Therapeutic Uses of Bacterial Subunit Toxins

1
Division of Molecular Medicine, Research Institute, Hospital for Sick Children, Toronto, ON M5G 1X8, Canada
2
Departments of Laboratory Medicine & Pathobiology, and Biochemistry, University of Toronto, Toronto, ON M5S 1A8, Canada
Toxins 2021, 13(6), 378; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060378
Submission received: 12 April 2021
/
Revised: 17 May 2021
/
Accepted: 18 May 2021
/
Published: 26 May 2021
(This article belongs to the Special Issue Application of Protein Toxins as Cell Biological and Pharmacological Tools)
Abstract
:The B subunit pentamer verotoxin (VT aka Shiga toxin-Stx) binding to its cellular glycosphingolipid (GSL) receptor, globotriaosyl ceramide (Gb3) mediates internalization and the subsequent receptor mediated retrograde intracellular traffic of the AB5 subunit holotoxin to the endoplasmic reticulum. Subunit separation and cytosolic A subunit transit via the ER retrotranslocon as a misfolded protein mimic, then inhibits protein synthesis to kill cells, which can cause hemolytic uremic syndrome clinically. This represents one of the most studied systems of prokaryotic hijacking of eukaryotic biology. Similarly, the interaction of cholera AB5 toxin with its GSL receptor, GM1 ganglioside, is the key component of the gastrointestinal pathogenesis of cholera and follows the same retrograde transport pathway for A subunit cytosol access. Although both VT and CT are the cause of major pathology worldwide, the toxin–receptor interaction is itself being manipulated to generate new approaches to control, rather than cause, disease. This arena comprises two areas: anti neoplasia, and protein misfolding diseases. CT/CTB subunit immunomodulatory function and anti-cancer toxin immunoconjugates will not be considered here. In the verotoxin case, it is clear that Gb3 (and VT targeting) is upregulated in many human cancers and that there is a relationship between GSL expression and cancer drug resistance. While both verotoxin and cholera toxin similarly hijack the intracellular ERAD quality control system of nascent protein folding, the more widespread cell expression of GM1 makes cholera the toxin of choice as the means to more widely utilise ERAD targeting to ameliorate genetic diseases of protein misfolding. Gb3 is primarily expressed in human renal tissue. Glomerular endothelial cells are the primary VT target but Gb3 is expressed in other endothelial beds, notably brain endothelial cells which can mediate the encephalopathy primarily associated with VT2-producing E. coli infection. The Gb3 levels can be regulated by cytokines released during EHEC infection, which complicate pathogenesis. Significantly Gb3 is upregulated in the neovasculature of many tumours, irrespective of tumour Gb3 status. Gb3 is markedly increased in pancreatic, ovarian, breast, testicular, renal, astrocytic, gastric, colorectal, cervical, sarcoma and meningeal cancer relative to the normal tissue. VT has been shown to be effective in mouse xenograft models of renal, astrocytoma, ovarian, colorectal, meningioma, and breast cancer. These studies are herein reviewed. Both CT and VT (and several other bacterial toxins) access the cell cytosol via cell surface ->ER transport. Once in the ER they interface with the protein folding homeostatic quality control pathway of the cell -ERAD, (ER associated degradation), which ensures that only correctly folded nascent proteins are allowed to progress to their cellular destinations. Misfolded proteins are translocated through the ER membrane and degraded by cytosolic proteosome. VT and CT A subunits have a C terminal misfolded protein mimic sequence to hijack this transporter to enter the cytosol. This interface between exogenous toxin and genetically encoded endogenous mutant misfolded proteins, provides a new therapeutic basis for the treatment of such genetic diseases, e.g., Cystic fibrosis, Gaucher disease, Krabbe disease, Fabry disease, Tay-Sachs disease and many more. Studies showing the efficacy of this approach in animal models of such diseases are presented.
Keywords:
retrograde transport; neoplastic Gb3 expression; endoplasmic reticulum associated degradation; protein misfolding diseasesKey Contribution: Shiga/verotoxin and toxin conjugates can be used in animal models as a therapy to target human tumour cells and their neovasculature. A subunit inactivated cholera toxin can be used as an exogenous competitive inhibitor of endoplasmic reticulum associated degradation in models of genetic protein misfolding diseases to rescue the deficiency disease phenotype.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The Verocytotoxin (VT, Shiga toxin: Stx) was first shown to be the cause of the hemolytic uremic syndrome (HUS) in 1985 by Karmali [1]. HUS was primarily a potentially fatal acute pediatric renal disease of unknown origin with a triad of symptoms: renal glomerular infarct, thrombocytopenia and anemia [2]. VT is a family of A-B5 subunit E. coli toxins [3] in which VT1 and VT2 are primarily associated with HUS [4,5]. Gastrointestinal infection with VT producing E. coli initially causes bloody diarrhea and the resulting systemic toxemia leads to renal disease some days later. VT2 is more commonly associated with human disease [6,7] and is additionally associated with encephalopathy [8].
The receptor of the VT B subunit pentamer is the glycosphingolipid, globotriaosyl ceramide, Gb3: Galα 1-4 galβ1-4Glc-cer [9]. This is the only VT receptor to mediate cytopathology [10,11]. VT1 shows a higher Gb3 binding affinity than VT2 [12]. Gb3 is highly expressed in human renal glomerular endothelial cells [13]. Membrane GSLs, together with cholesterol, are major components of lipid rafts which are membrane areas of increased ‘order’ [14] which serve as foci of transmembrane signalling [15] and portals for microbial–host cell interactions [16,17]. VT bound Gb3 is internalized by clathrin-dependent [18,19] and independent [20,21,22] mechanisms. Pentameric VT binding can cluster cell surface Gb3 to induce membrane curvature, invagination and vesicle scission [21,22,23]. Internalized VT is trafficked to endosomes and thence, via retrograde transport, to the Golgi and then ER. VT-> ER targeting is required for pathogenesis and requires that the cell surface bound Gb3 is within lipid rafts [24]. Non-raft Gb3 bound VT is, in contrast, internalized and transported to lysosomes [24]. This sorting corresponds with the presence of Gb3 in rafts in human renal glomerular endothelial cells, the site of primary pathology [25], but not in renal tubular epithelial cells [13] (which are affected only in later HUS stages [13]). The microvascular endothelial cell raft and non-raft Gb3 fatty acid composition has recently been found to vary, with saturated species restricted to the raft and unsaturated species to the non-raft fraction [26]. Moreover, different Gb3 fatty acid binding preferences of distinct Gb3 binding ligands can result in differential raft membrane Gb3 clustering [27].
In the ER, the A and B subunits separate [28] and a C-terminal A subunit sequence, which mimics an unfolded protein [29] and can insert in the ER membrane [30], becomes exposed. This ‘misfolded peptide’ recruits the ERAD (host cell quality control ER associated degradation) machinery [31] to transit the A subunit through the ER membrane via the ERAD dislocon [32] to the cytosol. Here, the A subunit avoids proteasomal degradation by mechanisms which prevent lysine ubiquitinylation [31] and inhibits protein synthesis via an RNA N-glycanase activity [33]. A similar trafficking pathway is followed by cholera toxin A subunit [34]. CT binds the widely expressed GSL, GM1 ganglioside, and similarly undergoes (raft dependent [35]) retrograde transport to the ER [36]. The cytosolic transit requires the chaperone-mediated unfolding of the A subunit and the threading of the linearized subunit through the membrane dislocon (retrotranslocon) to the cytosol where the A subunit refolds. This is unlike natural misfolded proteins which are unfolded in the ER and retrotranslocated through the dislocon via ubiqitinylation, to the cytosolic proteosome for degradation [37]. The VT and CT A subunits escape degradation by incompletely understood mechanisms which may be lipid-raft-dependent [31], and refold in the cytoplasm to effect cytopathology.
The exact composition and ATP dependent action mechanism of the dislocon are still a matter of debate [31,38] and varies as a function of substrate. Reverse transit of the Sec61 translocon was initially considered the primary mechanism [39]. While Sec61 retains a central role in current translocation models [31,40,41,42], more recent studies show more complex mechanisms. The Derlin proteins [43,44], p97 ATPase [45], Hrd 1 ubiquitin ligase [46,47], and SEL1 adaptor protein [48] are established components (37). Recently, Hrd 1 alone has been shown—as predicted [46]—to form pores in model membranes [49]. Both VT [50] and CT hijack the dislocon [51] and indeed, CT has been frequently used a tool to investigate ERAD [31,52,53].
In addition to inhibition of protein synthesis, VTs have been shown to induce apoptosis [54,55,56,57,58,59,60,61,62]. This can be due to the pentameric B subunit binding Gb3 [63,64,65], a property shared by anti-Gb3 [66], or require the intact holotoxin [62,67]. VT1 induces apoptosis in many human cancer cell lines [68,69,70,71,72], but VT-induced ER stress can also activate survival pathways [73].
2. Gb3 Is a Cancer Marker
Prior to the discovery of the VT receptor role for Gb3, Gb3 was considered only a minor GSL in biosynthesis of the major red cell GSL, globoside, Gb4 [74]. Gb4 is part of the P blood group in which Gb3 is Pk [75]. Its sole claim to fame was that of being the BLA-Burkitt lymphoma antigen [76,77]. Gb3 was shown to be CD77, a germinal center B cell antigen [78] required for B cell apoptosis [79]. However, even at these early stages, Gb3 became associated with lymphoid malignancy [80].
Gb3 is upregulated in many human cancers [81,82]. These include breast [72,83], ovarian [84,85], Burkitt’s lymphoma [63], hairy cell leukemia [86], megakaryoblastic leukemia [87], myeloma [88], post-transplant lymphoproliferative disease [89], renal [90], colorectal [65,91], gastric [70,92], testicular [90,93], prostate [90], pancreatic [94,95,96], sarcoma [97], glioma [71], astrocytoma [98,99] and meningioma [100].
Importantly, irrespective of the primary tumour Gb3 expression status, the proliferating endothelial cells of the tumour neovasculature also express Gb3, [101], and are, therefore, also targeted by VT in tumour xenograft models [100,102,103], to block tumour angiogenesis. VT binding has been proposed as a marker of tumour neovasculature [104] and tumour infiltrating blood vessels [81]. Anti Gb3 also prevents neoangiogenesis and tumour growth [105,106].
VT has been shown effective to prevent the growth of many Gb3 expressing human tumour xenografts in mice [65,70,90,98,100,104]. In these models, targeting of the neovasculature, in addition to the tumour per se, was observed [98,100,101]. In colon cancer, expression of Gb3 synthase was found to be sufficient to initiate metastases [65] and Gb3 expression correlates with lymphoma tumorigenicity [107].
2.1. Is This the Limit of Gb3 Detection?
Although many tumours express Gb3, this may represent an underestimate of potential tumour targeting. The receptor function of membrane Gb3 (and other GSLs) is complex [108] in that the lipid moiety [26,109] and the membrane environment [26,110] play a central role in Gb3 accessibility for ligand binding GSL. Differential tissue binding of VT and anti-Gb3 antibodies [111] and the differential detection of cell surface Gb3 by VTB and various antiGb3 Mabs [112] further indicate the importance of Gb3 membrane presentation. This has been termed cryptic GSL expression [113]. The carbohydrate moiety of GSLs can exist in a variety of conformations and this is affected by the GSL lipid moiety and relative plane of the membrane [114]. While the exact basis of this is not known in all cases, one situation has been mechanistically characterized. GSLs and cholesterol are the major component of lipid rafts [115]. In the membrane GSL-cholesterol complex, H-bonding results in a change in the GSL sugar conformation, from a membrane perpendicular to membrane parallel format [116,117]. In the parallel format, access for exogenous ligand binding is severely restricted. We have used the term ‘invisible GSLs’ to describe this phenomenon [118]. β-Methyl cyclodextrin (MCD) has been used to selectively extract cholesterol from cells in vitro [119] and in vivo [120]. Treatment of human tumour sections with MCD can markedly increase tumour GSL ‘exposure’ and the binding of VT1 (and other tumour associated GSL binding ligands) is greatly increased [121]. Less than 10% of tumour Gb3 may be available for VT binding without prior cholesterol extraction. Thus, MCD markedly increases membrane GSL exposure.
However, the fact that Gb3 needs to be within (cholesterol containing?) lipid rafts for VT cell killing [24] needs to be considered. A balance between cholesterol extraction for increased binding and potentially reduced ER trafficking needs to be achieved.
2.2. Cancer Stem Cells
Glycosphingolipid antigens have been used as markers of human stem cells [122,123,124] and cancer stem cells [125,126]. These are, for the most part, globoseries GSLs, and Gb3 has been defined as a marker of breast cancer, but not normal mammary, stem cells [127]. However, this has been contested [128]. Moreover, cancer stem cell express high levels of ABC transporters [129], which can be involved in GSL biosynthesis [130].
