Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease


1
Division of Nephrology and Hypertension, Vanderbilt University Medical Center, Nashville, TN 37232, USA
2
Division of Nephrology, Department of Medicine, Kurume University School of Medicine, Kurume 830-0011, Japan
*
Author to whom correspondence should be addressed.
Toxins 2021, 13(5), 361; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050361
Submission received: 31 March 2021
/
Revised: 17 May 2021
/
Accepted: 17 May 2021
/
Published: 19 May 2021
(This article belongs to the Special Issue Gut Microbiota Dynamics and Uremic Toxins)
Abstract
:Chronic kidney disease (CKD) is a public health concern that affects approximately 10% of the global population. CKD is associated with poor outcomes due to high frequencies of comorbidities such as heart failure and cardiovascular disease. Uremic toxins are compounds that are usually filtered and excreted by the kidneys. With the decline of renal function, uremic toxins are accumulated in the systemic circulation and tissues, which hastens the progression of CKD and concomitant comorbidities. Gut microbial dysbiosis, defined as an imbalance of the gut microbial community, is one of the comorbidities of CKD. Meanwhile, gut dysbiosis plays a pathological role in accelerating CKD progression through the production of further uremic toxins in the gastrointestinal tracts. Therefore, the gut-kidney axis has been attracting attention in recent years as a potential therapeutic target for stopping CKD. Trimethylamine N-oxide (TMAO) generated by gut microbiota is linked to the progression of cardiovascular disease and CKD. Also, advanced glycation endproducts (AGEs) not only promote CKD but also cause gut dysbiosis with disruption of the intestinal barrier. This review summarizes the underlying mechanism for how gut microbial dysbiosis promotes kidney injury and highlights the wide-ranging interventions to counter dysbiosis for CKD patients from the view of uremic toxins such as TMAO and AGEs.
Key Contribution: Gut microbiota dysbiosis causes chronic kidney disease through production of uremic toxins.
1. Introduction
Chronic kidney disease (CKD) has emerged as a major public health concern that affects an estimated 37 million people in the United States [1,2]. Not only is CKD the 9th leading cause of death in the U.S., but it also contributes to several diseases such as heart disease [3], stroke [4], cancer [5], and cognitive impairment [6], thereby increasing mortality rates in patients with CKD [7]. Therefore, preventing the new onset of CKD or inhibiting the progression of CKD is of paramount importance for public health. It has been shown that inappropriate activation of the renin-angiotensin system, sympathetic nerve activation, and upregulated inflammatory cytokines are implicated in the progression of CKD [8,9,10]. In addition to the traditional risk factors for CKD progression, recent studies have revealed that gut microbial dysbiosis, defined as an “imbalance” in the gut microbial community, is also one of the key contributors to accelerating CKD progression [11].
Substances that are typically excreted or metabolized by the kidney accumulate as renal function declines. If the substances induce clinical symptoms of uremia, they are referred as to “uremic toxins.” Advanced glycation endproducts (AGEs), a heterogeneous group of molecules formed by a non-enzymatic reaction between reducing sugars and amino acids, lipids, and DNA, are considered as one of the representative uremic toxins [12]. AGEs accumulate in visceral organs crosslinking with matrix molecules and disrupting matrix-matrix and matrix-cell interactions, resulting in organ dysfunction in CKD [13]. Also, through the interaction with the receptor for AGEs (RAGE), AGEs promote further oxidative stress and stimulate several intracellular signaling molecules, leading to the production of inflammatory and profibrotic cytokines [14]. Another uremic toxin, trimethylamine N-oxide (TMAO), which is a degradation product of choline and L-carnitine, is shown to accumulate when renal function declines and correlates with enhanced cardiovascular mortality in CKD patients [15,16]. An elevated TMAO value is not only a predictor of cardiovascular disease, but it also promotes renal fibrosis, the main feature of CKD [17].
The human gut microbiota consists of approximately 1000 species and there are ~100 trillion bacteria in the gut, the genetic information of which is 150 times larger than that of the host [18]. With better understanding as technology advances, gut microbiota seems to not only maintain homeostasis, but also to be involved in the pathology of several human diseases including obesity [19], hypertension [20], cancer [21], depression [22], and cardiovascular diseases [23]. Further, recent studies have shown that gut microbiota dysbiosis including loss of beneficial microbes, expansion of pathobionts, and loss of microbiome diversity, is one of the common features in patients with CKD, possibly due to metabolic acidosis, accumulation of uremic substances in the intestine, volume overload-induced edema of intestinal epithelial cells, and frequent use of antibiotics and oral iron agents [24]. It has been found that the proportions of the bacteria possessing urease, uricase, and p-cresol- and indole-producing enzymes are increased in CKD, leading to uremic toxicity and systemic inflammation [25]. Many substances secreted by the microbiota become uremic toxins in the setting of CKD and can be absorbed into the body [26,27]. Thus, gut microbiota impairment plays a pathological role in the development of uremia in CKD patients and represents a potential therapeutic target for CKD progression.
A recent study has demonstrated that an AGEs-rich diet affects the gut microbiota [28], which may be linked to TMAO production [15]. Further, accumulation of AGEs was found in the gastrointestinal tract of CKD patients in association with gut microbiota dysbiosis, possibly leading to further progression of CKD [29]. Understanding the mutual relationship between gut microbiota dysbiosis and uremic toxins such as AGEs and TMAO is of great importance for preventing kidney injury and developing new therapeutic options for CKD patients. This review highlights the underlying mechanisms by which the reciprocal regulation of gut microbiota dysbiosis and uremic toxins drives CKD progression.
2. Gut Microbiota, AGEs, TMAO, and Inflammation
2.1. Dysbiosis in CKD
A number of studies have shown that CKD affects the distribution and makeup of gut microbiota, which is linked to dysbiosis [30]. Although the results of clinical studies may vary depending on the ethnicity, medical treatment, and lifestyle, it is generally accepted that bacterial species that produce urease (Proteobacteria, Clostridiaceae, and Actinomycetales), uricase (Actinomycetales and Proteobacteria), and indole or p-cresol forming enzymes (Clostridiaceae and Prevotellaceae) thrive, whereas species containing butyrate-forming enzymes (Roseburia, Faecalibacterium, Clostridium, and Coprococcus) are reduced in CKD [25]. Urease-producing bacteria including Clostridiaceae are known to hydrolyze urea in the gut to form ammonia, which, in turn, is converted to ammonium hydroxide. The high burdens of ammonia and ammonium hydroxide in the gut are associated with the alteration of gut microbiota and disruption of the intestinal epithelial tight junction [11]. Besides this, indole- or p-cresol-forming enzymes are responsible for metabolizing tryptophan and tyrosine into indole and p-cresol, respectively, in the gut. Then, in the liver, indole and p-cresol are converted to indoxyl sulfate (IS) and p-cresol sulfate (PCS), well-documented uremic toxins [31]. Uremic toxins are substances that are usually filtered and excreted by the kidneys, whereas as glomerular filtration rate (GFR) declines in CKD, these compounds accumulate and exert their deleterious effects on various organs. In fact, the serum concentration of IS and PCS are positively correlated with serum creatinine, a marker of GFR, and the patients with a later stage of CKD showed a higher concentration of IS or PCS. Thus, in addition to the loss of the ability to remove uremic toxins due to decreased GFR, the expansion of such bacterial species producing uremic toxins in the gut of CKD is linked to further accumulation of uremic toxins in the body.
