915. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Skretting, G.; Van Deurs, B.; Sandvig, K. Internalization of cholera toxin by different endocytic mechanisms. J. Cell Sci. 2001, 114, 3737–3747. [Google Scholar]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Wernick, N.L.B.; Chinnapen, D.J.-F.; Cho, J.A.; Lencer, W.I. Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.P. Cholera. Am. J. Med. 1998, 104, 386–394. [Google Scholar] [CrossRef]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Genet. 2021, 19, 37–54. [Google Scholar] [CrossRef]
- Duan, Q.; Xia, P.; Nandre, R.; Zhang, W.; Zhu, G. Review of Newly Identified Functions Associated with the Heat-Labile Toxin of Enterotoxigenic Escherichia coli. Front. Cell. Infect. Microbiol. 2019, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Jobling, M.G.; Holmes, R.K. Heat-Labile Enterotoxins. EcoSal Plus 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhong, Z.; Luo, Y.; Cox, E.; Devriendt, B. Heat-Stable Enterotoxins of Enterotoxigenic Escherichia coli and Their Impact on Host Immunity. Toxins 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudrak, B.; Kuehn, M.J. Heat-Labile Enterotoxin: Beyond G M1 Binding. Toxins 2010, 2, 1445–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taxt, A.; Aasland, R.; Sommerfelt, H.; Nataro, J.; Puntervoll, P. Heat-Stable Enterotoxin of Enterotoxigenic Escherichia coli as a Vaccine Target. Infect. Immun. 2010, 78, 1824–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albano, F.; Thompson, M.R.; Orrù, S.; Scaloni, A.; Musetta, A.; Pucci, P.; Guarino, A. Structural and Functional Features of Modified Heat-Stable Toxins Produced by Enteropathogenic Klebsiella Cells. Pediatr. Res. 2000, 48, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.P.; Neill, R.J.; O’Brien, A.D.; Holmes, R.K.; Newland, J.W. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol. Lett. 1987, 44, 109–114. [Google Scholar] [CrossRef]
- Bryan, A.; Youngster, I.; McAdam, A.J. Shiga Toxin Producing Escherichia coli. Clin. Lab. Med. 2015, 35, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global Incidence of Human Shiga Toxin–ProducingEscherichia coliInfections and Deaths: A Systematic Review and Knowledge Synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licznerska, K.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Gąsior, T.; Węgrzyn, A.; Węgrzyn, G. Oxidative Stress in Shiga Toxin Production by Enterohemorrhagic Escherichia coli. Oxidative Med. Cell. Longev. 2015, 2016, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.S.; Ng, T.B. Shiga toxins: From structure and mechanism to applications. Appl. Microbiol. Biotechnol. 2015, 100, 1597–1610. [Google Scholar] [CrossRef]
- Lukyanenko, V.; Malyukova, I.; Hubbard, A.; Delannoy, M.; Boedeker, E.; Zhu, C.; Cebotaru, L.; Kovbasnjuk, O. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1140–C1149. [Google Scholar] [CrossRef] [Green Version]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Liver Physiol. 2009, 296, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Sandvig, K.; Garred, Ø.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; Van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef]
- Joseph, A.; Cointe, A.; Kurkdjian, P.M.; Rafat, C.; Hertig, A. Shiga Toxin-Associated Hemolytic Uremic Syndrome: A Narrative Review. Toxins 2020, 12, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Guo, Q.; Goff, H.D.; Lapointe, G. Oligosaccharides: Structure, Function and Application. Encycl. Food Chem. 2019, 202–207. [Google Scholar] [CrossRef]
- Seeberger, P.H. Monosaccharide Diversity. In Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2017; Chapter 2. [Google Scholar]
- Liu, J.; Yang, S.; Li, X.; Yan, Q.; Reaney, M.J.T.; Jiang, Z. Alginate Oligosaccharides: Production, Biological Activities, and Potential Applications. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1859–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, M.B.; Struszczyk-Swita, K.; Li, X.; Szczęsna-Antczak, M.; Daroch, M. Enzymatic Modifications of Chitin, Chitosan, and Chitooligosaccharides. Front. Bioeng. Biotechnol. 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muanprasat, C.; Chatsudthipong, V. Chitosan oligosaccharide: Biological activities and potential therapeutic applications. Pharmacol. Ther. 2017, 170, 80–97. [Google Scholar] [CrossRef]
- Ibrahim, O.O. Functional Oligo-saccharides: Chemicals Structure, Manufacturing, Health Benefits, Applications and Regulations. J. Food Chem. Nanotechnol. 2018, 4, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J.; Michel, C. How to manipulate the microbiota: Prebiotics. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2016; Volume 902, pp. 119–142. [Google Scholar]