3. Verotoxin as a Cancer Targeting Tool
The extremely high binding affinity of the B subunit pentamer for membrane Gb3 (>10−9 M [131,132]) and the widespread overexpression of Gb3 in human cancers makes verotoxin an attractive antineoplastic targeting tool.
3.1. B Subunit Conjugates
Due to the concern that VT holotoxin is central in the etiology of HUS, several anti-proliferative drugs have been conjugated to the nontoxic VT B subunit to increase tumour selective cytotoxicity [92,133,134,135]. This has been combined with delivery of a prodrug which requires subsequent activation [133,134], since, if side effects are a problem for the therapeutic use of the holotoxin, changing the toxic moiety delivered should not affect targeting and or side effects. In addition, B subunit conjugates have been developed to provide tools for in vivo tumour imaging [83,91,101,103]. Fluorescent VT/VTB has long been used to image Gb3 positive cells in vitro [20,136]. B subunit conjugates containing additional C terminal peptides retain native Gb3 binding [137]. The addition of a C terminal cysteine residue to the B subunit has facilitated the generation of such conjugates via disulfide linkage [138]. Such linker conjugates will be cleaved by reduction in the tumour ER [135,139,140] to allow specific ER cargo delivery [141]. A peptide which promotes ER retrotranslocon transit can be coupled to increase cytosolic cargo access [142,143].
In a different approach, Lactococcus bacteria have been transfected to express surface VTB as a potential antineoplastic drug carrier [144].
3.2. Native VT1 in Cancer Therapy
3.2.1. Potency
The use of the native VT1 toxin provides a far more potent antineoplastic approach than coupling drugs to the B subunit. Although the Gb3 binding affinity is in the nanomolar range, VT1 has proven cytotoxic in vitro at a dose of 10−13–10−17 M, [146,147], up to eight orders of magnitude lower. It has been proposed, and consistent evidence reported [148], that the catalytic activity of one cytosolic A subunit molecule is sufficient to kill a cell.
3.2.2. Risk
The question is ‘does the antineoplastic use of the VT holotoxin pose a risk for HUS?’. To address this question, firstly the demographics of VTEC HUS need to be considered. HUS is primarily a disease of the young and the elderly [149]. Thus, there is an age window. Secondly, pathology is more closely associated with VT2 producing EHEC than VT1 [150,151]. Thirdly, can VT1 alone cause HUS? In this regard, the baboon is the only appropriate model. In the baboon primate model, an i.v. bolus dose of 100 ng/kg VT1 was sufficient to induce the classical symptoms of HUS [152,153]. However, if this same VT1 dose was divided into four which were administered every 12 h for 48 h, no subsequent symptoms of HUS were observed [153]. At equal doses, only VT2 induced pathology [154]. Thus, there is a ‘safe’ sterile VT1 i.v dose which can be repeated for four days (at least).
VT1 antineoplastic treatment may temporarily dysregulate erythropoiesis [155,156] but if necessary, transfusion is a clear option.
In the in vivo context of gastrointestinal EHEC infection and presumed systemic verotoxemia, the situation is more complex. Bacterial LPS [157,158] and various cytokines induced by the infection [159,160,161,162] can upregulate cellular synthesis of Gb3 [163,164,165], increasing VT susceptibility and the number of cells killed by VT [161,166]. Indeed, LPS coadministration increased VT pathology in the baboon [167]. In addition, VT can induce the expression of such cytokines [168,169,170]. Thus, HUS is the long term (3–7 days) culmination of a ‘perfect storm’ arising from the initial gastrointestinal EHEC infection. This provides a therapeutic opportunity for a sterile VT1 bolus. Furthermore, Gb3 receptor expression within lipid rafts in normal cells is required for cytopathology and tumour cells have higher levels of lipid rafts [171]. Finally, HUS is rarely fatal whereas cancer, e.g., pancreatic, always is. Any potential circulating VT-induced renal pathology may prove treatable [172,173]
Intratumoural VT1 administration into a Gb3 expressing tumour provides a further means to reduce risk. Although circulating VT will concentrate in a Gb3 positive tumour [92,103], high affinity receptor binding within the tumour following judicious i.t. administration, will localize the toxin to the tumour and limit systemic access. In human tumour xenograft models, intratumoural VT injection can eliminate the tumour, which indicates the toxin spreads throughout the tumour from the injection site. Derivatized VTB can be used to image Gb3 expressing tumours in vivo [101,174] and could thereby serve to accurately guide intratumoural treatment and to monitor response efficacy.
3.2.3. Efficacy
Comparison of the antineoplastic efficacy of B subunit-drug conjugates and native VT1 holotoxin have not been made. However, tumour xenografts were reduced by 50% by an optimized B subunit-p53 inhibitor complex after 6× 10 mg/kg administrations every 48 h [143], whereas a single 4µg/kg VT1 treatment eliminated a xenograft tumour within 7 days [102]. In these first tumour xenograft studies, we determined that a single intratumoural dose of 1 µg VT1/50 mm diameter tumour was sufficient to reduce the astrocytoma size by 50% within 2 days with tumour elimination after about a week [102]
4. Verotoxin Interaction with Lymphoid Cells
4.1. As an Immunogen Carrier
Since human germinal center B cells [175] and more importantly, dendritic cells [176] express Gb3, verotoxin B subunit [138,177] or holotoxin [178] can be used as a carrier to more effectively deliver immunogens to the cytosol for proteasomal processing. Peptides thus generated, can readily transit back to the ER via the TAP peptide transporter [138,179] for loading onto MHC-1 within the ER for anterograde transport and cell surface antigen presentation [176,180,181]. ER antigen targeting facilitates dendritic cell proteasomal antigen processing and is, therefore, a more efficient immunization protocol [182]. This procedure was utilized to develop a cancer vaccine against the Her2/neu breast tumour antigen [181]. VTB subunit is an effective murine nasal mucosa immunogen vector [183,184] to elicit resident memory T cells and antitumour antibodies. Significantly, VT1 was found to bind extensively to mouse nasal turbinates [185] which, if reflective of the human nasal mucosa, would provide a unique immunoantineoplastic carrier.
4.2. Effects on Lymphoid Cells
The binding of VT to B lymphocytes is complex. Gb3 is also known as CD77 [186], a differentiation antigen expressed on germinal centre B cells [79] associated with apoptosis [187]. Due to immunoglobulin isotype switching in germinal centres [188] and the inhibition of IgG/A production by VT in vitro, we suggested this was a basis for the failure to develop long-lived immunity to VT after infection [189]. Despite this early work, the effect of VT on the immune system remains largely unknown. VT treated monocytes release proinflammatory cytokines [190] which may be enhanced by neutrophil VT delivery [191].
Recent reports show VT can have a surprising caspase-mediated anti-inflammatory activity against LPS [192] in macrophages and in mice. It may, therefore, be possible eventually, to use VT or derivative, as an anti-inflammatory or to identify anti-inflammatory targets. Interestingly, macrophage Gb3 is not expressed in lipid rafts [24] and VT retrograde transport to the ER does not occur. This lack of VT susceptibility may permit this (and other? e.g., [193]—see below) A subunit, holotoxin effects.
5. Verotoxin A Subunit Redirection of Intracellular Traffic
In a recent interesting and perhaps landmark study [193], the catalytic A subunit inactivated holotoxoid of VT2a(Stx2a) was found to partially undergo a differential intracellular traffic route (as opposed to the retrograde ER transport of all other verotoxin family members) from the cell surface to the lysosome for degradation, consistent with the reported more varied intracellular trafficking of the VT2 toxin family [136]. This lysosomal trafficking pathway is normally followed for VT binding non-raft cell surface Gb3 [24], which would suggest that this VT2a toxoid is able to selectively bind non-raft cell surface Gb3. In addition, the inactivated VT2a holotoxin was able to redirect the traffic of amyloid precursor protein (APP) to the lysosome for degradation in treated cells also. This intracellular misdirection of APP prevented the subsequent cellular release of the amyloid-β peptide, and thereby may provide a novel basis for the exogenous therapy of Alzheimer’s disease, particularly since VT2 is more frequently associated with the encephalopathogenic features of VT producing E. coli infections [8].
The mechanistic basis of such an approach is worthy of consideration. The Gb3 binding and subsequent intracellular traffic is B subunit dependent. Why is the (inactivated) A subunit required? Perhaps this is related to VT2 A subunit binding serum amyloid P [194] and the relationship between amyloid P and β amyloid [195,196,197,198]. Retrograde traffic to [24,199,200], and anterograde traffic from [201] the ER, are cholesterol dependent and the interactions between cholesterol and GSLs [202,203,204] can oppositely affect the GSL binding of VT [117] and β amyloid [116,205,206,207]. GSL biosynthesis promotes APP and β amyloid secretion [208] and both membrane GSLs and cholesterol regulate the β secretase which generates β amyloid [209].
6. Cholera Toxin as a Targeted ER Associated Degradation (ERAD) Blockade
Both VT and CT hijack endoplasmic reticulum associated degradation (ERAD) for A subunit cytosolic access. ERAD is the cellular quality control mechanism by which nascent polypeptides are screened for correct three dimensional folding [210]. It involves their sampling by various ER chaperones [211,212] and folding enzymes [213], unfolding/refolding opportunities [214], unfolding [215], ubiquitinylation [216], ER-cytosolic transfer [217] and proteasomal degradation [218]. Protein misfolding is central in many genetic diseases, and in those with small mutations that do not ablate function, ERAD plays a key role in initiating/amplifying deficiency disease symptoms [219].
Intracellular Plumbing
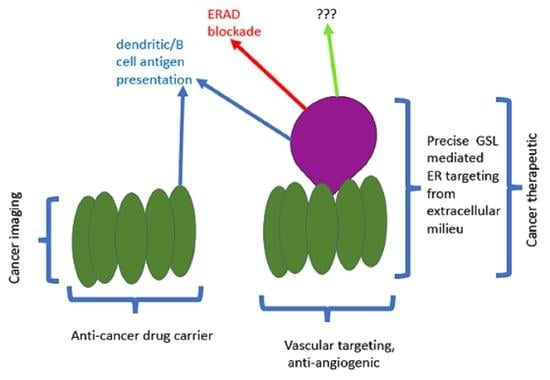
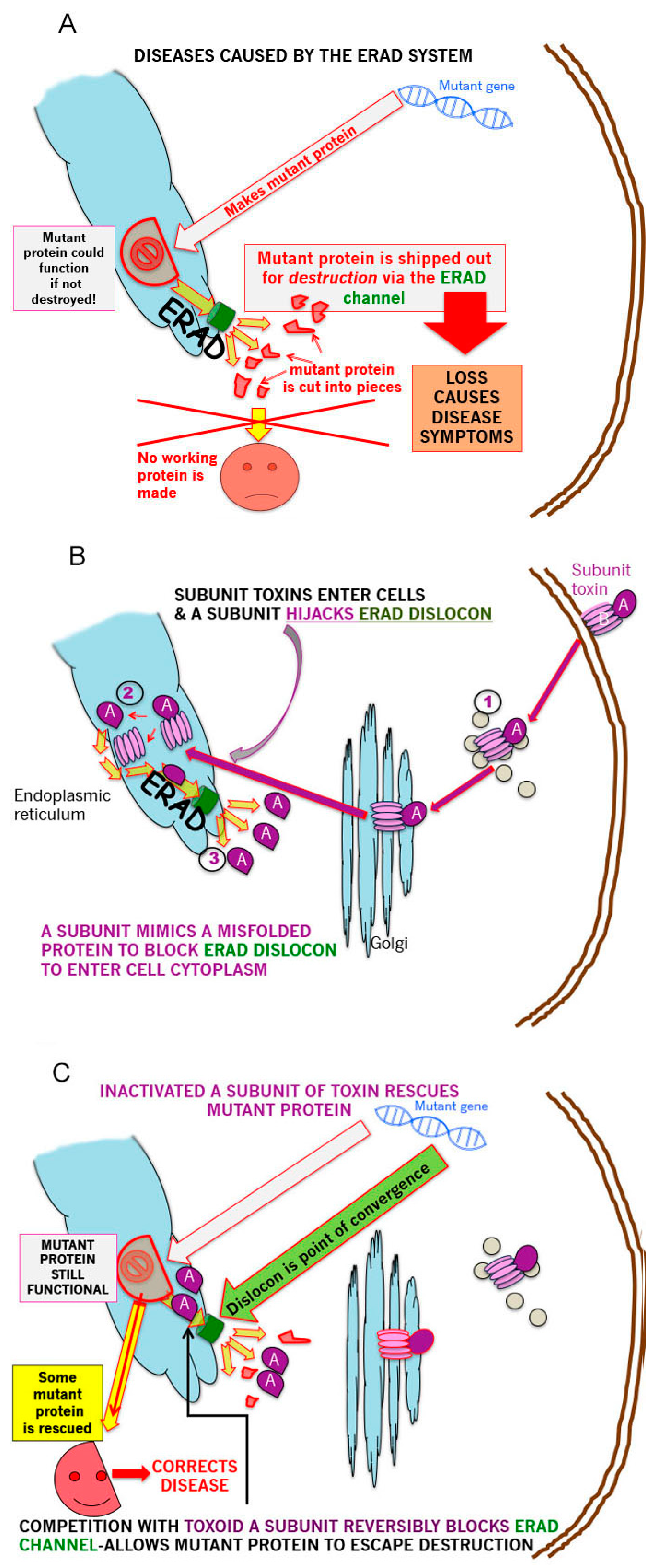
The GSL mediated retrograde transit of exogenous CT (GM1 ganglioside receptor), (VT or other subunit toxins) to the ER to deliver the A subunit, via the ERAD retrotranslocon, (dislocon) to the cytosol, represents a major opportunity to carry out precisely targeted, intracellular plumbing to stop (slow) the ‘leak’ of mutant partially misfolded proteins into ERAD and thereby ameliorate the symptoms of multiple genetic protein misfolding deficiency diseases this degradation causes. This is represented schematically in the 3 panels of Figure 1—panel A shows the basic problem for ERAD-dependent protein misfolding diseases; panel B, the pathway by which subunit toxins hijack ERAD; panel C shows the means by which this hijacking can be turned to advantage to partially correct disease symptoms.