The possible mechanisms for how the gut microbiota change in CKD are as follows: (1) accumulated uremic toxins perturb the composition of gut microbiota [11,32]; (2) metabolic acidosis; (3) therapeutic agents such as oral iron supplements [33], chelating agents [34], antibiotics [35], and proton-pump inhibitors [36]; (4) reduced bowel movement due to CKD–induced edema and ischemia; (5) lack of dietary fiber intake [37]; (6) prolonged intestinal transit time [24]. A recent investigation using microbiome analysis showed that the proportions of Eggerthella lenta and Fusobacterium nucleatum were increased in patients with end-stage renal disease (ESRD) when compared with healthy individuals, and fecal transplantation with the two species into rats with CKD exacerbated renal fibrosis and increased the production of uremic toxins [38], supporting the gut-kidney axis theory. Several systematic reviews and meta-analyses have also demonstrated that probiotic, prebiotic, and synbiotic supplements in the CKD population improve a circulating inflammation marker (C-reactive protein), oxidative stress markers (malondialdehyde, glutathione, and total antioxidant capacity), and lipids profiles [39,40]. Indeed, Rossi et al. conducted a randomized, double-blind trial to investigate the efficacy of synbiotic interventions in patients with CKD stage 4–5, and determined that with synbiotic supplementation, an enrichment of Bifidobacterium and depletion of Ruminococcaceae were observed with a decrease in the serum concentrations of IS and PCS [41].
CKD-induced dysbiosis is thought to accelerate the progression of CKD to ESRD through the production of uremic toxins, giving rise to a vicious cycle between CKD and dysbiosis. Once ESRD is established, uremic toxins still remain a major problem and source of uremic symptoms as they are not cleared by blood purification therapies, such as dialysis. Although an infusion of ibuprofen [42] or reduction of blood pH [43] are shown to increase the clearance of protein-bound uremic toxins during single dialysis treatment, no specific therapeutic option to enhance the removal of protein-bound uremic toxins has yet been established [44]. Thus, creating new interventions targeting the production process of uremic toxins is promising and needs to be further investigated.
2.2. Disruption of Gut Epithelial Barrier in CKD
Disruption of the gut epithelial barrier, referred to as a “leaky gut”, is another intestinal complication related to CKD [45]. The buildup of uremic toxins is thought to weaken the intestinal epithelial barrier, allowing bacterial components derived from intestinal bacteria, such as DNA, lipopolysaccharide (LPS), and endotoxins, to flow into systemic circulation through the leaky gut. IS induces systemic inflammation, leading to cellular injury in vascular endothelial cells, and promotes arteriosclerosis with an increase in thrombus formation (Figure 1) [46]. Kikuchi et al. demonstrated that a high serum concentration of phenyl sulfate, a protein-bound uremic solute, was associated with the progression of albuminuria in 362 patients with diabetic kidney disease (DKD) [47]. Besides endothelial dysfunction, IS induces cardiac fibrosis, thus affecting cardiac function [48]. AST-120, an oral charcoal adsorbent, was shown to attenuate cardiac hypertrophy and cardiac fibrosis by inhibiting the absorbance of uremic toxins in a rodent CKD model [49]. A meta-analysis concluded that serum IS and PCS levels strongly correlate with overall mortality in CKD patients [50]. The serum concentration of phenylacetylglutamine, a recently identified colonic microbial metabolite from amino acid fermentation, also predicts the prevalence of cardiovascular diseases [51]. These findings provide a link between the multiple organ damage seen in CKD patients and gut microbiota-derived uremic toxins.
As CKD progresses, endotoxins such as LPS flow into the body and function as pathogen-associated molecular patterns (PAMPs) [52]. A number of clinical investigations have demonstrated that individuals with a high concentration of serum endotoxins are more likely to develop cardiovascular diseases (CVD) [53]. Also, PAMPs and damage-associated molecular patterns (DAMPs) are involved in the progression of several types of kidney diseases including DKD through interaction with pattern recognition receptors such as RAGE and the toll-like receptor (TLR) [54]. In fact, injection with LPS worsened kidney function in streptozotocin-induced diabetic mice, and upregulation of the vascular endothelial growth factor (VEGF) in the PTECs of a diabetic kidney was further enhanced by the administration of LPS [55]. Recently, Nakano et al. revealed interesting findings, using intravital imaging by 2-photon microscopy, that LPS disrupts the tight junction of PTECs and induces paracellular leakage of filtered molecules and interstitial accumulation of extracellular fluid, leading to oliguria [56]. Interestingly, tubule-specific deletion of TLR-4, an LPS-binding receptor, retained the efficacy of intravenous fluid treatment in LPS-induced acute kidney injury (AKI) [56]. Although it is still not clear whether this theory is applicable to intestinal epithelial cells, the idea could provide an explanation for the prevalence of the “leaky gut” in CKD.
2.3. Gut Dysbiosis and Inflammation
There are many overlapping processes that drive the progression of CKD to ESRD such as renin-angiotensin system activation, sympathetic hyperactivity, mitochondrial dysfunction, and so on [8,9,10]. In addition to the traditional risk factors, subclinical inflammation can be considered one of the contributors to the progression of CKD; thus, this section focuses on the links between dysbiosis, uremic toxins, and inflammation in CKD. The subclinical inflammation in CKD is characterized by an increase in inflammatory markers such as cytokines, acute-phase proteins, and adhesion molecules [59]. Chronic subclinical inflammation in CKD can be the result of increased production and decreased elimination of proinflammatory cytokines [60], gut dysbiosis [45], reactive oxygen species (ROS), metabolic acidosis [61], and alteration of adipose tissue metabolism [62], or a combination of these factors. Besides an increase in uremic toxin-producing bacteria, a concomitant reduction in the species generating beneficial short-chain fatty acids (SCFAs) including butyrate is one of the characteristics of CKD-related dysbiosis [63,64]. SCFAs are saturated fatty acids with a chain length ranging from one to six carbon atoms, produced by fermentation of dietary fiber in the colon. Recent studies provide evidence for gut microbiota producing the SCFAs influencing the severity of AKI by regulating the immune and inflammatory responses [65]. Mishima et al. demonstrated that intestinal SCFA production was dramatically reduced in germ-free mice, which worsened adenine-induced kidney injury [66]. Meanwhile, supplementing with exogenous SCFAs not only reduced ROS, but also suppressed cytokines and chemokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and monocyte chemoattractant protein-1 (MCP-1) [66]. Intriguingly, in vitro experiments showed that SCFAs prevented dendric cells’ maturation with inhibition of CD4+ and CD8+ T cell proliferation [67], indicating that SCFAs seem to directly modulate immune cells’ function and regulate cytokine/chemokine production. In addition to CKD-related dysbiosis characterized by lack of SCFA-producing bacteria, dietary restriction of fermentable fiber such as potassium-rich fruits and vegetables in CKD leads to the depletion of the bacteria that convert indigestible carbohydrates to SCFAs [63,64]. The most recent investigations demonstrated that dietary intake of sulfur-containing amino acids inhibited the function of tryptophanase, a secreted enzyme that catalyzes the degradation of tryptophan to indole, pyruvate, and ammonia, which resulted in a reduction in uremic toxin production, leading to the prevention of tubular injury in an adenine-induced CKD model [68]. Similarly, a number of studies have shown the efficacy of interventions targeting dysbiosis for slowing down CKD progression. Dietary supplementation with resistant starch—which is a carbohydrate that resists digestion in the small intestine and ferments in the large intestine, acting as a prebiotic—ameliorated kidney injury in a type 2 diabetic rodent model [69] and a CKD rat model [70]. Lubiprostone, a chloride channel activator that can improve the symptoms of constipation, was also shown to prevent tubular injury and GFR decline with increases in proportions of Lactobacillus and Prevotella in an adenine-induced CKD model [71]. Thus, a clinical trial to examine the inhibitory efficacy of lubiprostone (UMIN000023850) is currently underway.
With respect to AKI, Jang et al. showed that structural injury and functional decline following ischemic reperfusion injury (IRI) were more severe in germ-free mice with enhanced CD8+ T cells trafficking into the injured kidney. Reconstituting microbiota by adding fecal material from control mice to the germ-free mice attenuated histological damage, which led the authors to conclude that regulatory T cells may not be stimulated or mature in germ-free mice during development, thus amplifying renal damage [72]. In contrast, Emal et al. demonstrated that treatment with broad-spectrum antibiotics like ampicillin, metronidazole, neomycin, and vancomycin reduced microbial diversity and protected mice against IRI-induced kidney injury. This was achieved by suppressing the maturation status of F4/80+ renal-resident macrophages, well-known immune cells involved in the detection, phagocytosis, and destruction of bacteria and other harmful organisms, as well as by reducing the release of chemokines. Fecal material transplantation from control mice to antibiotic-treated mice abolished this protective effect [73]. Although previous animal data depend on whether germ-free animals or broad-spectrum antibiotic treatment was utilized for the removal of gut microbiota, the gut microbiota seem to be associated with inflammation, leading to modulation of the outcome of kidney injury in AKI, as well.
