Dispersive Liquid–Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development, Optimization and Validation



Abstract
:1. Introduction
2. Results and Discussion
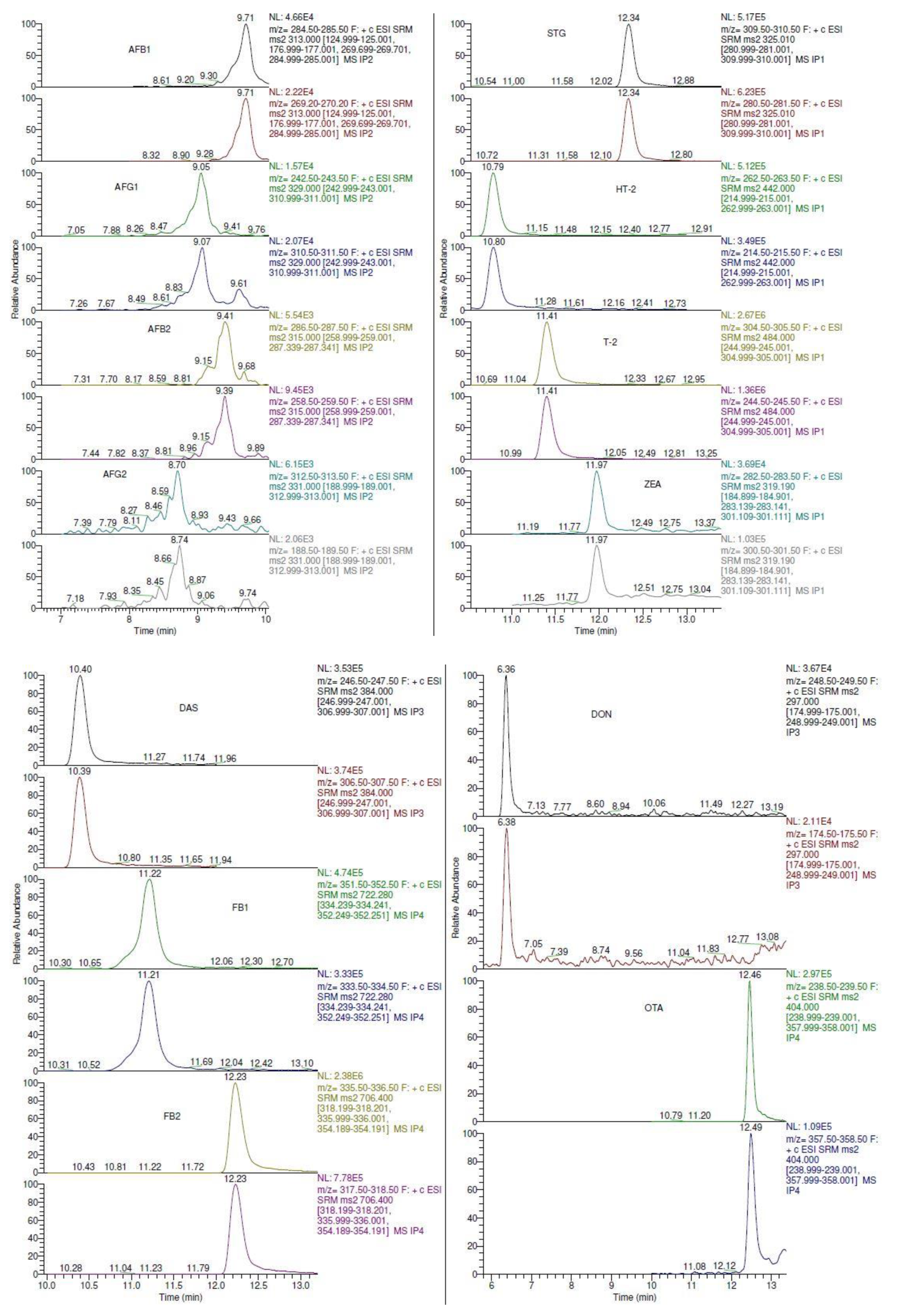
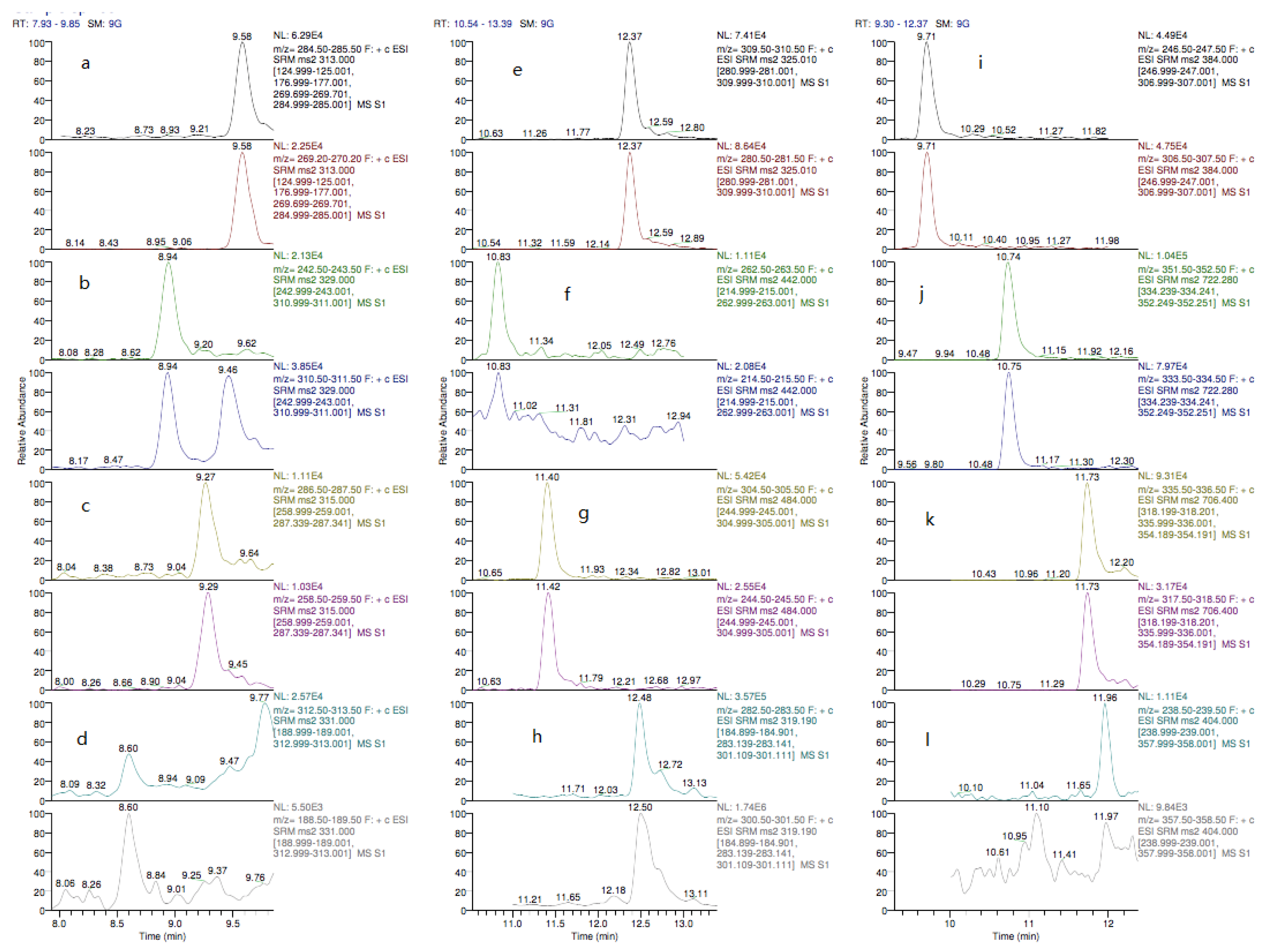
2.1. Liquid Chromatography Mass Spectrometry (LC-MS/MS) Optimization
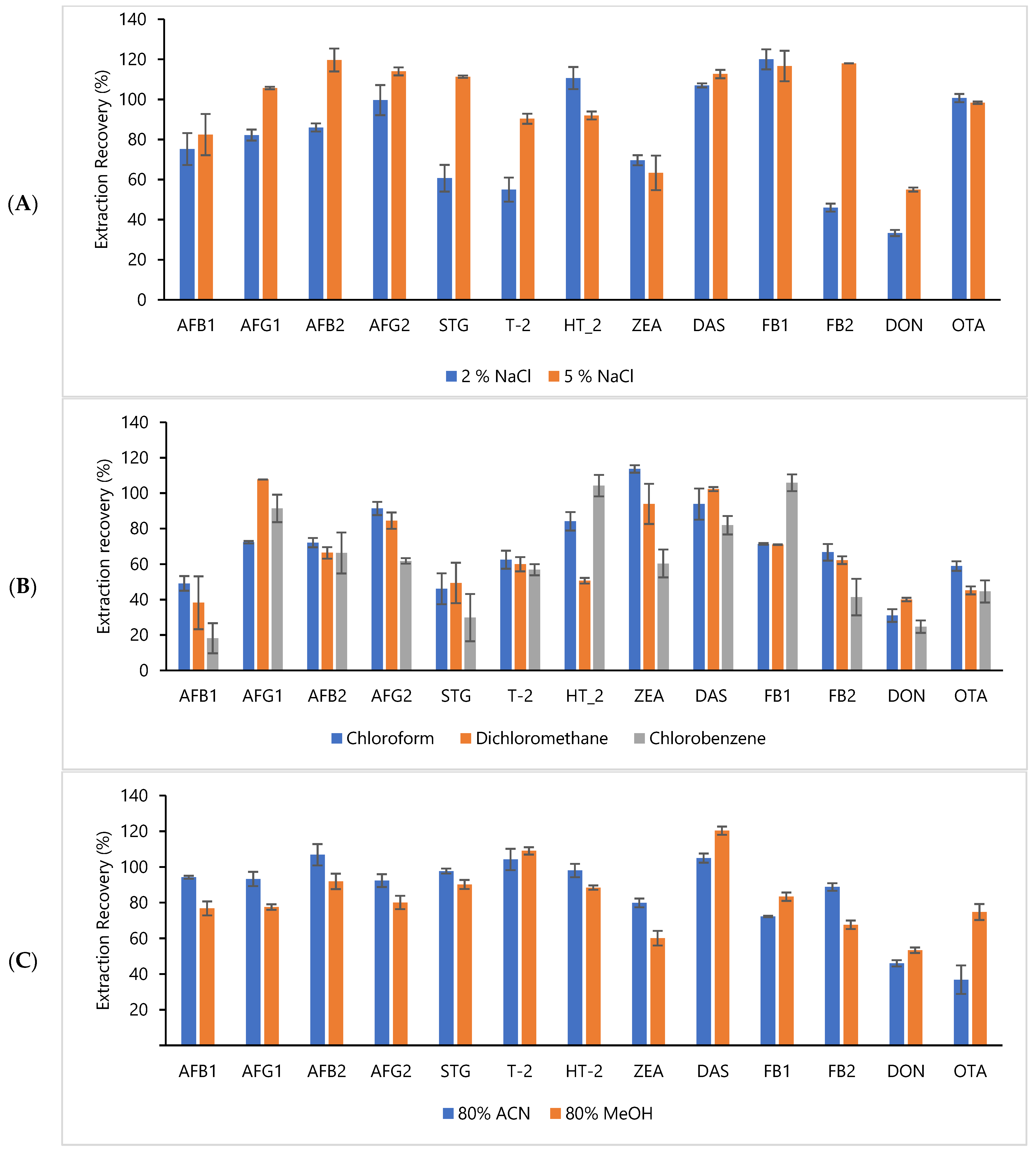
2.2. Dispersive Liquid–Liquid Microextraction (DLLME)
Selection of Dispersive Solvent, Extraction Solvent and Addition of Salt for Extraction of Mycotoxin in Rice Bran
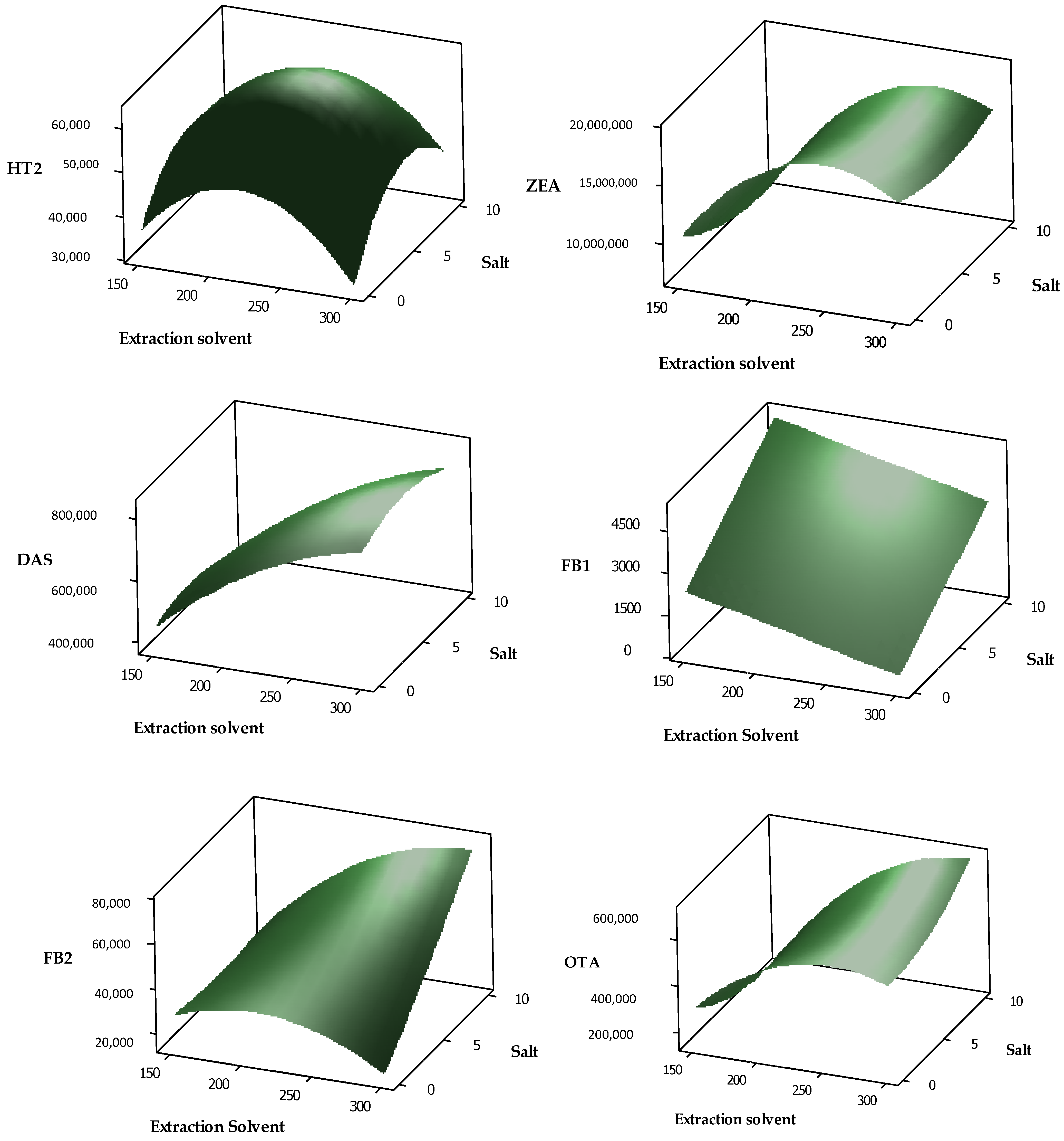
2.3. Optimising of DLLME by Box-Behnken Approach and Response Optimization Using Composite Desirability
2.4. Validation
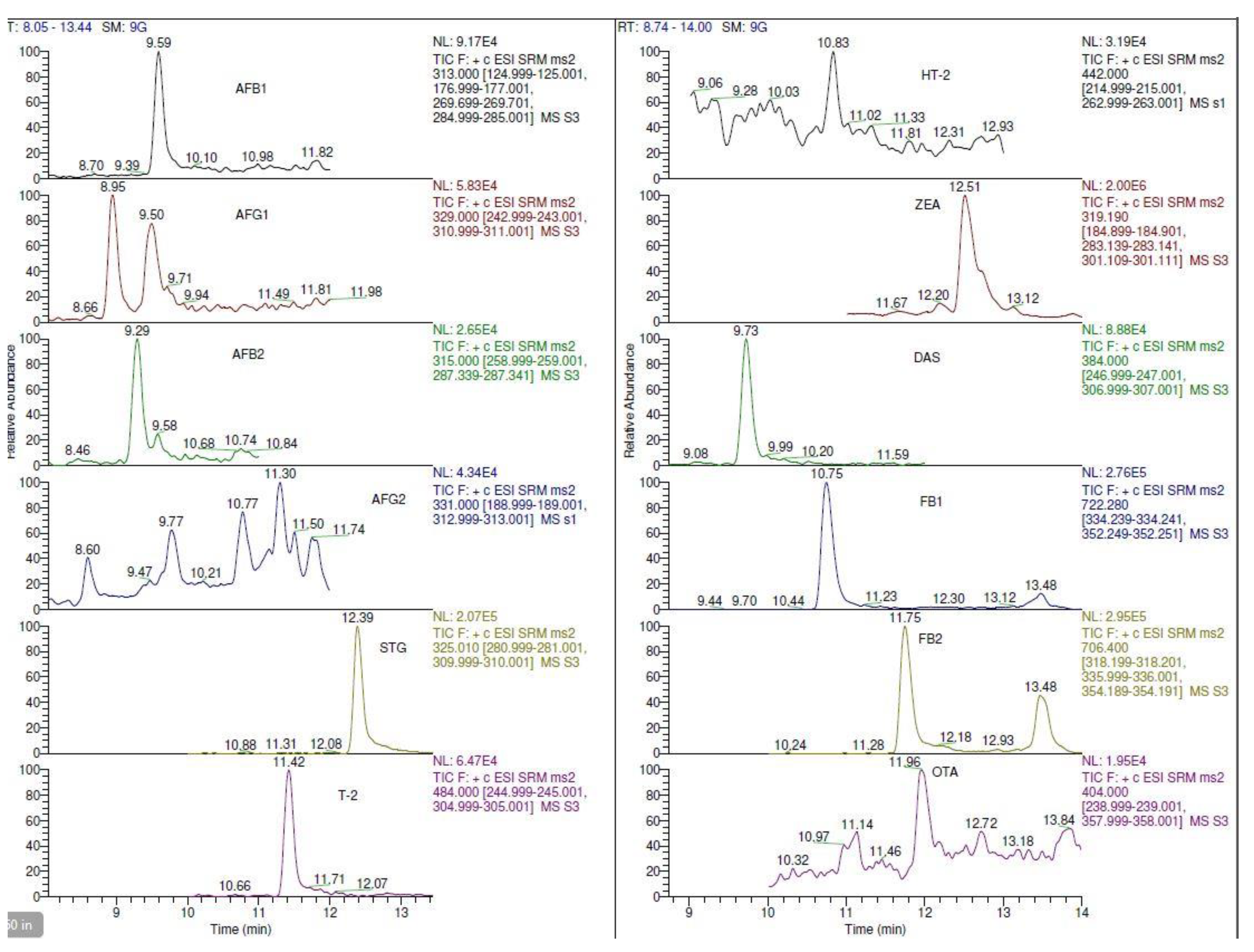
2.4.1. Specificity and Matrix-Effects (ME)
2.4.2. Limit of Detection (LOD), Limit of Quantitation (LOQ) and Linearity
2.4.3. Accuracy, Precision and Recovery
2.4.4. Occurrence of Multi-Mycotoxin in Industrial and Commercial Samples
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrumental and Analytical Conditions
4.3. Sample Preparation
4.3.1. Extraction Procedure
4.3.2. Dispersive Liquid–Liquid Microextraction Procedure
4.3.3. Selection of Dispersive Solvent, Extraction Solvent and Addition of Salt
4.3.4. Response Surface Methodology Using Box–Behnken Design
4.3.5. Response Optimization
4.4. Validation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohd Esa, N.; Ling, T.B. By-products of rice processing: An overview of health benefits and applications. J. Rice Res. 2013, 1, 1–11. [Google Scholar] [CrossRef]
- Sharif, M.K.; Butt, M.S.; Anjum, F.M. Rice bran: A novel functional ingredient rice bran. Food Sci. Nutr. 2014, 54, 807–816. [Google Scholar] [CrossRef]
- Alauddin, M.; Islam, J.; Shirakawa, H.; Koseki, T.; Ardiansyah; Komai, M. Rice bran as a functional food: An overview of the conversion of rice bran into a superfood/functional food. In Superfood and Functional Food—An Overview of Their Processing and Utilization; Waisundara, V., Shiomi, N., Eds.; InTechOpen: London, UK, 2017; pp. 291–305. [Google Scholar]