As long as the mutant protein retains a fraction of the wildtype activity, ‘plugging’ the ER dislocon should result in increased ERAD escape and the delivery of sufficient protein of sufficient function to the final target site, to reverse disease symptoms. Inactivation of the catalytic activity of the A subunit generates a holotoxin which will deliver a benign A subunit to the dislocon which when thus occupied, should be unable to simultaneously translocate misfolded nascent mutant protein to the proteosome for degradation. This is a titratable system and, therefore, up to a limit, it should be possible to ‘dial in’ the required level of mutant protein ERAD escape. It was previously calculated that it took 50 molecules of toxin to accumulate in the ER to ‘generate’ 1 A subunit in the cytosol [148]. This could indicate a slow A subunit translocation rate, which would be beneficial in this scenario, or poor unfolded protein mimicry, which would not. To prolong A subunit dislocon residence time, a C terminal polyleucine ‘stop transfer’ sequence was also added [220]. It should be noted that the use of disease-specific pharmacological chaperones in such misfolding diseases [221,222] is compatible as a combination therapy.
7. Treatment for Protein Misfolding Diseases
7.1. F508delta CFTR
The F508delta CFTR mutation is the most common cause of cystic fibrosis [223]. This single amino acid deletion results in the partial misfolding and destabilization of the CFTR chloride transporter [224,225] and its removal by ERAD [226]. Even wildtype CFTR itself is partially subject to ERAD [227]. F508delta CFTR retains significant chloride transport activity in vitro [228].
We first demonstrated the efficacy of this novel toxoid approach by treating HeLa cells expressing the F508delta CFTR mutation [220]. In these cells, none of the mature glycosylated CFTR (band c-lactosamine glycosylated) could be detected by Western blot but rather a small fraction of the band b (high mannose glycosylated) immature form was present. Even in the presence of wildtype VT1, when protein synthesis was completely inhibited, increased levels of the mature CFTR were detected after 2 h treatment. VT1 containing an inactivated A subunit was similarly effective. Since Gb3 expression is highly tissue selective, we switched to cholera toxin, whose receptor is the widely distributed GM1 ganglioside, in order to develop a generally applicable approach to ERAD exacerbated protein misfolding diseases. A subunit inactivated CT (mCT) was able to promote F508delta CFTR ERAD escape and partially restore cellular CFTR function (chloride transport) [220].
7.2. N370S Glucocerebrosidase
In vitro, mCT was also able to rescue cellular mutant glucocerebroside (GBA) N370S [220], the misfolding of which and subsequent ERAD, is the cause of Gaucher Disease [229], in which the glycosphingolipid substrate, glucosyl ceramide accumulates widely [230].
In order to examine the feasibility of this approach in vivo, mouse models of F508delCFTR and N370S GBA were tested following mCT treatment.
7.3. Cystic Fibrosis Animal Model
Although the F508delCFTR mouse does not fully reflect the lung pathology typical of human CF [231,232], intestinal epithelial chloride transport is ablated and residual F508delCFTR-dependent saliva production in this model, provides a convenient assay of systemic CFTR function [233,234], commonly used as an in vivo assay of potentially therapeutic drugs [235,236,237].
In preliminary experiments, we measured the saliva production in a single male F508delCFTR mouse and then treated the mouse i.p. with mCT (400 ng/25 gm mouse) every 48 h for 2 weeks. Our contention was that cell turnover in tissues in vivo could be less than in cells in culture, and that beneficial effect might therefore be accumulative. After the two-week treatment period, the CFTR dependent saliva production increased 5-fold. Following a subsequent two-week interval, we measured saliva production again and gave another two-week treatment course. The initial elevated saliva production was maintained and following the second course, production was increased a further threefold for a 15-fold increase overall.
These encouraging preliminary results led to the studies reported in Figure 2, in which 1–3 week old female F508delCFTR mice were treated with repeated i.p. mCT doses/48 h and CFTR dependent saliva production monitored periodically during the treatment. The results clearly show that saliva production is markedly increased during the first 2 week treatment (by ~300%). There was a lag period which was greater for older mice, but all reached the same approximate increased value by day 15. After this time, the dose was increased, and saliva production was further increased during treatment (by ~100%) to a plateau value seven fold higher than control mice levels, which was maintained during the latter 2 weeks of this second 1000 ng mCT/48 h treatment period. Leaving the mice for a subsequent ten days without further treatment was sufficient for saliva production in the F508delCFTR mice to return to the pretreatment control baseline. No significant effect on weight or adverse behaviour (e.g., ruffled fur) was observed during the course of treatment.
These data confirm that repeated mCT administration is accumulative and that the F508delCFTR saliva production can be rescued by mCT to levels in marked excess of those found in wild type mice, indicating that this is a potent therapeutic approach to CF.
8. Gaucher Disease Animal Model
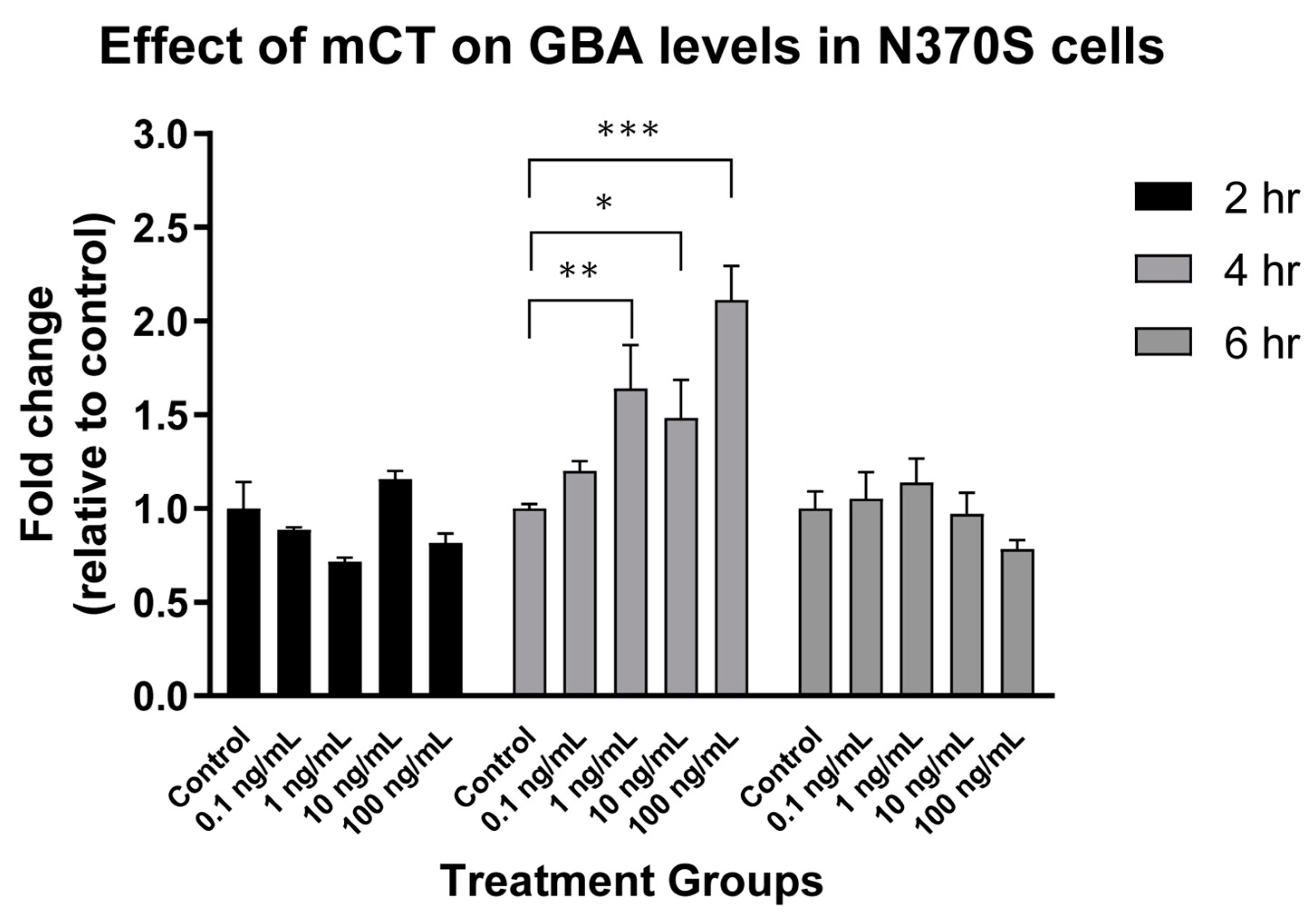
In our original studies, we also showed that mCT was a viable approach to the treatment of N370S GBA Gaucher Disease cells in vitro [220]. This was subsequently verified as a function of both mCT dose and time (Figure 3). N370S GBA activity showed a dose dependent increase after 4 h treatment but this was lost after 6 h treatment. Thus, the rescue of N370S GBA was time dependent in cultured cells.