2.4. AGEs–RAGE Axis and Inflammation
A non-enzymatic reaction between reducing sugars such as glucose and the α-amino group or lysine residue at the N-terminal of amino acids is an initial step in the synthesis of AGEs [74,75]. It is followed by the formation of a Schiff base and Amadori compounds, which in turn, undergo various reactions such as dehydration, condensation, oxidation, and reduction. AGEs are finally generated with intermolecular crosslinking in visceral organs. This was first reported by a French chemist, Louis-Camille Maillard, in 1912, and extensively studied in the area of food chemistry. However, AGEs have begun to receive greater attention after the discovery of hemoglobin A1C, a glycated hemoglobin and a marker for type 2 diabetes [76,77]. Since then, research studies have shown that proteins with a slow turnover rate in the body, like collagens exposed to reducing sugars for a long period, have lysine residues that are AGEs-modified, which induces protein polymerization, reduced solubility, and susceptibility to proteases. Thus, AGEs are rarely degraded, and crosslinking of AGEs leads to irreversible organ damage [78].
There are two main drivers of AGE generation, namely, glycation and oxidative stress. Pyrraline and crosslines, the major AGEs, are produced via a glycation-dependent pathway [79]. Thus, hyperglycemia in diabetes is closely associated with the production of AGEs. In recent years, oxidative stress has also been shown to promote the synthesis of AGEs. N(ε)-carboxymethyl lysine (CML), a well-established AGE, is generated after the oxidative cleavage of Amadori compounds or through the auto-oxidation of glucose [80]. In fact, CML accumulation has been observed in the glomeruli of DKD, as well as hypertensive nephropathy [81,82] and lupus nephritis, in humans and experimental rodents [83]. RAGE, the main receptor for AGEs, is a type I transmembrane receptor and is expressed on the surface of several kidney cells including PTECs, mesangial cells, podocytes, and endothelial cells [81,84,85,86]. In addition to the intrinsic kidney cells, RAGE is also expressed on the membrane of immune cells such as monocytes [87], macrophages [88], and T and B lymphocytes [89,90]. The interaction between RAGE and its ligands is shown to directly activate immune cells, causing inflammation and the development of autoimmune diseases [91]. Since AGEs are one of the ligands of RAGE, AGEs promote the secretion of proinflammatory cytokines, such as IL-1α, IL-6, and TNF-α, through the activation of NF-ĸB, leading to the amplification of systemic inflammation [92,93]. AGEs also delay spontaneous apoptosis of monocytes, thus, contributing to the development of inflammatory responses [94]. Considering that RAGE also recognizes a wide range of DAMPs [93], it is likely that DAMPs derived from gut microbiota bind to RAGE, inducing immune cells to migrate and proliferate. With respect to acute inflammation, the genetic deletion of RAGE attenuated the hepatic expression of pro-inflammatory cytokines with a better survival rate compared to wild-type mice in LPS/D-galN-induced acute liver injury [95]. The outcome of a clinical research study supported the concept that there is a positive correlation between the expression levels of RAGE and E-selectin, endothelin-1, and TNF-α in septic AKI patients [96]. In addition, RAGE was implicated in chronic inflammation. Durning et al. demonstrated that RAGE enhanced the inflammatory function of T cells, and that its increased levels in patients with type 1 diabetes may account for the chronic autoimmune response [97]. In another study, old RAGE knockout (KO) mice exhibited a decrease in the renal concentration of proinflammatory cytokines compared to old wild-type mice, and attenuated glomerulosclerosis, a marker of aging kidneys [98]. RAGE expressed on various cell types plays a central role in amplifying and prolonging inflammatory responses through engagement with AGEs (Figure 1).
RAGE is linked to the development of human autoimmune diseases such as systemic lupus erythematosus (SLE) [91] and arthritis [99]. Tian et al. demonstrated that HMGB-1 released from necrotic or dead cells in autoimmune diseases activates autoreactive B cells and plasmacytoid dendritic cells, and augments inflammatory cytokines secretion through TLR-9 and RAGE [100]. Clinical research also showed that the polymorphism of the RAGE gene is correlated with the severity of SLE and the progression of lupus nephritis [101]. In vitro data showed that peripheral blood mononuclear cells (PBMCs) incubated with serum from lupus nephritis patients enhance the secretion of INF-1α, which is reduced by RAGE-Fc or a monoclonal antibody to RAGE [100]. Thus, a number of studies provide clear evidence that RAGE is a key contributor to inflammation and can be a potential therapeutic target for regulating the massive inflammation in CKD.
2.5. TMAO and Inflammation
The dietary intake of TMAO precursors such as choline, betaine, and L-carnitine, is an important factor for TMAO production. Choline is higher in animal-derived food as compared to plant-based food on a per unit of weight basis, and specifically abundant in liver, eggs, beef, fish, pork, chicken, and milk [102]. Betaine is found in vegetables, animals, and microorganisms, and is a significant component in spinach, wheat, sugar beets, and shellfish [103,104]. L-carnitine is found in many animal products, but especially, red meat has high levels. Small amounts of L-carnitine are also found in chicken, milk and dairy products, fish, beans, and avocado. The L-carnitine content of red meat and fish is not affected by freezing or cooking [105]. These precursors are converted by gut microbiota into an intermediate compound known as trimethylamine (TMA) in the gastrointestinal tract, which is then absorbed and delivered to the liver. There, it undergoes oxidization by Flavin-dependent monooxygenase3 to generate TMAO [106], which is then transported to the brain, muscle, kidney, and intestine [107]. Since TMAO is excreted into the urine, the serum concentration of TMAO is known to be elevated as renal function declines [17], which correlates with an increased risk of cardiovascular disease in general, and especially in patients with CKD [108]. Thus, TMAO has been considered as a uremic toxin as well.
Perturbations of the intestinal microbiota composition in both human and experimental CKD have demonstrated a significant elevation of TMAO, which is linked to an increased burden of inflammation as well as renal disease progression [109]. Interestingly, high-salt and high-fat diets (HFDs), both of which are characteristics of the current Westernized diet, are more likely to alter the composition of microbiota and increase the serum concentration of TMAO (Figure 1) [110]. Therefore, the current generation may be at high risk of elevated TMAO and dysbiosis. A number of investigations have demonstrated that TMAO accelerates kidney injury. A high dietary intake of TMAO exacerbates tubular injury and promotes renal fibrosis, with an increase in phosphorylated Smad3 shown in a rodent CKD model [17]. Recent animal data also showed that HFD induces renal fibrosis, with a corresponding increase in circulating TMAO, which could be inhibited by the administration of 3,3-Dimethyl-1-butanol (DMB), a trimethylamine formation inhibitor [111]. Interestingly, DMB attenuated renal inflammation with a reduction of oxidative stress in mice fed with HFD [111]. Not only is TMAO a contributor to the progression of kidney injury, but it also amplifies inflammation by directly regulating the function of immune cells. In basic research, TMAO has been shown to aggravate the severity of graft-versus-host disease by promoting alloreactive T cell proliferation in a rodent model [112]. TMAO also promoted the migration of macrophages in a CD36-dependent pathway in vitro [113]. That is supported by the evidence that Apo-E KO mice injected with TMAO for eight weeks exhibited enhanced macrophage infiltration into the aortic root [114]. These data suggest that the direct effect of TMAO on the function of immune cells could be the missing link between gut microbiota dysbiosis and inflammation in CKD. A recent study identified that organic anion transporter-3 (OAT-3), a major transporter expressed on the basolateral membrane of PTECs, mediates the excretion of TMAO into the urine. Since flosemide, a loop diuretic often prescribed in the clinical setting, competitively binds to OAT-3, CKD patients treated with flosemide are at a high risk of TMAO accumulation [115]. Similarly, protein-bound uremic toxins including IS and PCS are also taken up through OAT-1 or OAT-3 and secreted by PTECs. Probenecid inhibits the function of OAT-1 and OAT-3 [116]; thus, frequent use of those medicines might cause the accumulation of TMAO or uremic toxins.