- Gul, K.; Yousuf, B.; Singh, A.K.; Singh, P.; Wani, A.A. Rice bran: Nutritional values and its emerging potential for development of functional food—A review. Bioact. Carbohydrates Diet. Fibre 2015, 6, 24–30. [Google Scholar] [CrossRef]
- Jayaraman, P.; Kalyanasundaram, I. Natural occurrence of toxigenic fungi and mycotoxins in rice bran. Mycopathologia 1990, 110, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Sempere Ferre, F.; Pilar Santamarina, M. Efficacy of Trichoderma harzianum in suppression of Fusarium culmorum. Ann. Microbiol. 2010, 60, 335–340. [Google Scholar] [CrossRef]
- Balendres, M.A.O.; Karlovsky, P.; Cumagun, C.J.R. Mycotoxigenic fungi and mycotoxins in agricultural crop commodities in the Philippines: A review. Foods 2019, 8, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scudamore, K.A.; Nawaz, S.; Hetmanski, M.T. Mycotoxins in ingredients of animal feeding stuffs: II. Determination of mycotoxins in maize and maize products. Food Addit. Contam. 1998, 15, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, T.V.P.; Kalyanasundaram, I. Prevalence of aflatoxin B1 in rice bran and some associated factors. Indian Phytopathol. 1999, 52, 129–133. [Google Scholar]
- Bueno, D.; Istamboulie, G.; Muñoz, R.; Marty, J.L. Determination of mycotoxins in food: A review of bioanalytical to analytical methods. Appl. Spectrosc. Rev. 2015, 50, 728–774. [Google Scholar] [CrossRef]
- Kunz, B.M.; Wanko, F.; Kemmlein, S.; Bahlmann, A.; Rohn, S.; Maul, R. Development of a rapid multi-mycotoxin LC-MS/MS stable isotope dilution analysis for grain legumes and its application on 66 market samples. Food Control 2020, 109, 1–10. [Google Scholar] [CrossRef]