- Aparecida, A.; Tomal, B.; Farinazzo, F.S.; Bachega, A.; Bosso, A.; Bonifácio Da Silva, J.; Suguimoto, H. Galactooligosacharides and Human Health Implications. Int. J. Nutr. Food Sci. 2019, 9. [Google Scholar]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Boehm, G.; Stahl, B.; Jelinek, J.; Knol, J.; Miniello, V.; Moro, G.E. Prebiotic carbohydrates in human milk and formulas. Acta Paediatr. 2007, 94, 18–21. [Google Scholar] [CrossRef]
- Wicinski, M.; Sawicka, E.; Gebalski, J.; Kubiak, K.; Malinovsi, B. Human Milk Oligosaccharides: Health Benefits, Potential Applications in Infant Formulas, and Pharmacology. Nutrients 2020, 12, 266. [Google Scholar] [CrossRef] [Green Version]
- Hoving, L.R.; van der Zande, H.J.P.; Pronk, A.; Guigas, B.; Willems van Dijk, K.; van Harmelen, V. Dietary yeast-derived mannan oligosaccharides have immune-modulatory properties but do not improve high fat diet-induced obesity and glucose intolerance. PLoS ONE 2018, 13, e0196165. [Google Scholar]
- Liu, X.; Wang, Q.; Cui, S.; Liu, H. A new isolation method of β-d-glucans from spent yeast Saccharomyces cerevisiae. Food Hydrocoll. 2008, 22, 239–247. [Google Scholar] [CrossRef]
- Baldassarre, S.; Babbar, N.; Van Roy, S.; Dejonghe, W.; Maesen, M.; Sforza, S.; Elst, K. Continuous production of pectic oligosaccharides from onion skins with an enzyme membrane reactor. Food Chem. 2018, 267, 101–110. [Google Scholar] [CrossRef]
- Babbar, N.; Dejonghe, W.; Gatti, M.; Sforza, S.; Elst, K. Pectic oligosaccharides from agricultural by-products: Production, characterization and health benefits. Crit. Rev. Biotechnol. 2015, 36, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-H.; Chou, L.-M.; Chien, Y.-W.; Chang, J.-S.; Lin, C.-I. Prebiotic Effects of Xylooligosaccharides on the Improvement of Microbiota Balance in Human Subjects. Gastroenterol. Res. Pr. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Aachary, A.A.; Prapulla, S.G. Xylooligosaccharides (XOS) as an Emerging Prebiotic: Microbial Synthesis, Utilization, Structural Characterization, Bioactive Properties, and Applications. Compr. Rev. Food Sci. Food Saf. 2010, 10, 2–16. [Google Scholar] [CrossRef]
- Ackerman, D.L.; Craft, K.M.; Doster, R.S.; Weitkamp, J.-H.; Aronoff, D.M.; Gaddy, J.A.; Townsend, S.D. Antimicrobial and Antibiofilm Activity of Human Milk Oligosaccharides against Streptococcus agalactiae, Staphylococcus aureus, and Acinetobacter baumannii. ACS Infect. Dis. 2018, 4, 315–324. [Google Scholar] [CrossRef]
- Asadpoor, M.; Peeters, C.; Henricks, P.A.J.; Varasteh, S.; Pieters, R.J.; Folkerts, G.; Braber, S. Anti-Pathogenic Functions of Non-Digestible Oligosaccharides In Vitro. Nutritiens 2020, 12, 1789. [Google Scholar] [CrossRef]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatek, J.; Krauss, H.; Ciechelska-Rybarczyk, A.; Bernatek, M.; Wojtyla-Buciora, P.; Sommermeyer, H. In-Vitro Growth Inhibition of Bacterial Pathogens by Probiotics and a Synbiotic: Product Composition Matters. Int. J. Environ. Res. Public Health 2020, 17, 3332. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Mandal, S.; Singh, K.S.; Patil, P.; Tomar, S.K. Synbiotic combination of Lactobacillus rhamnosus NCDC 298 and short chain fructooligosaccharides prevents enterotoxigenic Escherichia coli infection. LWT 2018, 98, 329–334. [Google Scholar] [CrossRef]
- Stefania, D.M.; Miranda, P.; Diana, M.; Claudia, Z.; Rita, P.; Donatella, P. Antibiofilm and Antiadhesive Activities of Different Synbiotics. J. Probiotics Health 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Fooks, L.J.; Gibson, G.R. In vitro investigations of the effect of probiotics and prebiotics on selected human intestinal pathogens. FEMS Microbiol. Ecol. 2002, 39, 67–75. [Google Scholar] [CrossRef]
- Lkhagvadorj, E.; Nagata, S.; Wada, M.; Bian, L.; Wang, C.; Chiba, Y.; Yamashiro, Y.; Shimizu, T.; Asahara, T.; Nomoto, K. Anti-infectious activity of synbiotics in a novel mouse model of methicillin-resistant Staphylococcus aureus infection. Microbiol. Immunol. 2010, 54, 265–275. [Google Scholar] [CrossRef]
- Kondepudi, K.K.; Ambalam, P.; Karagin, P.H.; Nilsson, I.; Wadström, T.; Ljungh, Å. A novel multi-strain probiotic and synbiotic supplement for prevention of Clostridium difficile infection in a murine model. Microbiol. Immunol. 2014, 58, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sosa, S.; Martín, M.-J.; Hueso, P. The Sialylated Fraction of Milk Oligosaccharides Is Partially Responsible for Binding to Enterotoxigenic and Uropathogenic Escherichia coli Human Strains. J. Nutr. 2002, 132, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Cravioto, A.; Tello, A.; Villafdn, H.; Ruiz, J.; Vedovo, S.; Neeser, J. Inhibition of Localized Adhesion of Enteropathogenic Escherichia coli to HEp-2 Cells by Immunoglobulin and Oligosaccharide Fractions of Human Colostrum and Breast Milk. J. Infect. Dis. 1991, 163, 1247–1255. [Google Scholar] [CrossRef]