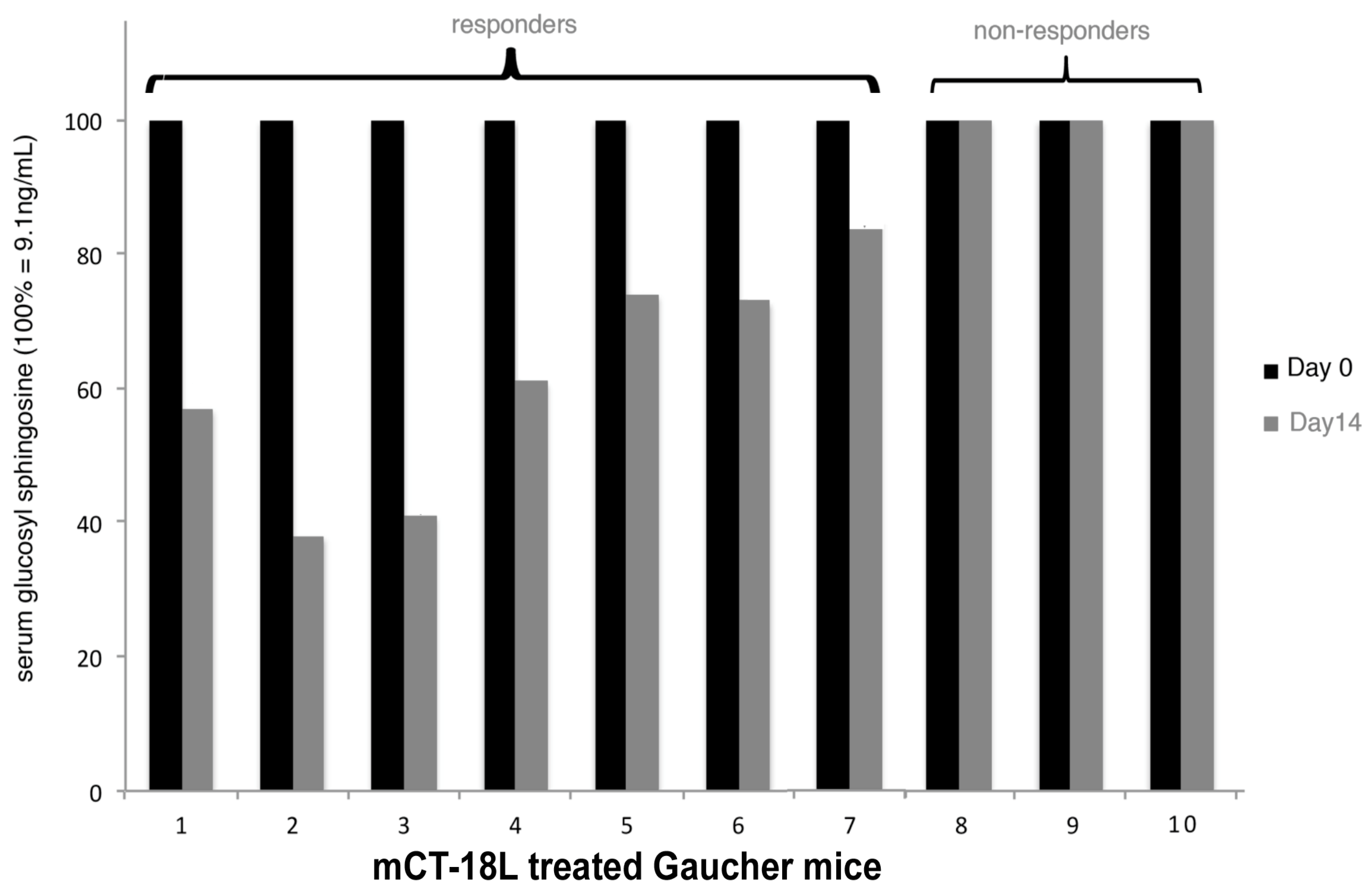
The mouse model of N370S GBA Gaucher disease [238] was used to access the in vivo therapeutic potential of mCT (Figure 4). Following from the first cystic fibrosis mouse study, we reasoned that repeated treatment might be accumulative and therefore employed the same initial treatment regime. Serial assays of GBA activity were not feasible and therefore the direct measurement of accumulated GSL was measured before and after treatment. In Gaucher disease, serum glucosyl sphingosine (psychosine) accumulation has been shown to be a more informative monitor of clinical disease [239,240,241]. We therefore measured levels of glucosyl sphingosine in the serum of each mouse and compared that with the level after treatment in the same mouse. In our original cell culture studies, we added a hydrophobic C terminal hydrophobic peptide sequence (equivalent to a -100 or 50% -i.e.,an 18 or 9-polyleucine-stop transfer sequence) to the inactivated A subunit to attempt to increase the retrotranslocon transit time and thereby increase the efficacy of ERAD blockade. This indeed proved more effective in cell culture [220]. It was mCT-18L (mCT with an 18 leucine C terminal tail) which was used to treat the Gaucher mice. mCT-18L was more difficult to purify and, therefore, was not used in the CF studies.
In seven of the ten treated mice, a decrease in glucosyl sphingosine was observed ranging from 20% to 60%. This suggested that there was an additive effect on repeated mCT i.p. injection, as seen for CF mice, but unlike the CF mice, as yet unknown factors can prevent an mCT response in 30% of Gaucher mice.
Nevertheless, in sum these animal model studies of partial misfolding diseases show the in vivo feasibility of using the A subunit inactivated cholera holotoxoid as a therapeutic approach to genetic protein misfolding diseases in which the mutant protein retains partial wildtype function but is degraded by ERAD, due to the mutation-induced partial misfolding. Depending on the extent of ERAD involvement, there are 40–60 such diseases [242].
The finding that wildtype CFTR was increased via holotoxoid ERAD blockade [220], is consistent with significant wildtype CFTR misfolding, even under normal conditions [188]. Thus, if CFTR (or any normal protein similarly subject to ERAD) were in short supply, for whatever reason, holotoxoid ERAD blockade might prove an effective remedy.
9. Future Studies
The ERAD pathway is also co-opted by several pathogenic viruses [243] and inactivated holotoxin may prove effective against such infections also. The ER is a central and highly dynamic compartment in cell metabolism. The ability to target exogenous proteins, etc., to this compartment by (cleavable) coupling them to VT/CT B subunits, offers a largely untapped resource to modify a large array of local metabolic pathways. Since the ER membrane is continuous with the nuclear envelope, some years ago, we coupled a DNA binding element to VTB as a means to transport genes to the ER-> nucleus [244]. Such outside-the-box concepts still have a place in scientific progress.
Funding
The commercial affiliation provided support in the form of partial industrial funding for two MITACS industrial/academic pdf fellowship awards.
Institutional Review Board Statement
The CF mouse studies were performed under contract with the animal facility at the Research Institute, Hospital for Sick Children protocol AUP# 27096 ‘Toxoid-based prevention of F508del CFTR degradation in mice’2013. The Gaucher mouse studies under contract with the University of British Columbia with ethics approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Lingwood is a founder of ERAD Therapeutics (eradtx.cpm) and has received travel support to present the new data reported herein.
References
- Karmali, M.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The Association between Idiopathic Hemolytic Uremic Syndrome and Infection by Verotoxin-Producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef]
- Jokiranta, T.S. HUS and atypical HUS. Blood 2017, 129, 2847–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, H.; Takeda, T. Escherichia coli Shiga toxin. J. Nat. Toxins 2000, 9, 299–313. [Google Scholar] [PubMed]
- Ray, P.E.; Liu, X.-H. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr. Nephrol. 2001, 16, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Andreoli, S. Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli (STEC HUS). Pediatr. Nephrol. 2019, 34, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between Virulence Factors of Shiga Toxin-ProducingEscherichia coli and Disease in Humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werber, D.; Fruth, A.; Buchholz, U.; Prager, R.; Kramer, M.H.; Ammon, A.; Tschäpe, H. Strong Association Between Shiga Toxin-Producing Escherichia coli O157 and Virulence Genes stx 2 and eae as Possible Explanation for Predominance of Serogroup O157 in Patients with Haemolytic Uraemic Syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 2003, 22, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Watahiki, M.; Isobe, J.; Kimata, K.; Shima, T.; Kanatani, J.-I.; Shimizu, M.; Nagata, A.; Kawakami, K.; Yamada, M.; Izumiya, H.; et al. Characterization of Enterohemorrhagic Escherichia coli O111 and O157 Strains Isolated from Outbreak Patients in Japan. J. Clin. Microbiol. 2014, 52, 2757–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [CrossRef]
- Okuda, T.; Numata, S.-I.; Ito, M.; Ohta, M.; Kawamura, K.; Wiels, J.; Urano, T.; Tajima, O.; Furukawa, K.; Furukawa, K. Targeted Disruption of Gb3/CD77 Synthase Gene Resulted in the Complete Deletion of Globo-series Glycosphingolipids and Loss of Sensitivity to Verotoxins. J. Biol. Chem. 2006, 281, 10230–10235. [Google Scholar] [CrossRef] [Green Version]
- Porubsky, S.; Luckow, B.; Bonrouhi, M.; Speak, A.; Cerundolo, V.; Platt, F.; Gröne, H.J. Glycosphingolipids Gb3 and iGb3. In vivo roles in hemolytic-uremic syndrome and iNKT cell function. Pathologe 2008, 29 (Suppl. 2), 297–302. [Google Scholar]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic Analysis of Binding between Shiga Toxin and Receptor Glycolipid Gb3Cer by Surface Plasmon Resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.; Proulx, F.; Lingwood, C.A. Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int. 2009, 75, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid Rafts as a Membrane-Organizing Principle. Science 2009, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.G.N. Lipid rafts generate digital-like signal transduction in cell plasma membranes. Biotechnol. J. 2012, 7, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Brumell, J.H.; Finlay, B. Microbial pathogenesis: Lipid rafts as pathogen portals. Curr. Biol. 2000, 10, R823–R825. [Google Scholar] [CrossRef] [Green Version]
- Heung, L.J.; Luberto, C.; Del Poeta, M. Role of Sphingolipids in Microbial Pathogenesis. Infect. Immun. 2006, 74, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utskarpen, A.; Massol, R.; Van Deurs, B.; Lauvrak, S.U.; Kirchhausen, T.; Sandvig, K. Shiga Toxin Increases Formation of Clathrin-Coated Pits through Syk Kinase. PLoS ONE 2010, 5, e10944. [Google Scholar] [CrossRef] [Green Version]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The A-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef]
- Khine, A.A.; Lingwood, C.A. Capping and receptor-mediated endocytosis of cell-bound verotoxin (shiga-like toxin) 1: Chemical identification of an amino acid in the B subunit necessary for efficient receptor glycolipid binding and cellular internalization. J. Cell. Physiol. 1994, 161, 319–332. [Google Scholar] [CrossRef]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.E.; Fraisier, V.; Florent, J.-C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nat. Cell Biol. 2007, 450, 670–675. [Google Scholar] [CrossRef]
- Renard, H.-F.; Simunovic, M.; Lemière, J.; Boucrot, E.; Garcia-Castillo, M.D.; Arumugam, S.; Chambon, V.; Lamaze, C.; Wunder, C.; Kenworthy, A.; et al. Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nat. Cell Biol. 2015, 517, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L. Shiga Toxin—A Model for Glycolipid-Dependent and Lectin-Driven Endocytosis. Toxins 2017, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga Toxin B-Subunit to Retrograde Transport Route in Association with Detergent-resistant Membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin triggers endothelial and podocyte injury: The role of complement activation. Pediatr. Nephrol. 2019, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Detzner, J.; Gloerfeld, C.; Pohlentz, G.; Legros, N.; Humpf, H.-U.; Mellmann, A.; Karch, H.; Müthing, J. Structural Insights into Escherichia coli Shiga Toxin (Stx) Glycosphingolipid Receptors of Porcine Renal Epithelial Cells and Inhibition of Stx-Mediated Cellular Injury Using Neoglycolipid-Spiked Glycovesicles. Microorganisms 2019, 7, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kociurzynski, R.; Makshakova, O.N.; Knecht, V.; Römer, W. Multiscale Molecular Dynamics Studies Reveal Different Modes of Receptor Clustering by Gb3-Binding Lectins. J. Chem. Theory Comput. 2021, 17, 2488–2501. [Google Scholar] [CrossRef]
- Sandvig, K.; Bergan, J.; Dyve, A.-B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon 2010, 56, 1181–1185. [Google Scholar] [CrossRef]
- Hazes, B.; Read, R. Accumulating Evidence Suggests That Several AB-Toxins Subvert the Endoplasmic Reticulum-Associated Protein Degradation Pathway To Enter Target Cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Saleh, M.T.; Ferguson, J.; Boggs, J.M.; Gariépy, J. Insertion and Orientation of a Synthetic Peptide Representing the C-Terminus of the A1Domain of Shiga Toxin into Phospholipid Membranes. Biochemistry 1996, 35, 9325–9334. [Google Scholar] [CrossRef]
- Nowakowska-Gołacka, J.; Sominka, H.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The Mammalian Endoplasmic Reticulum-Associated Degradation System. Cold Spring Harb. Perspect. Biol. 2012, 5, a013185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigotti, M.; Carnicelli, D.; Alvergna, P.; Mazzaracchio, R.; Sperti, S.; Montanaro, L. The RNA-N-glycosidase activity of Shiga-like toxin I: Kinetic parameters of the native and activated toxin. Toxicon 1997, 35, 1431–1437. [Google Scholar] [CrossRef]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar] [PubMed] [Green Version]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to ER. Mol. Biol. Cell. 2003, 14, 4783–4793. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Saslowsky, D. Raft trafficking of AB5 subunit bacterial toxins. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1746, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Lopata, A.; Kniss, A.; Löhr, F.; Rogov, V.V.; Dötsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef]
- Shi, J.; Hu, X.; Guo, Y.; Wang, L.; Ji, J.; Li, J.; Zhang, Z.-R. A technique for delineating the unfolding requirements for substrate entry into retrotranslocons during endoplasmic reticulum–associated degradation. J. Biol. Chem. 2019, 294, 20084–20096. [Google Scholar] [CrossRef]
- Wiertz, E.J.H.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec6l-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nat. Cell Biol. 1996, 384, 432–438. [Google Scholar] [CrossRef]
- Schäfer, A.; Wolf, D.H. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009, 28, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.-L.; Römisch, K. Proteasome 19S RP Binding to the Sec61 Channel Plays a Key Role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef] [PubMed]
- Römisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef]
- Bernardi, K.M.; Forster, M.L.; Lencer, W.I.; Tsai, B. Derlin-1 Facilitates the Retro-Translocation of Cholera Toxin. Mol. Biol. Cell 2008, 19, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballar, P.; Pabuççuoğlu, A.; Kose, F.A. Different p97/VCP complexes function in retrotranslocation step of mammalian Er-associated degradation (ERAD). Int. J. Biochem. Cell Biol. 2011, 43, 613–621. [Google Scholar] [CrossRef]
- Garza, R.M.; Sato, B.K.; Hampton, R.Y. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 2009, 284, 14710–14722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, E.M.; Kreft, S.G.; Greenblatt, W.H.; Swanson, R.; Hochstrasser, M. Aberrant substrate engagement of the ER translocon triggers degradation by the Hrd1 ubiquitin ligase. J. Cell Biol. 2012, 197, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E.; et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasic, V.; Denkert, N.; Schmidt, C.; Riedel, D.; Stein, A.; Meinecke, M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020, 22, 274–281. [Google Scholar] [CrossRef]
- Li, S.; Spooner, R.A.; Hampton, R.Y.; Lord, J.M.; Roberts, L.M. Cytosolic Entry of Shiga-Like Toxin A Chain from the Yeast Endoplasmic Reticulum Requires Catalytically Active Hrd1p. PLoS ONE 2012, 7, e41119. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of Protein Toxins into Mammalian Cells by Crossing the Endoplasmic Reticulum Membrane: Co-opting Basic Mechanisms of Endoplasmic Reticulum-Associated Degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar] [CrossRef]
- Teter, K.; Allyn, R.L.; Jobling, M.; Holmes, R.K. Transfer of the Cholera Toxin A1 Polypeptide from the Endoplasmic Reticulum to the Cytosol Is a Rapid Process Facilitated by the Endoplasmic Reticulum-Associated Degradation Pathway. Infect. Immun. 2002, 70, 6166–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, G.; Mikoryak, C.; Hayslett, T.; Bhat, A.; Draper, R.K. Cholera Toxin Up-Regulates Endoplasmic Reticulum Proteins That Correlate with Sensitivity to the Toxin. Exp. Biol. Med. 2008, 233, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Taga, S.; Carlier, K.; Mishal, Z.; Capoulade, C.; Mangeney, M.; Lecluse, Y.; Coulaud, D.; Tetaud, C.; Pritchard, L.L.; Tursz, T.; et al. Intracellular signaling events in CD77-mediated apoptosis of Burkitt’s lymphoma cells. Blood 1997, 90, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Uchida, H.; Kiyokawa, N.; Mori, T.; Sato, N.; Horie, H.; Takeda, T.; Fujimoto, J. Verotoxins induce apoptosis in human renal tubular epithelium derived cells. Kidney Int. 1998, 53, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Karpman, D.; Håkansson, A.; Perez, M.-T.R.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of Renal Cortical Cells in the Hemolytic-Uremic Syndrome: In Vivo and In Vitro Studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Koide, N.; Sugiyama, T.; Mori, I.; Yokochi, T. A Novel Caspase Dependent Pathway Is Involved in Apoptosis of Human Endothelial Cells by Shiga Toxins. Microbiol. Immunol. 2002, 46, 697–700. [Google Scholar] [CrossRef]
- Ching, J.C.Y.; Jones, N.L.; Ceponis, P.J.M.; Karmali, M.A.; Sherman, P.M. Escherichia coli Shiga-Like Toxins Induce Apoptosis and Cleavage of Poly(ADP-Ribose) Polymerase via In Vitro Activation of Caspases. Infect. Immun. 2002, 70, 4669–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, P.; Smith, S.J.; A Giembycz, M.; Rotondo, D.; Plevin, R. Verotoxin activates mitogen-activated protein kinase in human peripheral blood monocytes: Role in apoptosis and proinflammatory cytokine release. Br. J. Pharmacol. 2003, 140, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; et al. Shiga Toxin 2 Causes Apoptosis in Human Brain Microvascular Endothelial Cells via C/EBP Homologous Protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef]
- Debernardi, J.; Hollville, E.; Lipinski, M.; Wiels, J.; Robert, A. Differential role of FL-BID and t-BID during verotoxin-1-induced apoptosis in Burkitt’s lymphoma cells. Oncogene 2018, 37, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Mangeney, M.; A Lingwood, C.; Taga, S.; Caillou, B.; Tursz, T.; Wiels, J. Apoptosis induced in Burkitt’s lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. 1993, 53, 5314–5319. [Google Scholar]
- Nakagawa, I.; Nakata, M.; Kawabata, S.; Hamada, S. Regulated expression of the Shiga toxin B gene induces apoptosis in mammalian fibroblastic cells. Mol. Microbiol. 2002, 33, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tétaud, C.; Falguières, T.; Carlier, K.; Lécluse, Y.; Garibal, J.; Coulaud, D.; Busson, P.; Steffensen, R.; Clausen, H.; Johannes, L.; et al. Two distinct Gb3/CD77 signaling pathways leading to apoptosis are triggered by anti-Gb3/CD77 mAb and verotoxin-1. J. Biol. Chem. 2003, 278, 45200–45208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatelli, D.; Bortolotti, M.; Giorgi, F.M. Transcriptional network inference and master regulator analysis of the response to ribosome-inactivating proteins in leukemia cells. Toxicology 2020, 441, 152531. [Google Scholar] [CrossRef]
- Mori, T.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Sato, N.; Ohmi, K.; Nakajima, H.; Takeda, T.; et al. Globotriaosyl ceramide (CD77/Gb3) in the glycolipid-enriched membrane domain participates in B-cell receptor-mediated apoptosis by regulating lyn kinase activity in human B cells. Exp. Hematol. 2000, 28, 1260–1268. [Google Scholar] [CrossRef]
- Fujii, J.; Matsui, T.; Heatherly, D.P.; Schlegel, K.H.; Lobo, P.I.; Yutsudo, T.; Ciraolo, G.M.; Morris, R.E.; Obrig, T. Rapid Apoptosis Induced by Shiga Toxin in HeLa Cells. Infect. Immun. 2003, 71, 2724–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Peng, L.; Shen, M.; Xia, Y.; Li, Z.; He, N. Shiga-like toxin I exerts specific and potent anti-tumour efficacy against gastric cancer cell proliferation when driven by tumour-preferential Frizzled-7 promoter. Cell Prolif. 2019, 52, e12607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, D.; Johansson, A.; Grankvist, K.; Andersson, U.; Henriksson, R.; Bergström, P.; Brännström, T.; Behnam-Motlagh, P. Verotoxin-1 induction of apoptosis in Gb3-expressing human glioma cell lines. Cancer Biol. Ther. 2006, 5, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Johansson, D.; Kosovac, E.; Moharer, J.; Ljuslinder, I.; Brännström, T.; Johansson, A.; Behnam-Motlagh, P. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer 2009, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debernardi, J.; Pioche-Durieu, C.; Le Cam, E.; Wiels, J.; Robert, A. Verotoxin-1-Induced ER Stress Triggers Apoptotic or Survival Pathways in Burkitt Lymphoma Cells. Toxins 2020, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Larson, G.; Samuelsson, B.E. Blood Group Type Glycosphingolipids of Human Cord Blood Erythrocytes1. J. Biochem. 1980, 88, 647–657. [Google Scholar] [CrossRef]
- Kundu, S.K.; Evans, A.; Rizvi, J.; Glidden, H.; Marcus, D.M. A new pkphenotype in the p blood group system. Eur. J. Immunogenet. 1980, 7, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Wiels, J.; Fellous, M.; Tursz, T. Monoclonal antibody against a Burkitt lymphoma-associated antigen. Proc. Natl. Acad. Sci. USA 1981, 78, 6485–6488. [Google Scholar] [CrossRef] [Green Version]
- Fellous, M.; Wiels, J.; Cartron, J.P.; Tursz, T. A monoclonal antibody, specific for Burkitt’s lymphoma, is also a blood group Pk antibody. Dev. Boil. Stand. 1984, 57, 293–298. [Google Scholar]
- Balana, A.; Wiels, J.; Tetaud, C.; Tursz, T.; Mishal, Z. Induction of cell differentiation in burkitt lymphoma lines. BLA: A glycolipid marker of B-cell differentiation. Int. J. Cancer 1985, 36, 453–460. [Google Scholar] [CrossRef]
- Mangeney, M.; Richard, Y.; Coulaud, D.; Tursz, T.; Wiels, J. CD77: An antigen of germinal center B cells entering apoptosis. Eur. J. Immunol. 1991, 21, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, G.; Zeuthen, J. Distribution of the Burkitt’s-lymphoma-associated antigen (BLA) in normal human tissue and malignant lymphoma as defined by immunohistological staining with monoclonal antibody 38.13. J. Cancer Res. Clin. Oncol. 1987, 113, 78–86. [Google Scholar] [CrossRef]
- Bien, T.; Perl, M.; Machmüller, A.C.; Nitsche, U.; Conrad, A.; Johannes, L.; Müthing, J.; Soltwisch, J.; Janssen, K.-P.; Dreisewerd, K. MALDI-2 Mass Spectrometry and Immunohistochemistry Imaging of Gb3Cer, Gb4Cer, and Further Glycosphingolipids in Human Colorectal Cancer Tissue. Anal. Chem. 2020, 92, 7096–7105. [Google Scholar] [CrossRef]
- Zhang, T.; Van Die, I.; Tefsen, B.; Van Vliet, S.J.; Laan, L.C.; Zhang, J.; Dijke, P.T.; Wuhrer, M.; Belo, A.I. Differential O- and Glycosphingolipid Glycosylation in Human Pancreatic Adenocarcinoma Cells With Opposite Morphology and Metastatic Behavior. Front. Oncol. 2020, 10, 732. [Google Scholar] [CrossRef]
- Stimmer, L.; Dehay, S.; Nemati, F.; Massonnet, G.; Richon, S.; Decaudin, D.; Klijanienko, J.; Johannes, L. Human breast cancer and lymph node metastases express Gb3 and can be targeted by STxB-vectorized chemotherapeutic compounds. BMC Cancer 2014, 14, 916. [Google Scholar] [CrossRef] [PubMed]
- Arab, S.; Russel, E.; Chapman, W.B.; Rosen, B.; A Lingwood, C. Expression of the verotoxin receptor glycolipid, globotriaosylceramide, in ovarian hyperplasias. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1997, 9, 553–563. [Google Scholar]
- Farkas-Himsley, H.; Hill, R.P.; Rosen, B.; Arab, S.; Lingwood, C.A. The bacterial colicin active against tumor cells in vitro and in vivo is verotoxin 1. Proc. Natl. Acad. Sci. USA 1995, 92, 6996–7000. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Klock, J.; Macher, B. Neutral glycospingolipids in hairy cell leukemia. Biochemistry 1981, 20, 6505–6508. [Google Scholar] [CrossRef]
- Furukawa, K.; Yokoyama, K.; Sato, T.; Wiels, J.; Hirayama, Y.; Ohta, M.; Furukawa, K. Expression of the Gb3/CD77 synthase gene in megakaryoblastic leukemia cells: Implication in the sensitivity to verotoxins. J. Biol. Chem. 2002, 277, 11247–11254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacasse, E.C.; Bray, M.; Patterson, B.; Lim, W.M.; Perampalam, S.; Radvanyi, L.G.; Keating, A.; Stewart, A.K.; Buckstein, R.; Sandhu, J.S.; et al. Shiga-like toxin-1 receptor on human breast cancer, lymphoma, and myeloma and absence from CD34(+) hematopoietic stem cells: Implications for ex vivo tumor purging and autologous stem cell transplantation. Blood 1999, 94, 2901–2910. [Google Scholar]
- Arbus, G.S.; Grisaru, S.; Segal, O.; Dosch, M.; Pop, M.; Lala, P.; Nutikka, A.; A Lingwood, C. Verotoxin targets lymphoma infiltrates of patients with post-transplant lymphoproliferative disease. Leuk. Res. 2000, 24, 857–864. [Google Scholar] [CrossRef]
- Ishitoya, S.; Kurazono, H.; Nishiyama, H.; Nakamura, E.; Kamoto, T.; Habuchi, T.; Terai, A.; Ogawa, O.; Yamamoto, S. Verotoxin Induces Rapid Elimination of Human Renal Tumor Xenografts in SCID Mice. J. Urol. 2004, 171, 1309–1313. [Google Scholar] [CrossRef]