3. Possible Interplay between Gut Microbiota and Uremic Toxins in CKD
3.1. AGEs and Dysbiosis in CKD
In addition to the intracorporeal formation of AGEs, dietary intake is another main source of AGEs since modern foods contain relatively high amounts. Cooking temperature, duration, pH, and method are shown to influence the generation of new AGEs in foods [117,118]. Meats including beef and poultry and meat-derived products such as sausages and bacon processed at high, dry heats contain large amounts of AGEs. High-fat cheeses and spreads such as butter, cream cheese, and margarine are also among the products with high dietary AGEs [119]. Importantly, dietary intake of AGEs is positively correlated with AGE levels in serum [120]. Recent studies have demonstrated that not only serum AGEs but also dietary AGEs are likely to be deposited in the gastrointestinal tract tissue and lead to the disruption of the microbiota (Figure 1) [29]. Given that dysbiosis is related to CKD progression, excessive intake of AGEs is likely to advance the progression of CKD and its comorbidities such as nerve system disorder [121], bone mineral disorder [122], cardiovascular diseases [12], and sarcopenia [123], possibly via the induction of dysbiosis. Mastrocola et al. explored the impact of an AGEs-enriched diet on inflammation, as well as on the composition of gut microbiota in mice. In an AGEs-rich diet group, Nε-CML deposition and the upregulation of RAGE were observed in the ileum portion of the intestine and submandibular salivary glands, with an increase in systemic inflammation assessed by the levels of IL-1β, IL-17, and TNF-α [124]. Using fecal microbiota analysis, they found that high AGEs exposure in the intestine affected the gut microbiota population, characterized by the reduction of Anaerostipes, one of the butyrate-producing species [124]. Another investigation has demonstrated that a high-AGEs diet reduces the levels of saccharolytic bacteria such as Ruminococcaceae and Alloprevotella, which are related to SCFAs’ production. These findings suggest that intestinal AGEs exposure can reduce SCFA production, which affects the risk of the progression of kidney injury [125]. Further, high amounts of dietary AGEs exposure downregulate tight junction proteins in the epithelia, such as zonula occludens-1 and claudin-5 [125]. A recent study investigating intestinal permeability, assessed in vivo by the clearance of FITC-labelled dextran, clearly showed that the intake of high dietary AGEs disrupts the gut epithelial barrier, inducing a “leaky gut” in db/db mice, as shown with a type 2 diabetic mouse model [69]. Thus, an increase in the uptake of AGEs affects the composition of gut microbiota, leading to a “leaky gut” via the disruption of the tight junctions of intestinal epithelial cells, which may go on to affect the progression of CKD (Figure 1). The restriction of dietary AGEs, or removing AGEs pharmacologically in the gastrointestinal tract can, therefore, be a potential therapeutic intervention option to preserve gut microbiota ecology, and consequently, inhibit the progression of CKD.
Several strategies using anti-AGEs have been evaluated in recent years. A clinical study demonstrated that AST-120, known to adsorb uremic toxins in the gut leading to its fecal excretion, decreased the serum concentrations of both glycated AGEs and Nε-CML in patients undergoing hemodialysis [126]. A recent single-center, randomized, open-label crossover study has demonstrated that sevelamer carbonate, a non-absorbed phosphate-binding polymer, substantially reduced the concentration of serum AGEs, with a decrease in inflammation and oxidative stress markers in diabetic CKD patients [127,128]. On the other hand, a large proportion of dietary AGEs are degraded and metabolized by the colonic microflora [129]. Lactobacilli, a commercially available probiotic supplement, is capable of producing glyoxalase, the enzyme that degrades dietary AGEs [57]. Therapy targeting probiotics is thus promising for blocking the interaction between dysbiosis and enhanced AGEs accumulation in the CKD condition. Further studies using larger human cohorts are required to confirm this observation.
3.2. AGEs–RAGE Coordinates with TMAO to Progress CKD
Animal products such as red meat, eggs, and fish are major dietary sources of betaine and L-carnitine, precursors of TMAO. Cooking these products at high temperatures (grilling, broiling, and frying) leads huge amounts of AGEs-bound proteins to accumulate, leading to an increase in the concentration of TMAO as well as those of AGEs (Figure 1). Mitchell et al. demonstrated that high protein intake (1.6 g/kgBW/day) increases the circulating concentration of TMAO in the elderly [130]. A recent epidemiological investigation showed that the amount of protein intake correlates with serum Nε-CML levels (Table 1) [131]. Based on these findings, a low protein diet, recommended as per CKD guidelines in several countries, is expected to have a better outcome in reducing the accumulation of TMAO and AGEs in the body. Further, a few studies have shown that the population of Prevotella copri (P. copri), a gram-negative bacteria producing TMA (Table 1) [15], prospers due to an excess intake of AGEs [28,132], suggesting that AGEs may be associated with TMAO production by affecting the microbial composition in the gut. Tahara et al. also demonstrated that the serum AGEs/soluble RAGE (sRAGE) ratio has a positive correlation with serum TMAO concentration [133]. Given that sRAGE eliminates circulating AGEs by functioning as a decoy receptor [134], this finding indicates that endothelial cells in individuals with high circulating TMAO are more likely to be exposed to circulating AGEs, leading to endothelial dysfunction. An oral L-carnitine supplement is frequently used in hemodialysis patients who show carnitine deficiency due to insufficient carnitine synthesis and loss through dialysis in over 30 countries globally [135]. In fact, a clinical study by Adachi et al. showed a decrease in serum-free carnitine level in hemodialysis (HD) patients compared to healthy individuals, which was inversely correlated with skin AGE levels [136]. Thus, the L-carnitine supplement is shown to reduce skin AGE levels [137] with a decrease in vascular injury markers in HD patients (Table 1) [138]. However, it should be noted that there is a risk that oral L-carnitine supplementation can increase serum TMAO levels. Since intravenous L-carnitine administration does not affect TMAO levels, the method of L-carnitine administration should be considered depending on the patient’s condition. Taken together, these findings point to the possible implication of a link between AGEs and TMAO. However, further studies are required to understand the relationship and identify a therapeutic target.
4. Conclusions
The accumulation of uremic toxins is one of the main characteristics of advanced CKD and is linked to the further progression of kidney injury. Clinical symptoms of uremic syndrome including nausea, vomiting, muscle cramp, tremor, and loss of consciousness are caused by the accumulation of uremic toxins consequent to a failure of renal excretion. In addition to the inability to eliminate uremic toxins, CKD-related gut dysbiosis is recently suggested to produce more uremic toxins. Further, intake of dietary toxins such as AGEs and TMA precursors affects the composition of gut microbiota producing more uremic toxins and disruption of the epithelial barrier, leading to leaking of endotoxins, such as LPS, into the body. Thus, systematic studies aimed at teasing the interplay between these factors will pave the way to better understanding the relationship between all of these players in the progression of CKD.