- Coppa, G.V.; Zampini, L.; Galeazzi, T.; Facinelli, B.; Ferrante, L.; Capretti, R.; Orazio, G. Human Milk Oligosaccharides Inhibit the Adhesion to Caco-2 Cells of Diarrheal Pathogens: Escherichia coli, Vibrio cholerae, and Salmonella fyris. Pediatr. Res. 2006, 59, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Weichert, S.; Jennewein, S.; Hufner, E.; Weiss, C.; Borkowski, J.; Putze, J.; Schroten, H. Bioengineered 2′-fucosyllactose and 3-fucosyllactose inhibit the adhesion of Pseudomonas aeruginosa and enteric pathogens to human intestinal and respiratory cell lines. Nutr. Res. 2013, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Facinelli, B.; Marini, E.; Magi, G.; Zampini, L.; Santoro, L.; Catassi, C.; Monachesi, C.; Gabrielli, O.; Coppa, G.V. Breast milk oligosaccharides: Effects of 2′-fucosyllactose and 6′-sialyllactose on the adhesion of Escherichia coli and Salmonella fyris to Caco-2 cells. J. Matern.-Fetal Neonatal Med. 2019, 32, 2950–2952. [Google Scholar] [CrossRef]
- Manthey, C.F.; Autran, C.A.; Eckmann, L.; Bode, L. Human Milk Oligosaccharides Protect Against Enteropathogenic Escherichia coli Attachment In Vitro and EPEC Colonization in Suckling Mice. J. Pediatric Gastroenterol. Nutr. 2014, 58, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Tøndervik, A.; Sletta, H.; Klinkenberg, G.; Emanuel, C.; Onsøyen, E.; Myrvold, R.; Howe, R.A.; Walsh, T.R.; Hill, K.E.; et al. Overcoming Drug Resistance with Alginate Oligosaccharides Able To Potentiate the Action of Selected Antibiotics. Antimicrob. Agents Chemother. 2012, 56, 5134–5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.C.; Tavaria, F.K.; Soares, J.C.; Ramos, Ó.S.; Monteiro, M.J.; Pintado, M.E.; Malcata, F.X. Antimicrobial effects of chitosans and chitooligosaccharides, upon Staphylococcus aureus and Escherichia coli, in food model systems. Food Microbiol. 2008, 25, 922–928. [Google Scholar] [CrossRef]
- Rhoades, J.; Gibson, G.; Formentin, K.; Beer, M.; Rastall, R. Inhibition of the adhesion of enteropathogenic Escherichia coli strains to HT-29 cells in culture by chito-oligosaccharides. Carbohydr. Polym. 2006, 64, 57–59. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Park, P.J.; Kim, S.K. Antimicrobial effect of chitooligosaccharides produced by bioreactor. Carbohydr. Polym. 2001, 44, 71–76. [Google Scholar] [CrossRef]
- Sánchez, A.; Mengíbar, M.; Rivera-Rodríguez, G.; Moerchbacher, B.; Acosta, N.; Heras, A. The effect of preparation processes on the physicochemical characteristics and antibacterial activity of chitooligosaccharides. Carbohydr. Polym. 2017, 157, 251–257. [Google Scholar] [CrossRef]
- Laokuldilok, T.; Potivas, T.; Kanha, N.; Surawang, S.; Seesuriyachan, P.; Wangtueai, S.; Phimolsiripol, Y.; Regenstein, J.M. Physicochemical, antioxidant, and antimicrobial properties of chitooligosaccharides produced using three different enzyme treatments. Food Biosci. 2017, 18, 28–33. [Google Scholar] [CrossRef]
- Kumar, A.B.V.; Varadaraj, M.C.; Gowda, L.R.; Tharanathan, R.N. Characterization of chito-oligosaccharides prepared by chitosanolysis with the aid of papain and Pronase, and their bactericidal action against Bacillus cereus and Escherichia coli. Biochem. J. 2005, 391, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Shoaf, K.; Mulvey, G.L.; Armstrong, G.D.; Hutkins, R.W. Prebiotic Galactooligosaccharides Reduce Adherence of Enteropathogenic Escherichia coli to Tissue Culture Cells. Infect. Immun. 2006, 74, 6920–6928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Qiao, Y.; Peng, Q.; Gao, L.; Shi, B. Inhibitory effects of YCW and MOS from Saccharomyces cerevisiae on Escherichia coli and Salmonella pullorum adhesion to Caco-2 cells. Front. Biol. 2017, 12, 370–375. [Google Scholar] [CrossRef]
- Bouckaert, J.; MacKenzie, J.; De Paz, J.L.; Chipwaza, B.; Choudhury, D.; Zavialov, A.; Mannerstedt, K.; Anderson, J.; Pierard, D.; Wyns, L.; et al. The affinity of the FimH fimbrial adhesin is receptor-driven and quasi-independent of Escherichia coli pathotypes. Mol. Microbiol. 2006, 61, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Martinov, J.; Krstić, M.; Spasić, S.; Miletić, S.; Stefanović-Kojić, J.; Nikolić-Kokić, A.; Blagojević, D.; Spasojević, I.; Spasić, M.B. Apple pectin-derived oligosaccharides produce carbon dioxide radical anion in Fenton reaction and prevent growth of Escherichia coli and Staphylococcus aureus. Food Res. Int. 2017, 100, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Rhoades, J.; Manderson, K.; Wells, A.; Hotchkiss, A.T.; Gibson, G.R.; Formentin, K.; Beer, M.; Rastall, R.A. Oligosaccharide-Mediated Inhibition of the Adhesion of Pathogenic Escherichia coli Strains to Human Gut Epithelial Cells In Vitro. J. Food Prot. 2008, 71, 2272–2277. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-J.; Xia, J.-L.; Nie, Z.-Y.; Shan, Y. Pectic oligosaccharides hydrolyzed from orange peel by fungal multi-enzyme complexes and their prebiotic and antibacterial potentials. LWT 2016, 69, 203–210. [Google Scholar] [CrossRef]