- Falguières, T.; Maak, M.; von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.-F.; Robine, S.; Johannes, L.; Janssen, K.-P. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, P.E.; Maak, M.; Nitsche, U.; Perl, M.; Novotny, A.; Slotta-Huspenina, J.; Dransart, E.; Holtorf, A.; Johannes, L.; Janssen, K.-P. Gastric Adenocarcinomas Express the Glycosphingolipid Gb3/CD77: Targeting of Gastric Cancer Cells with Shiga Toxin B-Subunit. Mol. Cancer Ther. 2016, 15, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, C.; Fukushi, Y.; Satoh, M.; Saitoh, S.; Orikasa, S.; Nudelman, E.; Straud, M.; Hakomori, S.-I. Changes in glycolipid expression in human testicular tumor. Int. J. Cancer 1990, 45, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Souady, J.; Hülsewig, M.; Drmić-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga Toxin Receptor Gb3Cer/CD77: Tumor-Association and Promising Therapeutic Target in Pancreas and Colon Cancer. PLoS ONE 2009, 4, e6813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maak, M.; Nitsche, U.; Keller, L.; Wolf, P.; Sarr, M.; Thiebaud, M.; Rosenberg, R.; Langer, R.; Kleeff, J.; Friess, H.; et al. Tumor-Specific Targeting of Pancreatic Cancer with Shiga Toxin B-Subunit. Mol. Cancer Ther. 2011, 10, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storck, W.; Meisen, I.; Gianmoena, K.; Pläger, I.; Kouzel, I.U.; Bielaszewska, M.; Haier, J.; Mormann, M.; Humpf, H.-U.; Karch, H.; et al. Shiga toxin glycosphingolipid receptor expression and toxin susceptibility of human pancreatic ductal adenocarcinomas of differing origin and differentiation. Biol. Chem. 2012, 393, 785–799. [Google Scholar] [CrossRef]
- Li, S.C.; Kundu, S.K.; Degasperi, R.; Li, Y.T. Accumulation of globotriaosylceramide in a case of leiomyosarcoma. Biochem. J. 1986, 240, 925–927. [Google Scholar] [CrossRef]
- Arab, S.; Murakami, M.; Dirks, P.; Boyd, B.; Hubbard, S.L.; Lingwood, C.A.; Rutka, J.T. Verotoxins inhibit the growth of and induce apoptosis in human astrocytoma cells. J. Neuro Oncol. 1998, 40, 137–150. [Google Scholar] [CrossRef]
- Gariépy, J. The use of Shiga-like toxin 1 in cancer therapy. Crit. Rev. Oncol. 2001, 39, 99–106. [Google Scholar] [CrossRef]
- Salhia, B.; Rutka, J.T.; Lingwood, C.; Nutikka, A.; Van Furth, W.R. The Treatment of Malignant Meningioma with Verotoxin. Neoplasia 2002, 4, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Couture, O.; Dransart, E.; Dehay, S.; Nemati, F.; Decaudin, D.; Johannes, L.; Tanter, M. Tumor Delivery of Ultrasound Contrast Agents Using Shiga Toxin B Subunit. Mol. Imaging 2011, 10, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Arab, S.; Rutka, J.; Lingwood, C. Verotoxin induces apoptosis and the complete, rapid, long-term elimination of human astrocytoma xenografts in nude mice. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1999, 11, 33–39. [Google Scholar]
- Viel, T.; Dransart, E.; Nemati, F.; Henry, E.; Thézé, B.; Decaudin, D.; Lewandowski, D.; Boisgard, R.; Johannes, L.; Tavitian, B. In Vivo Tumor Targeting by the B-Subunit of Shiga Toxin. Mol. Imaging 2008, 7, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Heath-Engel, H.M.; Lingwood, C.A. Verotoxin sensitivity of ECV304 cells in vitro and in vivo in a xenograft tumour model: VT1 as a tumour neovascular marker. Angiogenesis 2003, 6, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Desselle, A.; Chaumette, T.; Gaugler, M.-H.; Cochonneau, D.; Fleurence, J.; Dubois, N.; Hulin, P.; Aubry, J.; Birklé, S.; Paris, F. Anti-Gb3 Monoclonal Antibody Inhibits Angiogenesis and Tumor Development. PLoS ONE 2012, 7, e45423. [Google Scholar] [CrossRef] [Green Version]
- Birklé, S.; Desselle, A.; Chaumette, T.; Gaugler, M.-H.; Cochonneau, D.; Fleurence, J.; Dubois, N.; Hulin, P.; Aubry, J.; Paris, F. Inhibition of tumor angiogenesis by globotriaosylceramide immunotargeting. Oncoimmunology 2013, 2, e23700. [Google Scholar] [CrossRef] [Green Version]
- Junqua, S.; Larsen, A.K.; Wils, P.; Mishal, Z.; Wiels, J.; Le Pecq, J.-B. Decreased accessibility of globotriaosylceramide associated with decreased tumorigenicity in Burkitt’s lymphoma variants induced by immunoselection. Cancer Res. 1989, 49, 6480–6486. [Google Scholar]
- Sibold, J.; Ahadi, S.; Werz, D.B.; Steinem, C. Chemically synthesized Gb3 glycosphingolipids: Tools to access their function in lipid membranes. Eur. Biophys. J. 2021, 50, 109–126. [Google Scholar] [CrossRef]
- Kiarash, A.; Boyd, B.; Lingwood, C. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [CrossRef]
- Arab, S.; Lingwood, C.A. Influence of phospholipid chain length on verotoxin/globotriaosyl ceramide binding in model membranes: Comparison of a surface bilayer film and liposomes. Glycoconj. J. 1996, 13, 159–166. [Google Scholar] [CrossRef]
- Chark, D.; Nutikka, A.; Trusevych, N.; Kuzmina, J.; Lingwood, C. Differential carbohydrate epitope recognition of globotriaosyl ceramide by verotoxins and a monoclonal antibody. Role in human renal glomerular binding. JBIC J. Biol. Inorg. Chem. 2004, 271, 405–417. [Google Scholar] [CrossRef]
- Kim, M.; Binnington, B.; Sakac, D.; Fernandes, K.R.; Shi, S.P.; Lingwood, C.A.; Branch, D.R. Comparison of detection methods for cell surface globotriaosylceramide. J. Immunol. Methods 2011, 371, 48–60. [Google Scholar] [CrossRef]
- Greenshields, K.N.; Halstead, S.K.; Zitman, F.M.; Rinaldi, S.; Brennan, K.M.; O’Leary, C.; Chamberlain, L.H.; Easton, A.; Roxburgh, J.; Pediani, J.; et al. The neuropathic potential of anti-GM1 autoantibodies is regulated by the local glycolipid environment in mice. J. Clin. Investig. 2009, 119, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Nyholm, P.-G.; Pascher, I. Steric presentation and recognition of the saccharide chains of glycolipids at the cell surface: Favoured conformations of the saccharide-lipid linkage calculated using molecular mechanics (MM3). Int. J. Biol. Macromol. 1993, 15, 43–51. [Google Scholar] [CrossRef]
- Hooper, N.M. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (Review). Mol. Membr. Biol. 1999, 16, 145–156. [Google Scholar] [CrossRef]
- Yahi, N.; Aulas, A.; Fantini, J. How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer’s beta amyloid peptide (Abeta1-40). PLoS ONE 2010, 5, e9079. [Google Scholar] [CrossRef]
- Lingwood, D.; Binnington, B.; Róg, T.; Vattulainen, I.; Grzybek, M.; Coskun, Ü.; ALingwood, C.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef]
- Mahfoud, R.; Manis, A.; Binnington, B.; Ackerley, C.; Lingwood, C.A. A Major Fraction of Glycosphingolipids in Model and Cellular Cholesterol-containing Membranes Is Undetectable by Their Binding Proteins. J. Biol. Chem. 2010, 285, 36049–36059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Christian, A.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Matencio, A.; Navarro-Orcajada, S.; González-Ramón, A.; García-Carmona, F.; López-Nicolás, J.M. Recent advances in the treatment of Niemann pick disease type C: A mini-review. Int. J. Pharm. 2020, 584, 119440. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Binnington, B.; Ngan, B.; Chadwick, K.; Fleshner, N.; A Lingwood, C. Cholesterol masks membrane glycosphingolipid tumor-associated antigens to reduce their immunodetection in human cancer biopsies. Glycobiology 2013, 23, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.W. Human teratocarcinoma stem cells: Glycolipid antigen expression and modulation during differentiation. J. Cell. Biochem. 1987, 35, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Bosnakovski, D.; Figueiredo, C.A.; Visser, J.W.; Perlingeiro, R.C.R. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood 2006, 109, 1743–1751. [Google Scholar] [CrossRef]
- Muramatsu, T.; Muramatsu, H. Carbohydrate antigens expressed on stem cells and early embryonic cells. Glycoconj. J. 2004, 21, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-L.; Hung, J.-T.; Cheung, S.K.C.; Lee, H.-Y.; Chu, K.-C.; Li, S.-T.; Lin, Y.-C.; Ren, C.-T.; Cheng, T.-J.R.; Hsu, T.-L.; et al. Carbohydrate-based vaccines with a glycolipid adjuvant for breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 2517–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.-Y.; Khoo, K.-H.; Li, S.-C.; Li, Y.-T.; et al. Ceramide Glycosylation by Glucosylceramide Synthase Selectively Maintains the Properties of Breast Cancer Stem Cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-J.; Ding, Y.; Levery, S.B.; Lobaton, M.; Handa, K.; Hakomori, S.-I. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 4968–4973. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Lala, P.; Ito, S.; Lingwood, C.A. Transfection of MDCK cells with the MDR1 gene results in a major increase in globotriaosyl ceramide and cell sensitivity to verocytotoxin: Role of P-gp in glycolipid biosynthesis. J. Biol. Chem. 2000, 275, 6246–6251. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.; Lingwood, C. Apparent cooperativity in multivalent verotoxin globotriaosyl ceramide binding: Kinetic and saturation binding experiments with radiolabelled verotoxin [125I]-VT1. Biochim. Biophys. Acta 2000, 1501, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga Toxin Binding to Glycolipids and Glycans. PLoS ONE 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Alaoui, A.; Schmidt, F.; Amessou, M.; Sarr, M.; Decaudin, D.; Florent, J.C.; Johannes, L. Shiga toxin-mediated retrograde delivery of a topoisomerase I inhibitor prodrug. Angew. Chem. Int. Ed. Engl. 2007, 46, 6469–6472. [Google Scholar] [CrossRef] [PubMed]
- Amessou, M.; Carrez, D.; Patin, D.; Sarr, M.; Grierson, D.S.; Croisy, A.; Tedesco, A.C.; Maillard, P.; Johannes, L. Retrograde delivery of photosensitizer (TPPp-O-beta-GluOH)3 selectively potentiates its photodynamic activity. Bioconjug. Chem. 2008, 19, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Batisse, C.; Dransart, E.; Sarkouh, R.A.; Brulle, L.; Bai, S.K.; Godefroy, S.; Johannes, L.; Schmidt, F. A new delivery system for auristatin in STxB-drug conjugate therapy. Eur. J. Med. Chem. 2015, 95, 483–491. [Google Scholar] [CrossRef]
- Tam, P.; Mahfoud, R.; Nutikka, A.; Khine, A.A.; Binnington, B.; Paroutis, P.; Lingwood, C. Differential Intracellular Trafficking and Binding of Verotoxin 1 and Verotoxin 2 to Globotriaosylceramide-containing Lipid Assemblies. J. Cell Physiol. 2008, 216, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Hagnerelle, X.; Plisson, C.; Lambert, O.; Marco, S.; Rigaud, J.L.; Johannes, L.; Lévy, D. Two-dimensional structures of the Shiga toxin B-subunit and of a chimera bound to the glycolipid receptor Gb3. J. Struct. Biol. 2002, 139, 113–121. [Google Scholar] [CrossRef]
- Haicheur, N.; Bismuth, E.; Bosset, S.; Adotevi, O.; Warnier, G.; Lacabanne, V.; Regnault, A.; Desaymard, C.; Amigorena, S.; Ricciardi-Castagnoli, P.; et al. The B Subunit of Shiga Toxin Fused to a Tumor Antigen Elicits CTL and Targets Dendritic Cells to Allow MHC Class I-Restricted Presentation of Peptides Derived from Exogenous Antigens. J. Immunol. 2000, 165, 3301–3308. [Google Scholar] [CrossRef] [Green Version]
- El Alaoui, A.; Schmidt, F.; Sarr, M.; Decaudin, D.; Florent, J.C.; Johannes, L. Synthesis and properties of a mitochondrial peripheral benzodiazepine receptor conjugate. Chem. Med. Chem. 2008, 3, 1687–1695. [Google Scholar] [CrossRef]
- Patel, C.; Saad, H.; Shenkman, M.; Lederkremer, G.Z. Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD. Cells 2020, 9, 2138. [Google Scholar] [CrossRef]
- Luginbuehl, V.; Meier, N.; Kovar, K.; Rohrer, J. Intracellular drug delivery: Potential usefulness of engineered Shiga toxin subunit B for targeted cancer therapy. Biotechnol. Adv. 2018, 36, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ryou, J.-H.; Sohn, Y.-K.; Hwang, D.-E.; Kim, H.-S. Shiga-like toxin-based high-efficiency and receptor-specific intracellular delivery system for a protein. Biochem. Biophys. Res. Commun. 2015, 464, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Ryou, J.; Sohn, Y.; Hwang, D.; Park, W.; Kim, N.; Heo, W.; Kim, M.-Y.; Kim, H.-S. Engineering of bacterial exotoxins for highly efficient and receptor-specific intracellular delivery of diverse cargos. Biotechnol. Bioeng. 2016, 113, 1639–1646. [Google Scholar] [CrossRef]