- De Ruyck, K.; Huybrechts, I.; Yang, S.; Arcella, D.; Claeys, L.; Abbeddou, S.; De Keyzer, W.; De Vries, J.; Ocken, M.; Ruprich, J.; et al. Mycotoxin exposure assessments in a multi-center European validation study by 24-hour dietary recall and biological fluid sampling. Environ. Int. 2020, 137, 1–20. [Google Scholar] [CrossRef]
- Arce-López, B.; Lizarraga, E.; Flores-Flores, M.; Irigoyen, Á.; González-Peñas, E. Development and validation of a methodology based on Captiva EMR-lipid clean-up and LC-MS/MS analysis for the simultaneous determination of mycotoxins in human plasma. Talanta 2020, 206, 1–10. [Google Scholar] [CrossRef]
- Abdullah Al Faris, N.; Zaidan Al Tamimi, J.; Al Othman, Z.A.; Fahad Al Qahtani, S.; Wabaidur, S.M.; Ghfar, A.A.; Saleh Aldayel, T. Analysis of aflatoxins in foods retailed in Saudi Arabia using immunoaffinity column cleanup and high-performance liquid chromatography-fluorescence detection. J. King Saud Univ. Sci. 2020, 32, 1437–1443. [Google Scholar] [CrossRef]
- Sun, D.; Qiu, N.; Zhou, S.; Lyu, B.; Zhang, S.; Li, J.; Zhao, Y.; Wu, Y. Development of sensitive and reliable UPLC-MS/MS methods for food analysis of emerging mycotoxins in China total diet study. Toxins 2019, 11, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsharif, A.; Choo, Y.M.; Tan, G.H. Detection of five mycotoxins in different food matrices in the Malaysian market by using validated liquid chromatography electrospray ionization triple quadrupole mass spectrometry. Toxins 2019, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Sharmili, K.; Jinap, S.; Sukor, R. Development, optimization and validation of QuEChERS based liquid chromatography tandem mass spectrometry method for determination of multimycotoxin in vegetable oil. Food Control 2016, 70, 152–160. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N.; López-García, I.; Hernández-Córdoba, M. Dispersive liquid-liquid microextraction in food analysis. A critical review. Anal. Bioanal. Chem. 2013, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tan, Z.C.; Huang, K.C.; Wen, Y.; Ying Li, X.; Long Zhao, J.; Liu, C.L. A vortex-assisted dispersive liquid-liquid microextraction followed by UPLC-MS/MS for simultaneous determination of pesticides and aflatoxins in herbal tea. Molecules 2019, 24, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Luan, C.; Wang, L.; Wang, S.; Shao, L. Simultaneous determination of six mycotoxins in peanut by high-performance liquid chromatography with a fluorescence detector. J. Sci. Food Agric. 2017, 97, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Pallarés, N.; Carballo, D.; Ferrer, E.; Fernández-Franzón, M.; Berrada, H. Mycotoxin dietary exposure assessment through fruit juices consumption in children and adult population. Toxins 2019, 11, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, G.B.; de Castro Gomes Vieira, C.M.; Orlando, R.M.; Faria, A.F. Simultaneous determination of fumonisins B1 and B2 in different types of maize by matrix solid phase dispersion and HPLC-MS/MS. Food Chem. 2017, 233, 11–19. [Google Scholar] [CrossRef]
- Escrivá, L.; Manyes, L.; Font, G.; Berrada, H. Mycotoxin analysis of human urine by LC-MS/MS: A comparative extraction study. Toxins 2017, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Qiu, L.; Zhang, H.; Sun, J.; Hu, X.; Wang, B. Optimization for the production of deoxynivalenol and zearalenone by Fusarium graminearum using response surface methodology. Toxins 2017, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Han, C.; Chen, S.; Li, L.; Zong, J.; Zeng, J.; Mei, G. Response surface methodology for the optimization of ultrasound-assisted extraction of tetrodotoxin from the liver of takifugu pseudommus. Toxins 2018, 10, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.S.; Wan, J.Z.; Ding, W.J.; Wu, X.F.; Xie, Z.Y. Multi-responses extraction optimization combined with high-performance liquid chromatography-diode array detection-electrospray ionization-tandem mass spectrometry and chemometrics techniques for the fingerprint analysis of Aloe barbadensis Miller. J. Pharm. Biomed. Anal. 2015, 107, 131–140. [Google Scholar] [CrossRef]