- Li, S.; Li, T.; Zhu, R.; Wang, N.; Song, Y.; Wang, S.; Guo, M. Antibacterial Action of Haw Pectic Oligosaccharides. Int. J. Food Prop. 2013, 16, 706–712. [Google Scholar] [CrossRef]
- Benhabiles, M.; Salah, R.; Lounici, H.; Drouiche, N.; Goosen, M.; Mameri, N. Antibacterial activity of chitin, chitosan and its oligomers prepared from shrimp shell waste. Food Hydrocoll. 2012, 29, 48–56. [Google Scholar] [CrossRef]
- Kyoon, H.; Young, N.; Ho, S.; Meyers, S.P. Antibacterial activity of chitosans and chitosan oligomers with different molecular weights. Int. J. Food Microbiol. 2002, 74, 65–72. [Google Scholar]
- Zheng, L.-Y.; Zhu, J.-F. Study on antimicrobial activity of chitosan with different molecular weights. Carbohydr. Polym. 2003, 54, 527–530. [Google Scholar] [CrossRef]
- Lee, J.-H.; Shim, J.S.; Lee, J.S.; Kim, M.-K.; Chung, M.-S.; Kim, K.H. Pectin-like acidic polysaccharide from Panax ginseng with selective antiadhesive activity against pathogenic bacteria. Carbohydr. Res. 2006, 341, 1154–1163. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Q.; Wang, X.; Li, R.; Qiao, H.; Ma, P.; Sun, Q.; Zhang, H. Antibacterial activity of xanthan-oligosaccharide against Staphylococcus aureus via targeting biofilm and cell membrane. Int. J. Biol. Macromol. 2020, 153, 539–544. [Google Scholar] [CrossRef]
- Piotrowski, M.; Wultańska, D.; Obuch-Woszczatyński, P.; Pituch, H. Fructooligosaccharides and mannose affect Clostridium difficile adhesion and biofilm formation in a concentration-dependent manner. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Di, R.; Vakkalanka, M.S.; Onumpai, C.; Chau, H.K.; White, A.; Rastall, R.A.; Yam, K.; Hotchkiss, A.T. Pectic oligosaccharide structure-function relationships: Prebiotics, inhibitors of Escherichia coli O157:H7 adhesion and reduction of Shiga toxin cytotoxicity in HT29 cells. Food Chem. 2017, 227, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Almagro-Moreno, S.; Pruss, K.; Taylor, R.K. Intestinal Colonization Dynamics of Vibrio cholerae. PLoS Pathog. 2015, 11, e1004787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.-K.; Kim, K.-Y.; Yoo, Y.-J.; Oh, S.-J.; Choi, J.-H.; Kim, C.-Y. In vitro antimicrobial activity of a chitooligosaccharide mixture against Actinobacillus actinomycetemcomitans and Streptococcus mutans. Int. J. Antimicrob. Agents 2001, 18, 553–557. [Google Scholar] [CrossRef]
- Liu, H.; Du, Y.; Wang, X.; Sun, L. Chitosan kills bacteria through cell membrane damage. Int. J. Food Microbiol. 2004, 95, 147–155. [Google Scholar] [CrossRef]
- Reffuveille, F.; Josse, J.; Vallé, Q.; Mongaret, C.; Gangloff, S.C. Staphylococcus aureus Biofilms and their Impact on the Medical Field. In The Rise of Virulence and Antibiotic Resistance in Staphylococcus aureus; IntechOpen: London, UK, 2017. [Google Scholar]
- Tin, S.; Lim, C.; Sakharkar, M.; Sakharkar, K. Synergistic Combinations of Chitosans and Antibiotics in Staphylococcus aureus. Lett. Drug Des. Discov. 2010, 7, 31–35. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, H.K.; Koo, H.C.; Joo, Y.S.; Nam, H.M.; Park, Y.H.; Kang, M. Il The antibacterial and immunostimulative effect of chitosan-oligosaccharides against infection by Staphylococcus aureus isolated from bovine mastitis. Appl. Microbiol. Biotechnol. 2007, 75, 989–998. [Google Scholar] [CrossRef]
- Fei Liu, X.; Lin Guan, Y.; Zhi Yang, D.; Yao, K. DE Antibacterial Action of Chitosan and Carboxymethylated Chitosan. J. Appl. Polym. 2000, 79, 1324–1335. [Google Scholar] [CrossRef]
- Valcheva, R.; Dieleman, L.A. Prebiotics: Definition and protective mechanisms. Best Pr. Res. Clin. Gastroenterol. 2016, 30, 27–37. [Google Scholar] [CrossRef]
- Servin, A.L. Antagonistic activities of lactobacilli and bifidobacteria against microbial pathogens. FEMS Microbiol. Rev. 2004, 28, 405–440. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, A.; Clinthorne, J.F.; Rondini, E.A.; McCaskey, S.J.; Gurzell, E.A.; Langohr, I.M.; Gardner, E.M.; Fenton, J.I. Supplementation with Galacto-Oligosaccharides Increases the Percentage of NK Cells and Reduces Colitis Severity in Smad3-Deficient Mice. J. Nutr. 2012, 142, 1336–1342. [Google Scholar] [CrossRef]
- Ringel, Y. Using Probiotics in Gastrointestinal Disorders Original Contributions. Am. J. Gastroenterol. Suppl. 2012, 1, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Chin, J. Modulating immune responses with probiotic bacteria. Immunol. Cell Biol. 2000, 78, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Cherbut, C.; Michel, C.; Rard Lecannu, G. Biochemical and Molecular Actions of Nutrients the Prebiotic Characteristics of Fructooligosaccharides Are Necessary for Reduction of TNBS-Induced Colitis in Rats; National Institute for Agricultural Research, Gut Function and Human Nutrition Unit: Nantes, France, 2003; Volume 133. [Google Scholar]