- Plavec, T.; Zahirović, A.; Zadravec, P.; Sabotič, J.; Berlec, A. Lectin-Mediated Binding of Engineered Lactococcus lactis to Cancer Cells. Microorganisms 2021, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Mohseni Moghadam, Z.; Halabian, R.; Sedighian, H.; Behzadi, E.; Amani, J.; Imani Fooladi, A.A. Designing and Analyzing the Structure of DT-STXB Fusion Protein as an Anti-tumor Agent: An in Silico Approach. Iran J. Pathol. 2019, 14, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Daniel, T.O. Shiga toxin-endothelial cell interactions. In Recent Advances in Verocytotoxin-Producing Eshcerichia Coli Infections; Karmali, M.A., Goglio, A.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 317–324. [Google Scholar]
- Robinson, L.A.; Hurley, R.M.; Lingwood, C.; Matsell, D.G. Escherichia coli verotoxin binding to human paediatric glomerular mesangial cells. Pediatr. Nephrol. 1995, 9, 700–704. [Google Scholar] [CrossRef]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M. Treatment options for HUS secondary to Escherichia coli O157:H7. Kidney Int. 2009, 75, S62–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene varients:frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, H.; Friedrich, A.W.; Gerber, A.; Zimmerhackl, L.B.; Schmidt, M.A.; Bielaszewska, M. New Aspects in the Pathogenesis of Enteropathic Hemolytic Uremic Syndrome. Semin. Thromb. Hemost. 2006, 32, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the Baboon Responses to Shiga-Like Toxin. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Siegler, R.L.; Pysher, T.J.; Tesh, V.L.; Taylor, F.B. Response to Single and Divided Doses of Shiga Toxin-1 in a Primate Model of Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2001, 12, 1458–1467. [Google Scholar] [CrossRef]
- Siegler, R.L.; Obrig, T.G.; Pysher, T.J.; Tesh, V.L.; Denkers, N.D.; Taylor, F.B. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.; Lingwood, C. Synergistic effect of verotoxin and interferon-alpha on erythropoiesis. Cell. Mol. Boil. 2003, 49, 1363–1369. [Google Scholar]
- Betz, J.; Dorn, I.; Kouzel, I.U.; Bauwens, A.; Meisen, I.; Kemper, B.; Bielaszewska, M.; Mormann, M.; Weymann, L.; Sibrowski, W.; et al. Shiga toxin of enterohaemorrhagic Escherichia coli directly injures developing human erythrocytes. Cell. Microbiol. 2016, 18, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, M.; Alves-Rosa, F.; Rubel, C.; Fernandez, G.C.; Fernández-Alonso, G.; Alberto, F.; Rivas, M.; Isturiz, M. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin. Exp. Immunol. 2000, 119, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; van Haaften, W.C.E.; Tesh, V.L. Regulation of Proinflammatory Cytokine Expression by Shiga Toxin 1 and/or Lipopolysaccharides in the Human Monocytic Cell Line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef] [Green Version]
- Keusch, G.T.; Acheson, D.W.K.; Aaldering, L.; Erban, J.; Jacewicz, M.S. Comparison of the effects of Shiga-like toxin 1 on cytokine-and butyrate pretreated human umbilical and saphenous vein endothelial cells. J. Infect. Dis. 1996, 173, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Louise, C.B.; Tran, M.C.; Obrig, T.G. Sensitization of human umbilical vein endothelial cells to Shiga toxin: Involvement of protein kinase C and NF-kappaB. Infect. Immun. 1997, 65, 3337–3344. [Google Scholar] [CrossRef] [Green Version]
- Molostvov, G.; Morris, A.; Rose, P.; Basu, S. Interaction of cytokines and growth factor in the regulation of verotoxin-induced apoptosis in cultured human endothelial cells. Br. J. Haematol. 2001, 113, 891–897. [Google Scholar] [CrossRef]
- Stone, M.K.; Kolling, G.L.; Lindner, M.H.; Obrig, T.G. p38 Mitogen-Activated Protein Kinase Mediates Lipopolysaccharide and Tumor Necrosis Factor Alpha Induction of Shiga Toxin 2 Sensitivity in Human Umbilical Vein Endothelial Cells. Infect. Immun. 2007, 76, 1115–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stricklett, P.K.; Hughes, A.K.; Ergonul, Z.; Kohan, D.E. Molecular Basis for Up-Regulation by Inflammatory Cytokines of Shiga Toxin 1 Cytotoxicity and Globotriaosylceramide Expression. J. Infect. Dis. 2002, 186, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Clayton, F.; Pysher, T.J.; Lou, R.; Kohan, D.E.; Denkers, N.D.; Tesh, V.L.; Taylor , F.B., Jr.; Siegler, R.L. Lipopolysaccharide Upregulates Renal Shiga Toxin Receptors in a Primate Model of Hemolytic Uremic Syndrome. Am. J. Nephrol. 2005, 25, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Warnier, M.; Römer, W.; Geelen, J.; Lesieur, J.; Amessou, M.; Heuvel, L.V.D.; Monnens, L.; Johannes, L. Trafficking of Shiga toxin/Shiga-like toxin-1 in human glomerular microvascular endothelial cells and human mesangial cells. Kidney Int. 2006, 70, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Hariya, Y.; Shirakawa, S.; Yonekura, N.; Yokosawa, N.; Kohama, G.-I.; Fujii, N. Augmentation of Verotoxin-Induced Cytotoxicity/Apoptosis by Interferon Is Repressed in Cells Persistently Infected with Mumps Virus. J. Interf. Cytokine Res. 1999, 19, 479–485. [Google Scholar] [CrossRef]
- Siegler, R.L.; Pysher, T.J.; Lou, R.; Tesh, V.L.; Taylor , F.B., Jr. Response to Shiga Toxin-1, with and without Lipopolysaccharide, in a Primate Model of Hemolytic Uremic Syndrome. Am. J. Nephrol. 2001, 21, 420–425. [Google Scholar] [CrossRef]
- van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.H.; Armstrong, C.S.; Sakiri, R.; Tesh, V.L. Shiga Toxin-Induced Tumor Necrosis Factor Alpha Expression: Requirement for Toxin Enzymatic Activity and Monocyte Protein Kinase C and Protein Tyrosine Kinases. Infect. Immun. 2000, 68, 5183–5189. [Google Scholar] [CrossRef] [Green Version]
- Ohara, T.; Kojio, S.; Taneike, I.; Nakagawa, S.; Gondaira, F.; Tamura, Y.; Gejyo, F.; Zhang, H.-M.; Yamamoto, T. Effects of Azithromycin on Shiga Toxin Production by Escherichia coli and Subsequent Host Inflammatory Response. Antimicrob. Agents Chemother. 2002, 46, 3478–3483. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Percheron, L.; Gramada, R.; Tellier, S.; Salomon, R.; Harambat, J.; Llanas, B.; Fila, M.; Allain-Launay, E.; Lapeyraque, A.-L.; Leroy, V.; et al. Eculizumab treatment in severe pediatric STEC-HUS: A multicenter retrospective study. Pediatr. Nephrol. 2018, 33, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Takahashi, M.; Tamada, M.; Senda, M.; Hibino, M.; Shimizu, E.; Okuta, A.; Miyazawa, A.; Senda, T.; Nishikawa, K. Identification of a peptide motif that potently inhibits two functionally distinct subunits of Shiga toxin. Commun. Biol. 2021, 4, 538. [Google Scholar] [CrossRef]
- Janssen, K.-P.; Vignjevic, D.M.; Boisgard, R.; Falguières, T.; Bousquet, G.; Decaudin, D.; Dollé, F.; Louvard, D.; Tavitian, B.; Robine, S.; et al. In vivo Tumor Targeting Using a Novel Intestinal Pathogen-Based Delivery Approach. Cancer Res. 2006, 66, 7230–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloskey, N.; Pound, J.D.; Holder, M.J.; Williams, J.M.; Roberts, L.M.; Lord, J.M.; Gordon, J. The extrafollicular-to-follicular transition of human B lymphocytes: Induction of functional globotriaosylceramide (CD77) on high threshold occupancy of CD40. Eur. J. Immunol. 1999, 29, 3236–3244. [Google Scholar] [CrossRef]
- Vingert, B.; Adotevi, O.; Patin, D.; Jung, S.; Shrikant, P.; Freyburger, L.; Eppolito, C.; Sapoznikov, A.; Amessou, M.; Quintin-Colonna, F.; et al. The Shiga toxin B-subunit targets antigen in vivo to dendritic cells and elicits anti-tumor immunity. Eur. J. Immunol. 2006, 36, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Tartour, E.; Van der Bruggen, P.; Vantomme, V.; Joyeux, I.; Goud, B.; Fridman, W.H.; Johannes, L. Major histocompatibility complex class I presentation of exogenous soluble tumor antigen fused to the B-fragment of Shiga toxin. Eur. J. Immunol. 1998, 28, 2726–2737. [Google Scholar] [CrossRef]
- Noakes, K.L.; Teisserenc, H.T.; Lord, J.; Dunbar, P.; Cerundolo, V.; Roberts, L.M. Exploiting retrograde transport of Shiga-like toxin 1 for the delivery of exogenous antigens into the MHC class I presentation pathway. FEBS Lett. 1999, 453, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D.C.; Williams, D.B. ER quality control in the biogenesis of MHC class I molecules. Semin. Cell Dev. Biol. 2010, 21, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Vingert, B.; Freyburger, L.; Shrikant, P.; Lone, Y.-C.; Quintin-Colonna, F.; Haicheur, N.; Amessou, M.; Herbelin, A.; Langlade-Demoyen, P.; et al. B Subunit of Shiga Toxin-Based Vaccines Synergize with α-Galactosylceramide to Break Tolerance against Self Antigen and Elicit Antiviral Immunity. J. Immunol. 2007, 179, 3371–3379. [Google Scholar] [CrossRef]
- Tran, T.; Diniz, M.O.; Dransart, E.; Gey, A.; Merillon, N.; Lone, Y.C.; Godefroy, S.; Sibley, C.; Ferreira, L.C.; Medioni, J.; et al. A Therapeutic Her2/neu Vaccine Targeting Dendritic Cells Preferentially Inhibits the Growth of Low Her2/neu-Expressing Tumor in HLA-A2 Transgenic Mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4133–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.C.; Lord, J.M.; Roberts, L.M.; Tartour, E.; Johannes, L. 1st Class Ticket to Class I: Protein Toxins as Pathfinders for Antigen Presentation. Traffic 2002, 3, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.-F.; Freyburger, L.; Clement, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal Imprinting of Vaccine-Induced CD8+ T Cells Is Crucial to Inhibit the Growth of Mucosal Tumors. Sci. Transl. Med. 2013, 5, 172ra20. [Google Scholar] [CrossRef] [Green Version]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, D.; Ueno, T.; Takashima, S.; Ohta, K.; Miyawaki, T.; Suzuki, T.; Suzuki, Y. Establishment of a monoclonal antibody directed against Gb3Cer/CD77: A useful immunochemical reagent for a differentiation marker in Burkitt’s lymphoma and germinal centre B cells. Glycoconj. J. 1997, 14, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; Rousselet, G.; Taga, S.; Tursz, T.; Wiels, J. The fate of human CD77+ germinal center B lymphocytes after rescue from apoptosis. Mol. Immunol. 1995, 32, 333–339. [Google Scholar] [CrossRef]
- Liu, Y.J.; Malisan, F.; De Bouteiller, O.; Guret, C.; Lebecque, S.; Banchereau, J.; Mills, F.C.; E Max, E.; Martinez-Valdez, H. Within Germinal Centers, Isotype Switching of Immunoglobulin Genes Occurs after the Onset of Somatic Mutation. Immunity 1996, 4, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.; Madrid-Marina, V.; Estrov, Z.; Freedman, M.H.; Lingwood, C.A.; Dosch, H.-M. Expression of glycolipid receptors to Shiga-like toxin on human B lymphocytes: A mechanism for the failure of long-lived antibody response to dysenteric disease. Int. Immunol. 1990, 2, 1–8. [Google Scholar] [CrossRef]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Porcellini, E.; Galassi, E.; Valerii, M.C.; Spisni, E. Human monocytes stimulated by Shiga toxin 1a via globotriaosylceramide release proinflammatory molecules associated with hemolytic uremic syndrome. Int. J. Med. Microbiol. 2018, 308, 940–946. [Google Scholar] [CrossRef]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Barbieri, S.; Rocchi, L.; Arfilli, V.; Scavia, G.; Ricci, F.; Bontadini, A.; et al. Endothelial damage induced by Shiga toxins delivered by neutrophils during transmigration. J. Leukoc. Biol. 2010, 88, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Havira, M.S.; Ta, A.; Kumari, P.; Wang, C.; Russo, A.J.; Ruan, J.; Rathinam, V.A.; Vanaja, S.K. Shiga toxin suppresses noncanonical inflammasome responses to cytosolic LPS. Sci. Immunol. 2020, 5, eabc0217. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Watanabe-Takahashi, M.; Hamabata, T.; Furukawa, K.; Funamoto, S.; Nishikawa, K. A nontoxigenic form of Shiga toxin 2 suppresses the production of amyloid β by altering the intracellular transport of amyloid precursor protein through its receptor-binding B-subunit. Biochem. Biophys. Res. Commun. 2021, 557, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Helm, K.V.; Mulvey, G.L.; Armstrong, G.D. Serum Amyloid P Component Binding to Shiga Toxin 2 Requires Both A Subunit and B Pentamer. Infect. Immun. 2003, 71, 6075–6078. [Google Scholar] [CrossRef] [Green Version]