- Zhu, Q.; Kakino, K.; Nogami, C.; Ohnuki, K. An LC-MS/MS-SRM method for simultaneous quantification of four representative organosulfur compounds in garlic products. Food Anal. Methods 2016, 9, 3378–3384. [Google Scholar] [CrossRef]
- Azaiez, I.; Giusti, F.; Sagratini, G.; Mañes, J.; Fernández-Franzón, M. Multi-mycotoxins analysis in dried fruit by LC/MS/MS and a modified QuEChERS procedure. Food Anal. Methods 2014, 7, 935–945. [Google Scholar] [CrossRef]
- Di Mavungu, J.D.; Monbaliu, S.; Scippo, M.L.; Maghuin-Rogister, G.; Schneider, Y.-J.; Larondelle, Y.; Callebaut, A.; Robbens, J.; Van Peteghem, C.; De Saeger, S. LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit. Contam. 2009, 26, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Moraleja, A.; Font, G.; Mañes, J.; Ferrer, E. Development of a new method for the simultaneous determination of 21 mycotoxins in coffee beverages by liquid chromatography tandem mass spectrometry. Food Res. Int. 2015, 72, 247–255. [Google Scholar] [CrossRef]
- Paschoal, F.N.; de Azevedo Silva, D.; von Sperling de Souza, R.; Silva de Oliveira, M.; Augusto Alves Pereira, D.; Vitorino Carvalho de Souza, S. A rapid single-extraction method for the simultaneous determination of aflatoxins B1, B2, G1, G2, fumonisin B1, and zearalenone in corn meal by ultra-performance liquid chromatography tandem mass spectrometry. Food Anal. Methods 2017, 10, 1631–1644. [Google Scholar] [CrossRef]
- European Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Volume 221. 2002. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32002D0657&from=EN (accessed on 1 January 2020).