- He, Y.; Liu, S.; Kling, D.E.; Leone, S.; Lawlor, N.T.; Huang, Y.; Feinberg, S.B.; Hill, D.R.; Newburg, D.S. The human milk oligosaccharide 2′-fucosyllactose modulates CD14 expression in human enterocytes, thereby attenuating LPS-induced inflammation. Gut 2014, 65, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Akbari, P.; Braber, S.; Alizadeh, A.; Verheijden, K.A.T.; Schoterman, M.H.C.; Kraneveld, A.D.; Garssen, J.; Fink-gremmels, J. Galacto-oligosaccharides Protect the Intestinal Barrier by Maintaining the Tight Junction Network and Modulating the Inflammatory Responses after a Challenge with the Mycotoxin Deoxynivalenol in Human Caco-2 Cell. J. Nutr. 2015, 145, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Folkerts, J.; Folkerts, G.; Maurer, M.; Braber, S. Microbiota-dependent and-independent effects of dietary fibre on human health. Br. J. Pharmacol. 2020, 177, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeurink, P.V.; Van Esch, B.C.A.M.; Rijnierse, A.; Garssen, J.; Knippels, L.M.J. Mechanisms underlying immune effects of dietary oligosaccharides. Am. J. Clin. Nutr. 2013, 98, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Kleessen, B.; Blaut, M. Modulation of gut mucosal biofilms. Br. J. Nutr. 2005, 93, S35–S40. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta Biomembr. 2009, 1788, 872–891. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Possemiers, S.; Van De Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Kleessen, B.; Hartmann, L.; Blaut, M. Fructans in the diet cause alterations of intestinal mucosal architecture, released mucins and mucosa-associated bifidobacteria in gnotobiotic rats. Br. J. Nutr. 2003, 89, 597–606. [Google Scholar] [CrossRef]
- Plöger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.-D.; Gäbel, G.; Martens, H.; Shen, Z.; Günzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Fachi, J.L.; De, J.; Felipe, S.; Passariello, L.; Dos, A.; Farias, S.; Varga-Weisz, P.; Auré, M.; Vinolo, R. Butyrate Protects Mice from Clostridium difficile-Induced Colitis through an HIF-1-Dependent Mechanism. Cell Rep. 2019, 27, 750.e7–761.e7. [Google Scholar] [CrossRef] [Green Version]
- Bilotta, A.J.; Cong, Y. Gut microbiota metabolite regulation of host defenses at mucosal surfaces: Implication in precision medicine. Precis. Clin. Med. 2019, 2, 110–119. [Google Scholar] [CrossRef]
- Paassen, N.B.-V.; Vincent, A.; Puiman, P.J.; Van Der Sluis, M.; Bouma, J.; Boehm, G.; Van Goudoever, J.B.; Van Seuningen, I.; Renes, I.B. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: Implications for epithelial protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Holscher, H.D.; Davis, S.R.; Tappenden, K.A. Human Milk Oligosaccharides Influence Maturation of Human Intestinal Caco-2Bbe and HT-29 Cell Lines. J. Nutr. 2014, 144, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Prabhu, P.N.; Benefiel, A.C.; Miller, M.J.; Chow, J.; Davis, S.R.; Gaskins, H.R. Galacto-oligosaccharides may directly enhance intestinal barrier function through the modulation of goblet cells. Mol. Nutr. Food Res. 2015, 59, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Shimizu, K.; Nomoto, K.; Hamabata, T.; Ozawa, A.; Takeda, Y. Probiotic Bifidobacteria Protect Mice from Lethal Infection with Shiga Toxin-Producing Escherichia coli O157:H7. Infect. Immun. 2004, 72, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.B.; Leung, P.S. Intestinal water and electrolyte transport. In The Gastrointestinal System: Gastrointestinal, Nutritional and Hepatobiliary Physiology; Springer: Dordrecht, The Netherlands, 2014; pp. 107–134. ISBN 9789401787710. [Google Scholar]
- Muanprasat, C.; Wongkrasant, P.; Satitsri, S.; Moonwiriyakit, A.; Pongkorpsakol, P.; Mattaveewong, T.; Pichyangkura, R.; Chatsudthipong, V. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: Mechanism of action and potential applications in intestinal disorders. Biochem. Pharmacol. 2015, 96, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, H.R.; Smejkal, C.W.; Glister, C.; Kemp, F.; Van Den Heuvel, De Slegte, E. J.; Gibson, G.R.; Rastall, R.A. Sialyloligosaccharides inhibit cholera toxin binding to the GM1 receptor. Carbohydr. Res. 2008, 343, 2589–2594. [Google Scholar] [CrossRef]
- Idota, T.; Kawakami, H.; Murakami, Y.; Sugawara, M. Inhibition of Cholera Toxin by Human Milk Fractions and Sialyllactose. Biosci. Biotechnol. Biochem. 1995, 59, 417–419. [Google Scholar] [CrossRef]
- Sinclair, H.R.; De Slegte, J.; Gibson, G.R.; Rastall, R.A. Galactooligosaccharides (GOS) Inhibit Vibrio cholerae Toxin Binding to Its GM1 Receptor. J. Agric. Food Chem. 2009, 57, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Wands, A.M.; Mettlen, M.; Krishnamurthy, S.; Wu, H.; Kohler, J.J. Cell type and receptor identity regulate cholera toxin subunit B (CTB) internalization. Interface Focus 2019, 9, 20180076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wands, A.M.; Cervin, J.; Huang, H.; Zhang, Y.; Youn, G.; Brautigam, C.A.; Dzebo, M.M.; Björklund, P.; Wallenius, V.; Bright, D.K.; et al. Fucosylated Molecules Competitively Interfere with Cholera Toxin Binding to Host Cells. ACS Infect. Dis. 2018, 4, 758–770. [Google Scholar] [CrossRef]