Author Contributions
Writing—original draft preparation, K.T.; writing—review and edit, B.C.E., K.F., and C.R.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Figure was created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GFR | Glomerular filtration rate |
| CKD | Chronic kidney disease |
| ESRD | End-stage renal disease |
| AKI | Acute kidney injury |
| DKD | Diabetic kidney disease |
| SLE | Systemic lupus erythematosus |
| PTECs | Proximal tubular epithelial cells |
| PBMCs | Peripheral blood mononuclear cells |
| AGEs | Advanced glycation endproducts |
| RAGE | Receptor for advanced glycation endproducts |
| sRAGE | Soluble RAGE |
| N(ε)-CML | N(ε)-carboxymethyl lysine |
| TMAO | Trimethylamine N-oxide |
| TMA | Trimethylamine |
| IS | Indoxyl sulfate |
| PCS | p-cresol sulfate |
| LPS | Lipopolysaccharide |
| ROS | Reactive oxygen species |
| NF-ĸB | Nuclear factor-ĸB |
| TNF-α | Tumor necrosis factor-α |
| MCP-1 | Monocyte chemoattractant protein-1 |
| INF-1α | Interferon-1α |
| VEGF | Vascular endothelial growth factor |
| sICAM-1 | Soluble forms of intracellular adhesion molecule-1 |
| sVCAM-1 | Soluble forms of vascular cell adhesion molecule-1 |
| MDA | Malondialdehyde |
| TLR | Toll-like receptor |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| SCFAs | Short-chain fatty acids |
| DMB | 3:3-Dimethyl-1-butanol |
| HFD | High-fat diet |
| OAT | Organic anion transporter |
References
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Arici, M.; Collins, A.J.; Garcia, G.G.; Hemmelgarn, B.R.; Jafar, T.H.; Pecoits-Filho, R.; Sola, L.; Swanepoel, C.R.; Tchokhonelidze, I.; et al. Understanding kidney care needs and implementation strategies in low- and middle-income countries: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016, 90, 1164–1174. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Eng. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Seliger, S.L.; Gillen, D.L.; Longstreth, W.; Kestenbaum, B.; Stehman-Breen, C.O. Elevated risk of stroke among patients with end-stage renal disease. Kidney Int. 2003, 64, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Stengel, B. Chronic kidney disease and cancer: A troubling connection. J. Nephrol. 2010, 23, 253–262. [Google Scholar] [PubMed]
- Tamura, K.M.; Xie, D.; Yaffe, K.; Cohen, D.L.; Teal, V.; Kasner, S.E.; Messe, S.R.; Sehgal, A.R.; Kusek, J.; De Salvo, K.; et al. Vascular risk factors and cognitive impairment in chronic kidney disease: The Chronic Renal Insufficiency Cohort (CRIC) study. Clin. J. Am. Soc. Nephrol. 2011, 6, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldanha da Silva, A.A.; Prestes, T.R.R.; Lauar, A.O.; Finotti, B.B.; Silva, A.C.S. Renin Angiotensin System and Cytokines in Chronic Kidney Disease: Clinical and Experimental Evidence. Protein Pept. Lett. 2017, 24, 799–808. [Google Scholar] [CrossRef]
- Grassi, G.; Quarti-Trevano, F.; Seravalle, G.; Arenare, F.; Volpe, M.; Furiani, S.; Dell’Oro, R.; Mancia, G. Early Sympathetic Activation in the Initial Clinical Stages of Chronic Renal Failure. Hypertension 2011, 57, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Colombo, P.C.; Ganda, A.; Lin, J.; Onat, D.; Harxhi, A.; Iyasere, J.E.; Uriel, N.; Cotter, G. Inflammatory activation: Cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail. Rev. 2012, 17, 177–190. [Google Scholar] [CrossRef]
- Jazani, N.H.; Savoj, J.; Lustgarten, M.; Lau, W.L.; Vaziri, N.D. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic. Kidney Dis. 2019, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Elias, B.C.; Brooks, C.R.; Ueda, S.; Fukami, K. Uremic Toxin–Targeting as a Therapeutic Strategy for Preventing Cardiorenal Syndrome. Circ. J. 2019, 84, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Lisowska-Myjak, B. Uremic Toxins and Their Effects on Multiple Organ Systems. Nephron 2014, 128, 303–311. [Google Scholar] [CrossRef]
- Chung, A.C.; Zhang, H.; Kong, Y.Z.; Tan, J.J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.; Wang, Z.; Levison, B.S.; A Buffa, J.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.; Klein, A.L.; Hazen, S.L. Intestinal Microbiota-Dependent Phosphatidylcholine Metabolites, Diastolic Dysfunction, and Adverse Clinical Outcomes in Chronic Systolic Heart Failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut Microbiota-Dependent TrimethylamineN-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.; Rodriguez, V.; Vermali, R.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalar, B.; Haslberger, A.; Peterlin, B. The Role of Microbiota in Depression—A brief review. Psychiatr. Danub. 2018, 30, 136–141. [Google Scholar] [CrossRef]
- Bansilal, S.; Castellano, J.M.; Fuster, V. Withdrawn: Global burden of CVD: Focus on secondary prevention of cardiovascular disease. Int. J. Cardiol. 2016, 201, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of Urease- and Uricase-Containing, Indole- and p-Cresol-Forming and Contraction of Short-Chain Fatty Acid-Producing Intestinal Microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Barba, C.; Soulage, C.O.; Caggiano, G.; Glorieux, G.; Fouque, D.; Koppe, L. Effects of Fecal Microbiota Transplantation on Composition in Mice with CKD. Toxins 2020, 12, 741. [Google Scholar] [CrossRef]
- Li, D.Y.; Tang, W.W. Contributory Role of Gut Microbiota and Their Metabolites Toward Cardiovascular Complications in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 193–205. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhao, J.; Qu, W.; Zhang, Y.; Jia, B.; Fan, Z.; He, Q.; Li, J. Accumulation and effects of dietary advanced glycation end products on the gastrointestinal tract in rats. Int. J. Food Sci. Technol. 2018, 53, 2273–2281. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; Macfarlane, G. Formation of Phenolic and Indolic Compounds by Anaerobic Bacteria in the Human Large Intestine. Microb. Ecol. 1997, 33, 180–188. [Google Scholar] [CrossRef]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S.; Mathew, A.; Vaziri, N.D. Iron toxicity: Relevance for dialysis patients. Nephrol. Dial. Transplant. 2013, 29, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, A.; Yamamoto, S.; Oda, A.; Tanaka, K.; Kazama, J.J.; Saeki, T.; Yamazaki, H.; Ishioka, K.; Suzutani, T.; Narita, I. Effect of sucroferric oxyhydroxide on gastrointestinal microbiome and uremic toxins in patients with chronic kidney disease undergoing hemodialysis. Clin. Exp. Nephrol. 2020, 24, 725–733. [Google Scholar] [CrossRef]
- Hviid, A.; Svanström, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2010, 60, 49–54. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Wu, M.-J.; Chang, C.-S.; Cheng, C.-H.; Chen, C.-H.; Lee, W.-C.; Hsu, Y.-H.; Shu, K.-H.; Tang, M.-J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Zheng, H.J.; Guo, J.; Wang, Q.; Wang, L.; Wang, Y.; Zhang, F.; Huang, W.-J.; Zhang, W.; Liu, W.J.; Wang, Y. Probiotics, prebiotics, and synbiotics for the improvement of metabolic profiles in patients with chronic kidney disease: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2021, 61, 577–598. [Google Scholar] [CrossRef]