- La Barbera, G.; Capriotti, A.L.; Cavaliere, C.; Foglia, P.; Montone, C.M.; Chiozzi, R.Z.; Lagan, A. A rapid magnetic solid phase extraction method followed by liquid chromatography-tandem mass spectrometry analysis for the determination of mycotoxins in cereals. Toxins 2017, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Yang, X.; Zhao, X.; Bai, B.; Yao, C.; Liu, N.; Wang, J.; Zhou, C. Vortex-assisted dispersive liquid-liquid microextraction for the analysis of major Aspergillus and Penicillium mycotoxins in rice wine by liquid chromatography-tandem mass spectrometry. Food Control 2017, 73, 862–868. [Google Scholar] [CrossRef]
- McElhinney, C.; O’Kiely, P.; Elliott, C.; Danaher, M. Development and validation of an UHPLC-MS/MS method for the determination of mycotoxins in grass silages. Food Addit. Contam. 2015, 32, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, E.; Ibáñez, M.; Portolés, T.; Ripollés, C.; Sancho, J.V.; Yusà, V.; Marín, S.; Hernández, F. Development of sensitive and rapid analytical methodology for food analysis of 18 mycotoxins included in a total diet study. Anal. Chim. Acta 2013, 783, 39–48. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; González-Peñas, E. An LC–MS/MS method for multi-mycotoxin quantification in cow milk. Food Chem. 2017, 218, 378–385. [Google Scholar] [CrossRef]
- Remiro, R.; Irigoyen, A.; González-Peñas, E.; Lizarraga, E.; López de Cerain, A. Levels of ochratoxins in Mediterranean red wines. Food Control 2013, 32, 63–68. [Google Scholar] [CrossRef]
- Miró-Abella, E.; Herrero, P.; Canela, N.; Arola, L.; Borrull, F.; Ras, R.; Fontanals, N. Determination of mycotoxins in plant-based beverages using QuEChERS and liquid chromatography-tandem mass spectrometry. Food Chem. 2017, 229, 366–372. [Google Scholar] [CrossRef]
- Samples, C.; De Santis, B.; Debegnach, F.; Gregori, E.; Russo, S.; Marchegiani, F.; Moracci, G.; Brera, C. Development of a LC-MS/MS method for the multi-mycotoxin determination in composite cereal-based samples. Toxins 2017, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Kiontke, A.; Oliveira-Birkmeier, A.; Opitz, A.; Birkemeyer, C. Electrospray ionization efficiency is dependent on different molecular descriptors with respect to solvent pH and instrumental configuration. PLoS ONE 2016, 11, e0167502. [Google Scholar] [CrossRef]
- Johnson, D.; Boyes, B.; Orlando, R. The use of ammonium formate as a mobile-phase modifier for LC-MS/MS analysis of tryptic digests. J. Biomol. Tech. 2013, 24, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Rastrelli, L. Application of dispersive liquid-liquid microextraction for the determination of aflatoxins B1, B2, G1 and G2 in cereal products. J. Chromatogr. A 2011, 1218, 7648–7654. [Google Scholar] [CrossRef]
- Lai, X.; Ruan, C.; Liu, R.; Liu, C. Application of ionic liquid-based dispersive liquid-liquid microextraction for the analysis of ochratoxin A in rice wines. Food Chem. 2014, 161, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, K.; Han, T.; Chen, X. Preparation of multi-target Fusarium toxins (zearalenone, deoxynivalenol, T-2, and HT-2 toxins) immunoaffinity column using polystyrene-divinylbenzene as matrix. Food Anal. Methods 2017, 10, 2053–2059. [Google Scholar] [CrossRef]
- Lai, X.W.; Sun, D.L.; Ruan, C.Q.; Zhang, H.; Liu, C.L. Rapid analysis of aflatoxins B1, B2, and ochratoxin A in rice samples using dispersive liquid-liquid microextraction combined with HPLC. J. Sep. Sci. 2014, 37, 92–98. [Google Scholar] [CrossRef]
- Alsharif, A.M.A.; Tan, G.H.; Choo, Y.M.; Lawal, A. Liquid phase microextraction for analysis of mycotoxins in food samples. Res. J. Chem. Environ. Sci. 2015, 3, 5–21. [Google Scholar]
- Karami-Osboo, R.; Maham, M.; Miri, R.; AliAbadi, M.H.S.; Mirabolfathy, M.; Javidnia, K. Evaluation of dispersive liquid-liquid microextraction-HPLC-UV for determination of deoxynivalenol (DON) in wheat flour. Food Anal. Methods 2013, 6, 176–180. [Google Scholar] [CrossRef]
- Zor, Ş.D.; Peksel, A. Optimization of ultrasound-assisted extraction of antioxidants from Apium graveolens L. seeds using response surface methodology. J. Turk. Chem. Soc. 2017, 4, 915–930. [Google Scholar] [CrossRef]
- Moldoveanu, S.C.; David, V. Parameters that Characterize HPLC Analysis. In Essentials in Modern HPLC Separations; Elsevier: Amsterdam, The Netherlands, 2013; pp. 53–83. [Google Scholar]
- Belouafa, S.; Habti, F.; Benhar, S.; Belafkih, B.; Tayane, S.; Hamdouch, S.; Bennamara, A.; Abourriche, A. Statistical tools and approaches to validate analytical methods: Methodology and practical examples. Int. J. Metrol. Qual. Eng. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Yogendrarajah, P.; Van Poucke, C.; De Meulenaer, B.; De Saeger, S. Development and validation of a QuEChERS based liquid chromatography tandem mass spectrometry method for the determination of multiple mycotoxins in spices. J. Chromatogr. A 2013, 1297, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, A.L.; Cavaliere, C.; Foglia, P.; Samperi, R.; Stampachiacchiere, S.; Ventura, S.; Laganà, A. Multiclass analysis of mycotoxins in biscuits by high performance liquid chromatography-tandem mass spectrometry. Comparison of different extraction procedures. J. Chromatogr. A 2014, 1343, 69–78. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; González-Peñas, E. Development and validation of a high performance liquid chromatographic-mass spectrometry method for the simultaneous quantification of 10 trichothecenes in ultra-high temperature processed cow milk. J. Chromatogr. A 2015, 1419, 37–44. [Google Scholar] [CrossRef]
- González-Jartín, J.M.; Alfonso, A.; Rodríguez, I.; Sainz, M.J.; Vieytes, M.R.; Botana, L.M. A QuEChERS based extraction procedure coupled to UPLC-MS/MS detection for mycotoxins analysis in beer. Food Chem. 2019, 275, 703–710. [Google Scholar] [CrossRef] [PubMed]
- European Commission Commission Regulation (EC) No 401/2006 Laying down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. Volume 70. 2016. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32006R0401&from=EN (accessed on 30 January 2020).