- El-Hawiet, A.; Kitova, E.N.; Klassen, J.S. Recognition of human milk oligosaccharides by bacterial exotoxins. Glycobiology 2015, 25, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Pukin, A.V.; Branderhorst, H.M.; Sisu, C.; Weijers, C.A.G.M.; Gilbert, M.; Liskamp, R.M.J.; Visser, G.M.; Zuilhof, H.; Pieters, R.J. Strong Inhibition of Cholera Toxin by Multivalent GM1 Derivatives. ChemBioChem 2007, 8, 1500–1503. [Google Scholar] [CrossRef]
- Ommen, D.D.Z.-V.; Pukin, A.V.; Fu, O.; Van Ufford, L.H.Q.; Janssens, H.M.; Beekman, J.M.; Pieters, R.J. Functional Characterization of Cholera Toxin Inhibitors Using Human Intestinal Organoids. J. Med. Chem. 2016, 59, 6968–6972. [Google Scholar] [CrossRef]
- Haksar, D.; De Poel, E.; Van Ufford, L.H.C.Q.; Bhatia, S.; Haag, R.; Beekman, J.M.; Pieters, R.J. Strong Inhibition of Cholera Toxin B Subunit by Affordable, Polymer-Based Multivalent Inhibitors. Bioconjugate Chem. 2019, 30, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Haksar, D.; Van Ufford, L.Q.; Pieters, R.J. A hybrid polymer to target blood group dependence of cholera toxin. Org. Biomol. Chem. 2019, 18, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, G.H.; Albert, M.J.; Rahman, H.; Chowdhury, A.K. Short-chain fatty acids inhibit fluid and electrolyte loss induced by cholera toxin in proximal colon of rabbit in vivo. Dig. Dis. Sci. 1999, 44, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- El-Hawiet, A.; Kitova, E.N.; Kitov, P.I.; Eugenio, L.; Ng, K.K.; Mulvey, G.L.; Dingle, T.C.; Szpacenko, A.; Armstrong, G.D.; Klassen, J.S. Binding of Clostridium difficile toxins to human milk oligosaccharides. Glycobiology 2011, 21, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhard, R. Receptors and binding structures for clostridium difficile toxins A and B. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 406, pp. 79–96. [Google Scholar]
- Nguyen, T.T.H.; Kim, J.W.; Park, J.-S.; Hwang, K.H.; Jang, T.-S.; Kim, C.-H.; Kim, D. Identification of Oligosaccharides in Human Milk Bound onto the Toxin A Carbohydrate Binding Site of Clostridium difficile. J. Microbiol. Biotechnol. 2016, 26, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Koleva, P.T.; Valcheva, R.S.; Sun, X.; Gänzle, M.G.; Dieleman, L.A. Inulin and fructo-oligosaccharides have divergent effects on colitis and commensal microbiota in HLA-B27 transgenic rats. Br. J. Nutr. 2012, 108, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Mackie, R.I.; Fahey, G.C.; Cremin, J.C.; Garleb, K.A. Effect of Fiber Source on Short-Chain Fatty Acid Production and on the Growth and Toxin Production by Clostridium difficile. Scand. J. Gastroenterol. 1994, 29, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Veshnyakova, A.; Protze, J.; Rossa, J.; Blasig, I.E.; Krause, G.; Piontek, J. On the Interaction of Clostridium perfringens Enterotoxin with Claudins. Toxins 2010, 2, 1336–1356. [Google Scholar] [CrossRef] [Green Version]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of Tight Junction Permeability by Intestinal Bacteria and Dietary Components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Wrigley, D.M. Inhibition of Clostridium perfringens sporulation by Bacteroides fragilis and short-chain fatty acids. Anaerobe 2004, 10, 295–300. [Google Scholar] [CrossRef]
- Newburg, D.S.; Pickering, L.K.; McCluer, R.H.; Cleary, T.G. Fucosylated Oligosaccharides of Human Milk Protect Suckling Mice from Heat-Stabile Enterotoxin of Escherichia coli. J. Infect. Dis. 1990, 162, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Azar, S.S.; Stam, A.; Newburg, D.S. Oligosaccharides from Human Milk Block Binding and Activity of the Escherichia coli Heat-Stable Enterotoxin (STa) in T84 Intestinal Cells. J. Nutr. 1994, 124, 2358–2364. [Google Scholar] [CrossRef]
- Anand, S.; Mandal, S.; Tomar, S.K. Effect of Lactobacillus rhamnosus NCDC 298 with FOS in Combination on Viability and Toxin Production of Enterotoxigenic Escherichia coli. Probiotics Antimicrob. Proteins 2017, 11, 23–29. [Google Scholar] [CrossRef]
- Takashi, K.; Fujita, I.; Kobari, K. Effects of short chain fatty acids on the production of heat-labile enterotoxin from enterotoxigenic Escherichia coli. Jpn. J. Pharmacol. 1989, 50, 495–498. [Google Scholar] [CrossRef]