- Bakhtiary, M.; Morvaridzadeh, M.; Agah, S.; Rahimlou, M.; Christopher, E.; Zadro, J.R.; Heshmati, J. Effect of Probiotic, Prebiotic, and Synbiotic Supplementation on Cardiometabolic and Oxidative Stress Parameters in Patients With Chronic Kidney Disease: A Systematic Review and Meta-analysis. Clin. Ther. 2021, 43, e71–e96. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins during Hemodialysis Using a Binding Competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Etinger, A.; Kumar, S.R.; Ackley, W.; Soiefer, L.; Chun, J.; Singh, P.; Grossman, E.; Matalon, A.; Holzman, R.; Meijers, B. The effect of isohydric hemodialysis on the binding and removal of uremic retention solutes. PLoS ONE 2018, 13, e0192770. [Google Scholar]
- Lee, S.; Sirich, T.L.; Meyer, T.W. Improving Clearance for Renal Replacement Therapy. Kidney360 2021, 10, 340670002922021. [Google Scholar] [CrossRef]
- Anders, H.-J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Hung, S.; Kuo, K.; Wu, C.; Tarng, D. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Yonekura, Y.; Yamashita, Y.; Kono, K.; Nakai, K.; Goto, S.; Sugano, M.; Goto, S.; Fujieda, A.; Ito, Y.; et al. Anti-oxidative effect of AST-120 on kidney injury after myocardial infarction. Br. J. Pharmacol. 2016, 173, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Wu, V.; Wu, P.-C.; Wu, C.-J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef]
- Poesen, R.; Claes, K.; Evenepoel, P.; De Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-Derived Phenylacetylglutamine Associates with Overall Mortality and Cardiovascular Disease in Patients with CKD. J. Am. Soc. Nephrol. 2016, 27, 3479–3487. [Google Scholar] [CrossRef] [Green Version]
- Villard, A.; Boursier, J.; Andriantsitohaina, R. Bacterial and eukaryotic extracellular vesicles and non-alcoholic fatty liver disease: New players in the gut-liver axis? Am. J. Physiol. Liver Physiol. 2021, 320, 485. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.-C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating Endotoxemia: A Novel Factor in Systemic Inflammation and Cardiovascular Disease in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 6, 133–141. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Ortega, A.; Fernández, A.; Arenas, M.I.; López-Luna, P.; Muñóz-Moreno, C.; Arribas, I.; Olea, N.; García-Bermejo, L.; Lucio-Cazana, J.; Bosch, R.J. Outcome of Acute Renal Injury in Diabetic Mice with Experimental Endotoxemia: Role of Hypoxia-Inducible Factor-1α. J. Diabetes Res. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nakano, D.; Kitada, K.; Wan, N.; Zhang, Y.; Wiig, H.; Wararat, K.; Yanagita, M.; Lee, S.; Jia, L.; Titze, J.M.; et al. Lipopolysaccharide induces filtrate leakage from renal tubular lumina into the interstitial space via a proximal tubular Toll-like receptor 4–dependent pathway and limits sensitivity to fluid therapy in mice. Kidney Int. 2020, 97, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Masilamani, M.; Li, X.-M.; Sampson, H.A. The false alarm hypothesis: Food allergy is associated with high dietary advanced glycation end-products and proglycating dietary sugars that mimic alarmins. J. Allergy Clin. Immunol. 2017, 139, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2011, 26, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.M.; Kaskel, F. Update on Inflammation in Chronic Kidney Disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Bellocq, A.; Suberville, S.; Philippe, C.; Bertrand, F.; Perez, J.; Fouqueray, B.; Cherqui, G.; Baud, L. Low Environmental pH Is Responsible for the Induction of Nitric-oxide Synthase in Macrophages. J. Biol. Chem. 1998, 273, 5086–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, P.; Díez, J.J. Adipose tissue in renal disease: Clinical significance and prognostic implications. Nephrol. Dial. Transplant. 2010, 25, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S. The Kidney–Gut–Muscle Axis in End-Stage Renal Disease is Similarly Represented in Older Adults. Nutrients 2019, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaney, L.M.; Davies, O.G.; Selby, N.M. Gut microbial metabolites as mediators of renal disease: Do short-chain fatty acids offer some hope? Future Sci. 2019, 5, FSO384. [Google Scholar] [CrossRef] [Green Version]
- Rabb, H.; Griffin, M.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2015, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS–based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; De Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518–1524. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.; Sourris, K.; Thallas-Bonke, V.; Ziemann, M.; El-Osta, S.; Cooper, M.; Coughlan, M. SAT-301 Resistant Starch Ameliorates Advanced Glycation Endproduct-Induced Gut Dysbiosis and Albuminuria in a Mouse Model of Type 2 Diabetes. Kidney Int. Rep. 2019, 4, S134. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Liu, S.-M.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Farzaneh, S.H.; Kieffer, D.A.; Adams, S.H.; Martin, R.J. High Amylose Resistant Starch Diet Ameliorates Oxidative Stress, Inflammation, and Progression of Chronic Kidney Disease. PLoS ONE 2014, 9, e114881. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.R.; Gandolfo, M.T.; Ko, G.J.; Satpute, S.; Racusen, L.; Rabb, H. Early exposure to germs modifies kidney damage and inflammation after experimental ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2009, 297, F1457–F1465. [Google Scholar] [CrossRef] [Green Version]
- Emal, D.; Rampanelli, E.; Stroo, I.; Butter, L.M.; Teske, G.J.; Claessen, N.; Stokman, G.; Florquin, S.; Leemans, J.C.; Dessing, M.C. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2016, 28, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Henning, C.; Glomb, M.A. Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 2016, 33, 499–512. [Google Scholar] [CrossRef]
- de Oliveira, F.C.; Coimbra, J.S.; Oliveira, E.B.; Zuniga, A.D.G.; Rojas, E.E.R. Food Protein-polysaccharide Conjugates Obtained via the Maillard Reaction: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1108–1125. [Google Scholar] [CrossRef]
- Bunn, H.F.; Briehl, R.W. The interaction of 2,3-diphosphoglycerate with various human hemoglobins. J. Clin. Investig. 1970, 49, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Bunn, H.F.; Haney, D.N.; Kamin, S.; Gabbay, K.H.; Gallop, P.M. The biosynthesis of human hemoglobin A1c. Slow glycosylation of hemoglobin in vivo. J. Clin. Investig. 1976, 57, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, P.; Perichon, M.; Borot–Laloi, C.; Schaeverbeke, J.; Bakala, H. Accumulation of Advanced Glycation Endproducts in the Rat Nephron: Link with Circulating AGEs During Aging. J. Histochem. Cytochem. 1997, 45, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef]