- Schober, P.; Schwarte, L.A. Correlation coefficients: Appropriate use and interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aubry, A.F.; Cornelius, G.; Caporuscio, C.; Sleczka, B.; Ranasinghe, A.; Wang-Iverson, D.; Olah, T.; Jemal, M. Importance of mobile phase and injection solvent selection during rapid method development and sample analysis in drug discovery bioanalysis illustrated using convenient multiplexed LC-MS/MS. Anal. Methods 2010, 2, 375–381. [Google Scholar] [CrossRef]
- European Commission. EUR 24105 EN: Guidelines for Performance Criteria and Validation Procedures of Analytical Methods Used in Controls of Food Contact Materials; Publications Office of the European Union: Luxembourg, 2009. [Google Scholar]
- National Association of Testing Authorities General Accreditation Guidance—Validation and Verification of Quantitative and Qualitative Test Methods. 2018. Available online: https://www.nata.com.au/phocadownload/gen-accreditation-guidance/Validation-and-Verification-of-Quantitative-and-Qualitative-Test-Methods.pdf (accessed on 15 January 2020).
- Raposo, F.; Ibelli-Bianco, C. Performance parameters for analytical method validation: Controversies and discrepancies among numerous guidelines. Trends Anal. Chem. 2020, 129, 1–12. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Polarity | Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy (CE) | Tube Lens | Retention Time (min) |
|---|---|---|---|---|---|---|
| AFB1 | + | 313 | 285 | 21 | 94 | 9.7 |
| 269 | 25 | |||||
| AFG1 | + | 329 | 243 | 22 | 86 | 9.0 |
| 311 | 25 | |||||
| AFB2 | + | 315 | 287 | 25 | 95 | 9.4 |
| 259 | 28 | |||||
| AFG2 | + | 331 | 313 | 23 | 86 | 8.7 |
| 189 | 35 | |||||
| STG | + | 325 | 310 | 25 | 76 | 12.3 |
| 281 | 34 | |||||
| HT-2 | + | 442 | 263 | 20 | 70 | 10.7 |
| 215 | 20 | |||||
| T-2 | + | 484 | 305 | 20 | 70 | 11.4 |
| 245 | 20 | |||||
| ZEA | + | 319 | 283 | 8 | 86 | 11.9 |
| 301 | 10 | |||||
| DAS | + | 384 | 247 | 20 | 70 | 10.4 |
| 307 | 20 | |||||
| FB1 | + | 722 | 352 | 34 | 126 | 11.2 |
| 334 | 37 | |||||
| FB2 | + | 706 | 336 | 34 | 120 | 12.2 |
| 318 | 35 | |||||
| DON | + | 297 | 249 | 9 | 90 | 6.3 |
| 175 | 23 | |||||
| OTA | + | 404 | 239 | 25 | 90 | 12.4 |
| 358 | 20 |
| Target Analyte | Final Equation | R2 | ANOVA p-Value | |
|---|---|---|---|---|
| Model | Lack of Fit | |||
| AFB1 | −718,670 + 7156 A − 15,649 B + 24,688 C − 11.86 A2+ 697 B2 − 1369 C2 + 20.6 AB − 85.2 AC + 191 BC | 0.934 | 0.01 | 0.57 |
| AFG1 | 44,372 + 1536 A − 1881 B − 17,849 C − 2.41 A2 −188 B2 + 402 C2 + 12.5 AB + 53.7 AC − 221 BC | 0.946 | 0.01 | 0.43 |
| AFB2 | −59,869 + 1822 A + 120 B − 15,664 C − 3.79 A2 − 39 B2 + 126 C2 + 9.1 AB + 51.7 AC − 423 BC | 0.938 | 0.01 | 0.21 |
| AFG2 | −4299 + 513 A + 2432 B − 11,286 C − 1.016 A2 − 215 B2 + 359 C2 − 1.0 AB + 24.7 AC− 61 BC | 0.866 | 0.03 | 0.41 |
| STG | −995,532 + 22,381 A − 144,405 B − 141,900 C − 44.8 A2 + 5697 B2 + 2733 C2 + 232 AB + 26 AC + 6119 BC | 0.775 | 0.06 | 0.76 |
| T-2 | −112,185 + 3512 A − 16,011 B − 15,247 C − 6.71 A2 − 722 B2 − 274 C2 + 52.5 AB + 30.8 AC + 1289 BC | 0.954 | 0.00 | 0.30 |
| HT-2 | 58,093 + 768 A + 6157 B − 25,799 C − 2.66 A2 − 455 B2 + 728 C2 + 5.6 AB + 61.3 AC − 354 BC | 0.865 | 0.04 | 0.91 |
| ZEA | −28,806,469 + 407,356 A − 882,897 B − 475,031 C − 829 A2 + 43,282 B2 − 24,239 C2 + 2063 AB + 782 AC −27,500 BC | 0.854 | 0.02 | 0.31 |
| DAS | −43,534 + 6319 A + 32,240 B − 72,094 C − 10.44 A2 − 2154 B2 + 3002 C2 + 24 AB + 111 AC − 2973 BC | 0.883 | 0.01 | 0.82 |
| FB1 | −54,518 + 408 A + 12,533 B + 9554 C − 1.046 A2 + 266.0 B2 − 801 C2 − 25.53 AB + 9.50 AC − 1002 BC | 0.957 | 0.01 | 0.23 |
| FB2 | −724,670 + 9510 A − 151,680 B + 67,179 C − 25.6 A2 + 1067 B2 12,128 C2 + 439 AB + 209 AC + 11,944 BC + 209 AC + 11,944 BC | 0.829 | 0.04 | 0.75 |
| OTA | −700,860 + 12,471 A − 140,637 B − 22,763 C − 24.5 A2 + 2232 B2 − 4309 C2 + 213 AB + 7 AC + 7 AC + 11,293 BC | 0.807 | 0.05 | 0.81 |
| Target Analyte | Peak Height | |