- Olano-Martin, E.; Williams, M.R.; Gibson, G.R.; Rastall, R.A. Pectins and pectic-oligosaccharides inhibit Escherichia coli O157:H7 Shiga toxin as directed towards the human colonic cell line HT29. FEMS Microbiol. Lett. 2003, 218, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Di, R.; Kyu, E.; Shete, V.; Saidasan, H.; Kahn, P.C.; Tumer, N.E. Identification of amino acids critical for the cytotoxicity of Shiga toxin 1 and 2 in Saccharomyces Cerevisiae. Toxicon 2011, 57, 525–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, C.M.; Kostrzynska, M.; Ojha, S.; Thompson, S. The effect of probiotics and organic acids on Shiga-toxin 2 gene expression in enterohemorrhagic Escherichia coli O157:H7. J. Microbiol. Methods 2008, 73, 125–132. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Taylor, T.D.; Ohno, H.; Hattori, M. Acetate-producing bifidobacteria protect the host from enteropathogenic infection via carbohydrate transporters. Gut Microbes 2012, 3, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nat. Cell Biol. 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Zumbrun, S.D.; Melton-Celsa, A.R.; Smith, M.A.; Gilbreath, J.J.; Merrell, D.S.; O’Brien, A.D. Dietary choice affects Shiga toxin-producing Escherichia coli (STEC) O157:H7 colonization and disease. Proc. Natl. Acad. Sci. USA 2013, 110, E2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davie, J.R. Nutritional Proteomics in Cancer Prevention Inhibition of Histone Deacetylase Activity. J. Nutr. 2003, 133, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nat. Cell Biol. 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Fredholm, S.; Willerslev-Olsen, A.; Hansen, M.; Bonefeld, C.M.; Geisler, C.; Andersen, M.H.; Ødum, N.; Woetmann, A. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M.; Slavin, J. Nondigestible. Crit. Rev. Food Sci. Nutr. 2000, 40, 461–480. [Google Scholar] [CrossRef] [PubMed]
- De Toro-Martín, J.; Arsenault, B.J.; Després, J.-P.; Vohl, M.-C. Precision Nutrition: A Review of Personalized Nutritional Approaches for the Prevention and Management of Metabolic Syndrome. Nutritiens 2017, 9, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Non-Digestible Oligosaccharides | Structures |
|---|---|
| Alginate-oligosaccharides (AOS) |  |
| Chito-oligosaccharides/Chitosan-oligosaccharides (COS) |  |
| Fructo-oligosaccharides (FOS) |  |
| Galacto-oligosaccharides (GOS) |  |
| Human milk oligosaccharides (HMOs) |  |
| Isomalto-oligosaccharides (IMOS) |  |
| Mannan-oligosaccharides (MOS) |  |
| Pectic-oligosaccharides (POS) |  |
| Xylo-oligosaccharides (XOS) |  |
 ); fucose (Fuc,
); fucose (Fuc,  ); galactose(Gal,
); galactose(Gal,  ); galacturonic acid (GalA,
); galacturonic acid (GalA,  ); glucosamine (GlcN,
); glucosamine (GlcN,  ); N-acetylglucosamine (GlcNAc,
); N-acetylglucosamine (GlcNAc,  ); glucose (Glc,
); glucose (Glc,  ); guluronic acid (GulA,
); guluronic acid (GulA,  ); mannose (Man,
); mannose (Man,  ); mannuronic acid (ManA,
); mannuronic acid (ManA,  ); rhamnose (Rha,
); rhamnose (Rha,  ); sialic acid (Sia,
); sialic acid (Sia,  ); xylose (Xyl,
); xylose (Xyl,  ).
).| NDOs | Beneficial Bacteria | Bacteria Producing Enterotoxins | Mechanism of Action | Health Benefit | Ref. |
|---|---|---|---|---|---|
| FOS |
| Enteropathogenic Escherichia coli (EPEC), Clostridium difficile | Not investigated | Growth inhibition | [108] |
| FOS, inulin | Lactobacillus rhamnosus | Enterotoxigenic Escherichia coli (ETEC) | Decrease in cAMP and cGMP levels | Anti-virulence activity | [109] |
| FOS, inulin |
| Staphylococcus aureus, Escherichia coli | Production of metabolites by beneficial bacteria | Anti-biofilm, anti-adhesive, antimicrobial effect | [110] |
| FOS, XOS, inulin, mixtures of inulin:FOS (80:20 w/w) and FOS:XOS (50:50 w/w) |
| Escherichia coli | Production of SCFA | Growth inhibition of Escherichia coli | [111] |
| XOS |
| Clostridium perfringens | Production of SCFA | Growth suppression of Clostridium Perfringens | [103] |
| GOS | Bifidobacterium breve | Methicillin-resistant Staphylococcus aureus (MRSA) | High production of acetic acid (low pH) | Prevention of systemic MRSA infection | [112] |
| GOS, IMOS |
| Clostridium difficile | Stimulation of gut microflora | Protective effect against CDI | [113] |
| Bacteria | Toxins | NDOs | Mechanism of Action | Ref. |
|---|---|---|---|---|
| Escherichia coli | Stx, LTs, STs | Pooled HMOs, 3′SL, 6′SL, DSLNT, 6SLN, 3′S3FL, LST a | Inhibition of hemagglutination | [114] |
| Fucosylated HMOs | Anti-adhesive effect | [115] | ||
| Pooled HMOs, 3-FL, 3′SL, 6′SL, acidic and neutral (HMW and LMW) HMOs | Anti-adhesive effect | [116] | ||
| 2′FL, 3-FL | Anti-adhesive effect | [117] | ||
| 2′FL, 6′SL | Anti-adhesive effect | [118] | ||
| Pooled HMOs | Reduced enteropathogenic E. coli (EPEC) attachment to epithelial cells | [119] | ||
| AOS | Inhibition of biofilm formation | [120] | ||
| COS | Antimicrobial effect | [121] | ||
| Anti-adhesive effect | [122] | |||
| Antibacterial effect | [123,124] | |||
| (growth inhibition) | ||||
| Antioxidant and antimicrobial effect | [125] | |||
| Membrane disruption (growth inhibition) | [126] | |||
| GOS | Anti-adhesive effect | [127] | ||
| MOS | Anti-adhesive effect | [128,129] | ||