- Nagai, R.; Unno, Y.; Hayashi, M.C.; Masuda, S.; Hayase, F.; Kinae, N.; Horiuchi, S. Peroxynitrite induces formation of N( epsilon )-(carboxymethyl) lysine by the cleavage of Amadori product and generation of glucosone and glyoxal from glucose: Novel pathways for protein modification by peroxynitrite. Diabetes 2002, 51, 2833–2839. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamagishi, S.-I.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Taguchi, K.; Miyazono, Y.; Uemura, K.; Koike, K.; Kurokawa, Y.; Nakayama, Y.; Kaida, Y.; Shibata, R.; Tsuchimoto, A.; et al. AGEs–RAGE overexpression in a patient with smoking-related idiopathic nodular glomerulosclerosis. CEN Case Rep. 2018, 7, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Yamamoto, Y.; Nangaku, M.; Usuda, N.; Okamato, H.; Kurokawa, K.; Strihou, C.V.Y.D.; Yamamoto, H.; Miyata, T. A Severe Diabetic Nephropathy Model with Early Development of Nodule-Like Lesions Induced by Megsin Overexpression in RAGE/iNOS Transgenic Mice. Diabetes 2006, 55, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-κB Pathway. Biomed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef] [Green Version]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE Expression in Human T Cells: A Link between Environmental Factors and Adaptive Immune Responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef]
- Avalos, A.M.; Kiefer, K.; Tian, J.; Christensen, S.; Shlomchik, M.; Coyle, A.J.; Marshak-Rothstein, A. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmun. Rev. 2009, 43, 103–110. [Google Scholar] [CrossRef]
- Abdulahad, D.; Westra, J.; Limburg, P.; Kallenberg, C.; Bijl, M. HMGB1 in systemic lupus Erythematosus: Its role in cutaneous lesions development. Autoimmun. Rev. 2010, 9, 661–665. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Stern, D.M.; Schmidt, A.M. RAGE is a multiligand receptor of the immunoglobulin superfamily: Implications for homeostasis and chronic disease. Cell. Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Li, L.-M.; Hou, D.-X.; Guo, Y.-L.; Yang, J.-W.; Liu, Y.; Zhang, C.-Y.; Zen, K. Role of MicroRNA-214–Targeting Phosphatase and Tensin Homolog in Advanced Glycation End Product-Induced Apoptosis Delay in Monocytes. J. Immunol. 2011, 186, 2552–2560. [Google Scholar] [CrossRef]
- Weinhage, T.; Wirth, T.; Schütz, P.; Becker, P.; Lueken, A.; Skryabin, B.V.; Wittkowski, H.; Foell, D. The Receptor for Advanced Glycation Endproducts (RAGE) Contributes to Severe Inflammatory Liver Injury in Mice. Front. Immunol. 2020, 11, 1157. [Google Scholar] [CrossRef]
- Sadik, N.A.; Mohamed, W.A.; Ahmed, M.I. The association of receptor of advanced glycated end products and inflammatory mediators contributes to endothelial dysfunction in a prospective study of acute kidney injury patients with sepsis. Mol. Cell. Biochem. 2012, 359, 73–81. [Google Scholar] [CrossRef]
- Durning, S.P.; Preston-Hurlburt, P.; Clark, P.R.; Xu, D.; Herold, K.C.; Type 1 Diabetes TrialNet Study Group. The Receptor for Advanced Glycation Endproducts Drives T Cell Survival and Inflammation in Type 1 Diabetes Mellitus. J. Immunol. 2016, 197, 3076–3085. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Hofmann, M.; Drury, S.; Hudson, B.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.T.; Sirois, C.M.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Martens, H.; A Nienhuis, H.L.; Gross, S.; Van Der Steege, G.; Brouwer, E.; Berden, J.H.M.; De Sévaux, R.G.L.; Derksen, R.H.W.M.; E Voskuyl, A.; Berger, S.P.; et al. Receptor for advanced glycation end products (RAGE) polymorphisms are associated with systemic lupus erythematosus and disease severity in lupus nephritis. Lupus 2012, 21, 959–968. [Google Scholar] [CrossRef]
- Wiedeman, A.M.; Barr, S.I.; Green, T.J.; Xu, Z.; Innis, S.M.; Kitts, D.D. Dietary Choline Intake: Current State of Knowledge Across the Life Cycle. Nutrients 2018, 10, 1513. [Google Scholar] [CrossRef] [Green Version]
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2010, 34, 3–15. [Google Scholar] [CrossRef]
- Koc, H.; Mar, M.H.; Ranasinghe, A.; Swenberg, J.; Zeisel, S. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal. Chem. 2002, 74, 4734–4740. [Google Scholar] [CrossRef]
- Rigault, C.; Mazué, F.; Bernard, A.; Demarquoy, J.; Le Borgne, F. Changes in l-carnitine content of fish and meat during domestic cooking. Meat Sci. 2008, 78, 331–335. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nat. Cell Biol. 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laxson, C.J.; Condon, N.E.; Drazen, J.; Yancey, P.H. Decreasing urearatiotrimethylamine N-oxide ratios with depth in chondrichthyes: A physiological depth limit? Physiol. Biochem. Zool. 2011, 84, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2017, 32, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef]
- Bielinska, K.; Radkowski, M.; Grochowska, M.; Perlejewski, K.; Huc, T.; Jaworska, K.; Motooka, D.; Nakamura, S.; Ufnal, M. High salt intake increases plasma trimethylamine N-oxide (TMAO) concentration and produces gut dysbiosis in rats. Nutrition 2018, 54, 33–39. [Google Scholar] [CrossRef]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Li, D.Y.; Wang, Z.; Jia, X.; Yan, D.; Shih, D.M.; Hazen, S.L.; Lusis, A.J.; Tang, W.W. Loop Diuretics Inhibit Renal Excretion of Trimethylamine N-Oxide. JACC: Basic Transl. Sci. 2021, 6, 103–115. [Google Scholar] [CrossRef]
- Bush, K.T.; Singh, P.; Nigam, S.K. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight 2020, 5, e133817. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef]
- Lee, T.C.; Kimiagar, M.; Pintauro, S.J.; Chichester, C. Physiological and safety aspects of Maillard browning of foods. Prog. Food Nutr. Sci. 1981, 5, 243–256. [Google Scholar]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [Green Version]
- Koschinsky, T.; He, C.-J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [Green Version]
- Münch, G.; Gerlach, M.; Sian, J.; Wong, A.; Riederer, P. Advanced glycation end products in neurodegeneration: More than early markers of oxidative stress? Ann. Neurol. 1998, 44, S85–S88. [Google Scholar] [CrossRef] [PubMed]
- Mitome, J.; Yamamoto, H.; Saito, M.; Yokoyama, K.; Marumo, K.; Hosoya, T. Nonenzymatic Cross-Linking Pentosidine Increase in Bone Collagen and Are Associated with Disorders of Bone Mineralization in Dialysis Patients. Calcif. Tissue Int. 2011, 88, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.-I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of advanced glycation end products with sarcopenia and frailty in chronic kidney disease. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.; Ferreira, G.A.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef]