|---|---|---|
| Before Optimization | After Optimization | |
| AFB1 | 11,282 ± 1484 | 23,011 ± 1411 |
| AFG1 | 12,186 ± 477 | 21,256 ± 1636 |
| AFB2 | 5833 ± 928 | 10,256 ± 289 |
| AFG2 | 3245 ± 725 | 10,460 ± 122 |
| STG | 12,847 ± 588 | 87,240 ± 1939 |
| T-2 | 16,959 ± 3429 | 54,895 ± 1414 |
| HT-2 | 19,540 ± 896 | 11,042 ± 1416 |
| ZEA | 47,244 ± 2849 | 136,034 ± 13,576 |
| DAS | 6958 ± 7393 | 43,421 ± 3427 |
| FB1 | 3245 ± 382 | 76,253 ± 7302 |
| FB2 | 12,847 ± 790 | 37,199 ± 7099 |
| OTA | 16,959 ± 101 | 10,154 ± 1529 |
| Mycotoxin | LOD (ng g−1) | LOQ (ng g−1) | Linearity Range (ng g−1) | R2 |
|---|---|---|---|---|
| AFB1 | 0.5 | 1.5 | 0.5–1.5 | 0.992 |
| AFG1 | 0.5 | 1.5 | 0.5–1.5 | 0.992 |
| AFB2 | 1.0 | 3.0 | 1.5–4.5 | 0.992 |
| AFG2 | 1.0 | 3.0 | 1.5–4.5 | 0.994 |
| STG | 2.5 | 5.0 | 2.5–7.5 | 0.990 |
| T-2 | 2.5 | 5.0 | 2.5–7.5 | 0.990 |
| HT-2 | 2.5 | 5.0 | 2.5–7.5 | 0.992 |
| ZEA | 2.5 | 5.0 | 2.5–7.5 | 0.992 |
| DAS | 2.5 | 5.0 | 2.5–7.5 | 0.993 |
| FB1 | 50 | 150 | 50–150 | 0.990 |
| FB2 | 25 | 75 | 25–75 | 0.991 |
| OTA | 1.25 | 3.75 | 2.5–7.5 | 0.993 |
| Mycotoxin | Concentration (ng g−1) | Within Assay Precision (%) (n = 7) | Between Assay Precision (%) (n = 21) | Accuracy (%) (n = 21) | Recovery (%) ±SD (n = 7) |
|---|---|---|---|---|---|
| AFB1 | 1.5 | 11.6 | 11.3 | 113.7 (11.7) | 74.6 ± 0.1 |
| AFG1 | 1.5 | 11.1 | 9.1 | 115.1 (8.8) | 80.5 ± 0.0 |
| AFB2 | 3.0 | 15.3 | 15.1 | 112.5 (15.1) | 70.2 ± 0.1 |
| AFG2 | 3.0 | 16.2 | 16.1 | 112.8 (16.5) | 74.8 ± 0.2 |
| STG | 5.0 | 17.9 | 18.5 | 74.6 (13.5) | 99.4 ± 0.1 |
| T-2 | 5.0 | 13.1 | 7.9 | 119.1 (9.8) | 76.6 ± 0.2 |
| HT-2 | 5.0 | 14.3 | 14.5 | 108.6 (13.8) | 72.8 ± 0.9 |
| ZEA | 5.0 | 13.9 | 12.8 | 102.1 (1.1) | 95.1 ± 0.9 |
| DAS | 5.0 | 14.2 | 7.9 | 62.3 (4.9) | 90.1 ± 0.3 |
| FB1 | 150 | 5.6 | 12.0 | 101.0 (8.1) | 71.9 ± 0.8 |
| FB2 | 75 | 6.5 | 14.6 | 73.1 (7.1) | 82.5 ± 1.3 |
| OTA | 3.75 | 14.6 | 14.5 | 60.2 (14.6) | 87.0 ± 0.2 |
| Mean Concentration (ng g−1) ± SD (n = 2) | ||||||
|---|---|---|---|---|---|---|
| AFB1 | AFG1 | AFB2 | AFG2 | FB1 | FB2 | |
| S2 | 1.69 ± 0.17 | 0.11 ± 0.01 | 0.33 ± 0.07 | 6.49 ± 1.38 | 157.44 ± 0.52 | 28.3 ± 0.91 |
| S3 | 2.13 ± 0.50 | 0.10 ± 0.02 | 0.33 ± 0.01 | 2.70 ± 0.27 | n.d. | n.d. |
| S5 | 1.08 ± 0.22 | 0.08 ± 0.01 | 0.37 ± 0.17 | 6.19 ± 0.91 | n.d. | n.d. |
| S6 | 1.09 ± 0.29 | 0.12 ± 0.03 | 0.36 ± 0.04 | 8.07 ± 0.06 | n.d. | n.d. |
| S7 | 1.67 ± 0.02 | 0.08 ± 0.07 | 0.36 ± 0.07 | 2.34 ± 0.59 | n.d. | n.d. |
| S9 | 2.19 ± 0.21 | 0.07 ± 0.02 | 0.20 ± 0.05 | 1.45 ± 0.41 | n.d. | n.d. |
| S17 | 0.34 ± 0.02 | 0.10 ± 0.01 | 2.72 ± 0.71 | 2.85 ± 0.27 | 77.70 ± 0.62 | 75.56 ± 0.73 |
| S20 | 0.27 ± 0.04 | 0.12 ± 0.01 | 3.88 ± 1.60 | 5.41 ± 1.31 | n.d. | n.d. |
| S21 | 0.34 ± 0.04 | 0.12 ± 0.05 | 1.74 ± 0.22 | 2.76 ± 0.30 | n.d. | n.d. |
| S22 | 0.31 ± 0.07 | 0.14 ± 0.07 | 1.55 ± 0.96 | 6.39 ± 1.50 | n.d. | n.d. |
| Variables | Symbol | Range and Level | ||
|---|---|---|---|---|
| Low | Central | High | ||
| Volume of extraction solvent (µL) | A | 150 | 225 | 300 |
| Concentration of salt (%) | B | 0 | 5 | 10 |
| Volume of water (mL) | C | 3 | 6.5 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salim, S.A.; Sukor, R.; Ismail, M.N.; Selamat, J. Dispersive Liquid–Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development, Optimization and Validation. Toxins 2021, 13, 280. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13040280
Salim SA, Sukor R, Ismail MN, Selamat J. Dispersive Liquid–Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development, Optimization and Validation. Toxins. 2021; 13(4):280. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13040280
Chicago/Turabian StyleSalim, Sofiyatul Akmal, Rashidah Sukor, Mohd Nazri Ismail, and Jinap Selamat. 2021. "Dispersive Liquid–Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development, Optimization and Validation" Toxins 13, no. 4: 280. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13040280