| POS | Growth inhibition | [130] | ||
| Anti-adhesive effect | [131] | |||
| Antimicrobial effect | [132,133] | |||
| Vibrio cholerae | CT | Pooled HMOs | Anti-adhesive effect (neutral, HMW) | [116] |
| Isolated HMOs | No anti-adhesive effect (LMW) | [116] | ||
| COS, NAc-COS | Bactericidal effect | [134] | ||
| Bacillus cereus | Hbl, Nhe, CytK | COS | Antibacterial effect | [134,135] |
| (growth inhibition) | ||||
| Membrane disruption (growth inhibition) | [126] | |||
| Staphylococcus aureus | SEs | Pooled HMOs | Inhibition of biofilm formation | [105] |
| COS | Antibacterial effect | [123,134,136] | ||
| (growth inhibition) | ||||
| Antioxidant and antimicrobial effect | [125] | |||
| POS | Growth inhibition | [130] | ||
| Antimicrobial effect | [132] | |||
| Anti-adhesive effect | [137] | |||
| XOS | Antibacterial effect | [138] | ||
| Clostridium spp. | TcdA, TcdB, CDT, CPE, CPB | FOS | Anti-adhesive effect Inhibition of biofilm formation | [139] |
| Enterotoxins | NDOs/SCFA | Mechanism of Action | Health Benefit | Ref. |
|---|---|---|---|---|
| Cholera toxin | COS | Activation of AMPK | Reduction in CT-induced intestinal fluid secretion | [171] |
| Cholera toxin | SOS | Mimicking GM1 receptor | Anti-adhesive effect | [172] |
| Cholera toxin | Pooled HMOs | GM1 mimicking | Inhibition of sialyllactose on fluid accumulation induced by CT | [173] |
| Cholera toxin | GOS | Mimicking GM1 receptor | Anti-adhesive effect | [174] |
| Cholera toxin | 2′-FL | 2′-FL resembles fucosylated glycan epitopes | Inhibition of CTB binding | [176] |
| Cholera Toxin | 2′FL, LNnT, LNFP I, II, III | Binding with CTB | No inhibitory effect | [177] |
| Cholera toxin | Acetate, propionate, butyrate | Reduction in water and electrolyte secretion | Anti-diarrheal effects | [182] |
| Shiga toxins | 2′FL, LNnT, LNFP I, II, III | Binding with STX | No inhibitory effect | [177] |
| Shiga toxins | POS | POS mimic the interaction with the galabiose receptor | Inhibition of Stx | [195] |
| Shiga toxins | POS | Reduction in rRNA depurination | Reduction in Stx cytotoxicity | [140] |
| Shiga toxins | Acetic acid, Lactic acid | Decrease in pH | Reduction in stx2 gene expression | [169,197] |
| Shiga Toxins | Acetate | Reduction in transepithelial electrical resistance | Prevention of Stx translocation | [198,199] |
| Heat-stable enterotoxin (ST) | Fucosylated HMOs | not determined | Protective effect (suckling mice) | [191] |
| Heat-stable enterotoxin (ST) | Pooled HMOs, Fucosylated HMOs | Block of human guanylate cyclase activation by allosteric binding to the STa receptor | Protective effect (T84 cells) | [192] |
| Heat-labile enterotoxin (LT) | FOS | Bifidogenic effect with L. rhamnosus | Reduction in growth and toxin production | [193] |
| Heat-labile enterotoxin type 1 (LT-1) | 2′FL, LNnT, LNFP I, II, III | Binding with LTB | No inhibitory effect | [177] |
| Heat-labile enterotoxin (LT) | Acetic, propionic, butyric, valeric, caproic and heptylic acids | Effects on the biosynthesis of LT | Reduction in LT production | [194] |
| C. difficile toxin B (TcdB) | FOS, inulin | Inhibiting toxin-related gene expression | Anti-inflammatory effect | [186] |
| C. difficile toxins A (TcdA) and B (TcdB) | Pooled HMOs | Mimicking the structure of cellular receptors | Inhibition of toxins’ binding to cellular receptors | [185] |
| C. difficile toxins A (TcdA) and B (TcdB) | Acetate, propionate, butyrate | Decrease in pH | Prevention of growth and elaboration of toxin (colonization resistance) | [187] |
| C. difficile toxins A (TcdA) and B (TcdB) | Butyrate | Stabilization of HIF-1 | Anti-inflammatory effects and intestinal barrier function improvement | [164] |
| C. perfringens enterotoxin | Acetate, isobutyrate, isovalerate, succinate | Inhibition of toxin sporulation | Reduction in enterotoxin cytotoxicity | [190] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asadpoor, M.; Ithakisiou, G.-N.; Henricks, P.A.J.; Pieters, R.; Folkerts, G.; Braber, S. Non-Digestible Oligosaccharides and Short Chain Fatty Acids as Therapeutic Targets against Enterotoxin-Producing Bacteria and Their Toxins. Toxins 2021, 13, 175. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030175
Asadpoor M, Ithakisiou G-N, Henricks PAJ, Pieters R, Folkerts G, Braber S. Non-Digestible Oligosaccharides and Short Chain Fatty Acids as Therapeutic Targets against Enterotoxin-Producing Bacteria and Their Toxins. Toxins. 2021; 13(3):175. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030175
Chicago/Turabian StyleAsadpoor, Mostafa, Georgia-Nefeli Ithakisiou, Paul A. J. Henricks, Roland Pieters, Gert Folkerts, and Saskia Braber. 2021. "Non-Digestible Oligosaccharides and Short Chain Fatty Acids as Therapeutic Targets against Enterotoxin-Producing Bacteria and Their Toxins" Toxins 13, no. 3: 175. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030175