- Ueda, S.; Yamagishi, S.-I.; Takeuchi, M.; Kohno, K.; Shibata, R.; Matsumoto, Y.; Kaneyuki, U.; Fujimura, T.; Hayashida, A.; Okuda, S. Oral Adsorbent AST-120 Decreases Serum Levels of AGEs in Patients with Chronic Renal Failure. Mol. Med. 2006, 12, 180–184. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J.; Cai, W.; Goodman, S.; Pyzik, R.; Post, J.; Grosjean, F.; Woodward, M.; Striker, G.E. Effects of Sevelamer on HbA1c, Inflammation, and Advanced Glycation End Products in Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 934–942. [Google Scholar] [CrossRef]
- Kakuta, T.; Tanaka, R.; Hyodo, T.; Suzuki, H.; Kanai, G.; Nagaoka, M.; Takahashi, H.; Hirawa, N.; Oogushi, Y.; Miyata, T.; et al. Effect of Sevelamer and Calcium-Based Phosphate Binders on Coronary Artery Calcification and Accumulation of Circulating Advanced Glycation End Products in Hemodialysis Patients. Am. J. Kidney Dis. 2011, 57, 422–431. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.M.; Milan, A.M.; Mitchell, C.J.; Gillies, N.A.; D’Souza, R.F.; Zeng, N.; Ramzan, F.; Sharma, P.; O’Knowles, S.; Roy, N.C.; et al. Protein Intake at Twice the RDA in Older Men Increases Circulatory Concentrations of the Microbiome Metabolite Trimethylamine-N-Oxide (TMAO). Nutrients 2019, 11, 2207. [Google Scholar] [CrossRef] [Green Version]
- Brinkley, T.; Semba, R.D.; Kritchevsky, S.B.; Houston, D.K. Dietary protein intake and circulating advanced glycation end product/receptor for advanced glycation end product concentrations in the Health, Aging, and Body Composition Study. Am. J. Clin. Nutr. 2020, 112, 1558–1565. [Google Scholar] [CrossRef]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Tahara, A.; Tahara, N.; Yamagishi, S.-I.; Honda, A.; Igata, S.; Nitta, Y.; Bekki, M.; Nakamura, T.; Sugiyama, Y.; Sun, J.; et al. Ratio of serum levels of AGEs to soluble RAGE is correlated with trimethylamine-N-oxide in non-diabetic subjects. Int. J. Food Sci. Nutr. 2017, 68, 1013–1020. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Hurot, J.-M.; Cucherat, M.; Haugh, M.; Fouque, D. Effects of L-carnitine supplementation in maintenance hemodialysis patients: A systematic review. J. Am. Soc. Nephrol. 2002, 13, 708–714. [Google Scholar] [CrossRef]
- Adachi, T.; Fukami, K.; Yamagishi, S.-I.; Kaida, Y.; Ando, R.; Sakai, K.; Adachi, H.; Otsuka, A.; Ueda, S.; Sugi, K.; et al. Decreased serum carnitine is independently correlated with increased tissue accumulation levels of advanced glycation end products in haemodialysis patients. Nephrology 2012, 17, 689–694. [Google Scholar] [CrossRef]
- Fukami, K.; Yamagishi, S.; Sakai, K.; Kaida, Y.; Adachi, K.; Ando, R.; Okuda, S. Potential inhibitory effects of L-carnitine supplementation on tissue advanced glycation end products in patients with hemodialysis. Rejuvenation Res. 2013, 16, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Yamagishi, S.-I.; Sakai, K.; Kaida, Y.; Yokoro, M.; Ueda, S.; Wada, Y.; Takeuchi, M.; Shimizu, M.; Yamazaki, H.; et al. Oral L-Carnitine Supplementation Increases Trimethylamine-N-oxide but Reduces Markers of Vascular Injury in Hemodialysis Patients. J. Cardiovasc. Pharmacol. 2015, 65, 289–295. [Google Scholar] [CrossRef]
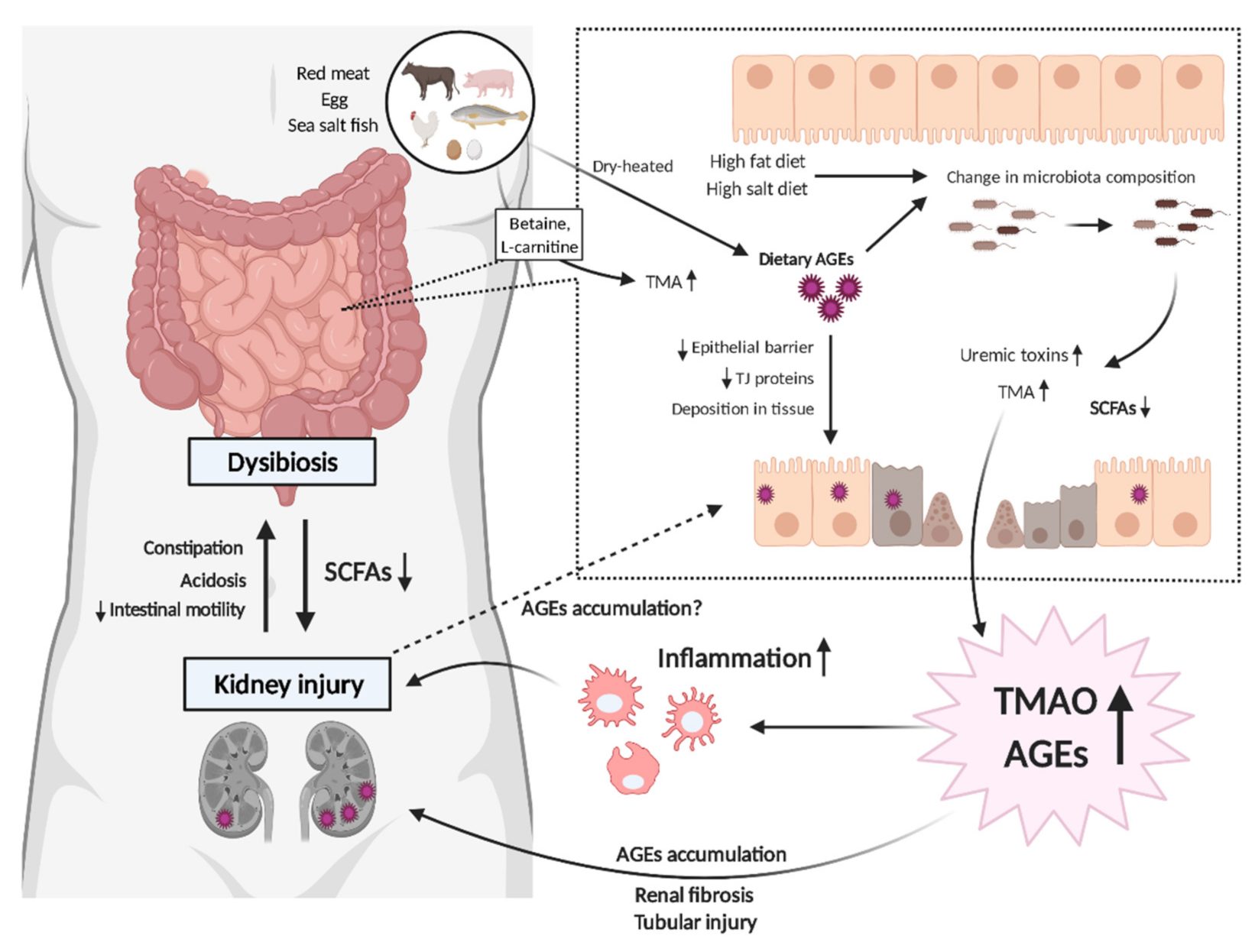
Figure 1.
Schematic of the mutual link between gut dysbiosis and uremic toxins in chronic kidney disease (CKD). Excess dietary intake of advanced glycation endproducts (AGEs) affects the composition of the gut microbiome, leading to further uremic toxin production, which results in kidney injury. AGEs deposition in the gastrointestinal tract also disrupts the epithelial barrier, allowing bacterial components and endotoxins to flow into the systemic circulation, which, in turn, leads to other organ damage. The high burdens of uremic toxins such as trimethylamine N-oxide (TMAO) or AGEs are linked to progressive tubular injury and renal fibrosis, which are associated with the development of CKD to end stage renal disease.
Figure 1.
Schematic of the mutual link between gut dysbiosis and uremic toxins in chronic kidney disease (CKD). Excess dietary intake of advanced glycation endproducts (AGEs) affects the composition of the gut microbiome, leading to further uremic toxin production, which results in kidney injury. AGEs deposition in the gastrointestinal tract also disrupts the epithelial barrier, allowing bacterial components and endotoxins to flow into the systemic circulation, which, in turn, leads to other organ damage. The high burdens of uremic toxins such as trimethylamine N-oxide (TMAO) or AGEs are linked to progressive tubular injury and renal fibrosis, which are associated with the development of CKD to end stage renal disease.


{kind=link}
Table 1.
Previous studies indicating possible correlation between AGEs and TMAO.
| Authors [Reference] (Number of Participants) | Intervention | Outcome |
|---|---|---|
| Mitchell et al. [130] (n = 20) | High protein diet (1.6 g/kgBW/day) | Serum TMAO↑ |
| Brinkley et al. [131] (n = 2439) | High protein diet (≥1.2 g/kgBW/day) | Serum Nε-CML↑, Serum sRAGE↑ |
| Yacoub et al. [28] (n = 20) | Restriction of dietary AGEs intake | Serum Nε-CML↓, Serum methylglyoxal-derivatives↓ Prevotella copri↓, Bifidobacterium animalis↓ Alistipes indistinctus↑, Clostridium citroniae↑ |
| Adachi et al. [136] (n = 204) | Observational study in healthy subjects (n = 75) and HD patients (n = 129) | Clostridium hathewayi↑, Ruminococcus gauvreauii↑ Serum-free carnitine inversely correlates with skin AGEs |
| Tahara et al. [133] | Observational study in non-diabetic subjects | Clostridium hathewayi↑, Ruminococcus gauvreauii↑ Serum-free carnitine inversely correlates with skin AGEs |
| Fukami et al. [137] (n = 102, HD patients) | Oral L-carnitine supplementation (900 mg/d), six months | Skin AGEs↓ Serum-free carnitine inversely correlates with the decrease in skin AGEs |
| Fukami et al. [138] (n = 31, HD patients) | Oral L-carnitine supplementation (900 mg/d), six months | Vascular injury markers (sICAM-1, sVCAM-1)↓ Oxidative stress marker (MDA)↓ Serum AGE tends to be decreased TMA↑, TMAO↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Taguchi, K.; Fukami, K.; Elias, B.C.; Brooks, C.R. Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins 2021, 13, 361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050361
AMA Style
Taguchi K, Fukami K, Elias BC, Brooks CR. Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease. Toxins. 2021; 13(5):361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050361
Chicago/Turabian StyleTaguchi, Kensei, Kei Fukami, Bertha C. Elias, and Craig R. Brooks. 2021. "Dysbiosis-Related Advanced Glycation Endproducts and Trimethylamine N-Oxide in Chronic Kidney Disease" Toxins 13, no. 5: 361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050361
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.