- Hamazaki, H. Ca(2+)-dependent binding of human serum amyloid P component to Alzheimer’s beta-amyloid peptide. J. Biol. Chem. 1995, 270, 10392–10394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamazaki, H. Amyloid P component promotes aggregation of Alzheimer’s beta-amyloid peptide. Biochem. Biophys. Res. Commun. 1995, 211, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Tennent, G.A.; Lovat, L.B.; Pepys, M.B. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc. Natl. Acad. Sci. USA 1995, 92, 4299–4303. [Google Scholar] [CrossRef] [Green Version]
- Pepys, M.B.; Herbert, J.; Hutchinson, W.L.; Tennent, G.A.; Lachmann, H.J.; Gallimore, J.R.; Lovat, L.B.; Bartfai, T.; Alanine, A.; Hertel, C.; et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nat. Cell Biol. 2002, 417, 254–259. [Google Scholar] [CrossRef]
- Reverter, M.; Rentero, C.; de Muga, S.V.; Alvarez-Guaita, A.; Mulay, V.; Cairns, R.; Wood, P.; Monastyrskaya, K.; Pol, A.; Tebar, F.; et al. Cholesterol transport from late endosomes to the Golgi regulates t-SNARE trafficking, assembly and function. Mol. Biol. Cell. 2011, 22, 4108–4123. [Google Scholar] [CrossRef]
- Lingwood, C. Verotoxin Receptor-Based Pathology and Therapies. Front. Cell. Infect. Microbiol. 2020, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Ridsdale, A.; Denis, M.; Gougeon, P.-Y.; Ngsee, J.K.; Presley, J.F.; Zha, X. Cholesterol Is Required for Efficient Endoplasmic Reticulum-to-Golgi Transport of Secretory Membrane Proteins. Mol. Biol. Cell 2006, 17, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Marks, D.L.; Pagano, R.E. Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol. 2002, 12, 605–613. [Google Scholar] [CrossRef]
- Puri, V.; Jefferson, J.R.; Singh, R.D.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Sphingolipid Storage Induces Accumulation of Intracellular Cholesterol by Stimulating SREBP-1 Cleavage. J. Biol. Chem. 2003, 278, 20961–20970. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, C. Is Cholesterol the Key Factor for Autism? Am. J. Biomed. Sci. Res. 2020, 7, 483–486. [Google Scholar]
- Levy, M.; Garmy, N.; Gazit, E.; Fantini, J. The minimal amyloid-forming fragment of the islet amyloid polypeptide is a glycolipid-binding domain. FEBS J. 2006, 273, 5724–5735. [Google Scholar] [CrossRef]
- Di Scala, C.; Yahi, N.; Lelievre, C.; Garmy, N.; Chahinian, H.; Fantini, J. Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s beta amyloid peptide. ACS Chem. Neurosci. 2013, 4, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s beta-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar] [CrossRef] [Green Version]
- Tamboli, I.Y.; Prager, K.; Barth, E.; Heneka, M.; Sandhoff, K.; Walter, J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the beta-amyloid precursor protein and amyloid beta-peptide. J. Biol. Chem. 2005, 280, 28110–28117. [Google Scholar] [CrossRef] [Green Version]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [Green Version]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum–Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef]
- Gething, M.-J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum–associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free. Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Benaroudj, N.; Tarcsa, E.; Cascio, P.; Goldberg, A.L. The unfolding of substrates and ubiquitin-independent protein degradation by proteasomes. Biochimie 2001, 83, 311–318. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Hebert, D.N.; Bernasconi, R.; Molinari, M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010, 21, 526–532. [Google Scholar] [CrossRef]
- Hoff, H.; Zhang, H.; Sell, C.; Giordano, A.; Romano, G. Protein Degradation Via the Proteosome. Cell Cycle Control Dysregulation Protoc. 2004, 285, 079–092. [Google Scholar] [CrossRef]
- Osaki, Y.; Saito, A.; Imaizumi, K. The degradation of mutant proteins by ERAD and the pathogenesis of diseases. Clin. Calcium 2018, 28, 1684–1689. [Google Scholar]
- Adnan, H.; Zhang, Z.; Park, H.-J.; Tailor, C.; Che, C.; Kamani, M.; Spitalny, G.; Binnington, B.; Lingwood, C. Endoplasmic Reticulum-Targeted Subunit Toxins Provide a New Approach to Rescue Misfolded Mutant Proteins and Revert Cell Models of Genetic Diseases. PLoS ONE 2016, 11, e0166948. [Google Scholar] [CrossRef]
- Castilla, J.; Rísquez, R.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Diaz, Y.; Mellet, C.O.; Fernández, J.M.G.; Castillón, S. Conformationally-locked N-glycosides: Exploiting long-range non-glycone interactions in the design of pharmacological chaperones for Gaucher disease. Eur. J. Med. Chem. 2015, 90, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Spano, V.; Montalbano, A.; Carbone, A.; Scudieri, P.; Galietta, L.J.V.; Barraja, P. An overview on chemical structures as DeltaF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibodeau, P.H.; Richardson, J.M., III; Wang, W.; Millen, L.; Watson, J.; Mendoza, J.L.; Du, K.; Fischman, S.; Senderowitz, H.; Lukacs, G.L.; et al. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2010, 285, 35825–35835. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Ward, C.L.; Kopito, R.R. Cotranslational Ubiquitination of Cystic Fibrosis Transmembrane Conductance Regulator in Vitro. J. Biol. Chem. 1998, 273, 7189–7192. [Google Scholar] [CrossRef] [Green Version]
- van Barneveld, A.; Stanke, F.; Tamm, S.; Siebert, B.; Brandes, G.; Derichs, N.; Ballmann, M.; Junge, S.; Tümmler, B. Functional analysis of F508del CFTR in native human colon. Biochim. Biophys. Acta 2010, 1802, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Bendikov-Bar, I.; Horowitz, M. Gaucher disease paradigm: From ERAD to co-morbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- van Doorninck, J.H.; French, P.J.; Verbeek, E.; Peters, R.H.; Morreau, H.; Bijman, J.; Scholte, B.J. A mouse model for the cystic fibrosis delta F508 mutation. Embo J. 1995, 14, 4403–4411. [Google Scholar] [CrossRef] [Green Version]
- Zeiher, B.G.; Eichwald, E.; Zabner, J.; Smith, J.J.; Puga, A.P.; McCray, P.B.; Capecchi, M.R.; Welsh, M.J.; Thomas, K.R. A mouse model for the delta F508 allele of cystic fibrosis. J. Clin. Investig. 1995, 96, 2051–2064. [Google Scholar] [CrossRef] [Green Version]
- Best, J.A.; Quinton, P.M.; Klemcke, H.G.; Vallet, J.L.; Christenson, R.K. Salivary secretion assay for drug efficacy for cystic fibrosis in mice. Exp. Physiol. 2005, 90, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droebner, K.; Sandner, P. Modification of the salivary secretion assay in F508del mice—The murine equivalent of the human sweat test. J. Cyst. Fibros. 2013, 12, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ciciriello, F.; Anjos, S.M.; Carissimo, A.; Liao, J.; Carlile, G.W.; Balghi, H.; Robert, R.; Luini, A.; Hanrahan, J.W.; et al. Ouabain Mimics Low Temperature Rescue of F508del-CFTR in Cystic Fibrosis Epithelial Cells. Front. Pharmacol. 2012, 3, 176. [Google Scholar] [CrossRef] [Green Version]
- Dhooghe, B.; Bouckaert, C.; Capron, A.; Wallemacq, P.; Leal, T.; Noel, S. Resveratrol increases F508del-CFTR dependent salivary secretion in cystic fibrosis mice. Biol. Open 2015, 4, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Robert, R.; Goepp, J.; Matthes, E.; Liao, J.; Kus, B.; Macknight, S.D.; Rotin, D.; Hanrahan, J.W.; Thomas, D.Y. Ibuprofen rescues mutant cystic fibrosis transmembrane conductance regulator trafficking. J. Cyst. Fibros. 2015, 14, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Sanders, A.; Hemmelgarn, H.; Melrose, H.L.; Hein, L.; Fuller, M.; Clarke, L.A. Transgenic mice expressing human glucocerebrosidase variants: Utility for the study of Gaucher disease. Blood Cells Mol. Dis. 2013, 51, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; Van Dussen, L.; Hollak, C.E.M.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; Van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.M.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; McDermott, B.K.; Chin, S.J.; Fletcher, J.M.; Fuller, M. Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 558–563. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Chen, Y.; Bellamy, W.P.; Seabra, M.C.; Field, M.C.; Ali, B.R. ER-associated protein degradation is a common mechanism underpinning numerous monogenic diseases including Robinow syndrome. Hum. Mol. Genet. 2005, 14, 2559–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, H.; Gou, Y.; Zook, A.; Lozano, M.M.; Dudley, J.P. ERAD and how viruses exploit it. Front. Microbiol. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchini, L.M.; Lingwood, C.A. A Verotoxin 1 B Subunit-Lambda CRO Chimeric Protein Specifically Binds Both DNA and Globotriaosylceramide (Gb3) to Effect Nuclear Targeting of Exogenous DNA in Gb3 Positive Cells. Exp. Cell Res. 2001, 269, 117–129. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hijacking the hijacker. (A). Genetic diseases in which a small (e.g., point) mutation induces partial protein misfolding while retaining a significant fraction of the wildtype protein function, are candidates for this approach. The misfolded mutant is selected by ER chaperones for degradation by the cytosolic proteosome. Such proteins are unfolded, ubiquinylated and translocated across the ER membrane via the ERAD channel—the dislocon (indicated in green). This cytosolic destruction precipitates/exacerbates the deficiency disease symptoms. (B). Several bacterial subunit toxins hijack the ERAD translocon by A subunit mimicry of an unfolded protein. The holotoxin undergoes (GSL-dependent for VT and CT) retrograde from the cell surface to the ER. Subunit separation (via PDI) then allows the A subunit to enter and transit the dislocon which can only transport one polypeptide at a time. (C). Thus, the exogenous (inactivated toxin A subunit) and endogenous (mutant misfolded protein) translocation substrates converge at the ER dislocon. A subunit occupancy of the dislocon will compete for mutant protein transit, and thereby allow a fraction of the mutant protein to avoid ERAD and traffic to its correct cellular address to rescue (at least in part) the deficiency disease phenotype.
Figure 1.
Hijacking the hijacker. (A). Genetic diseases in which a small (e.g., point) mutation induces partial protein misfolding while retaining a significant fraction of the wildtype protein function, are candidates for this approach. The misfolded mutant is selected by ER chaperones for degradation by the cytosolic proteosome. Such proteins are unfolded, ubiquinylated and translocated across the ER membrane via the ERAD channel—the dislocon (indicated in green). This cytosolic destruction precipitates/exacerbates the deficiency disease symptoms. (B). Several bacterial subunit toxins hijack the ERAD translocon by A subunit mimicry of an unfolded protein. The holotoxin undergoes (GSL-dependent for VT and CT) retrograde from the cell surface to the ER. Subunit separation (via PDI) then allows the A subunit to enter and transit the dislocon which can only transport one polypeptide at a time. (C). Thus, the exogenous (inactivated toxin A subunit) and endogenous (mutant misfolded protein) translocation substrates converge at the ER dislocon. A subunit occupancy of the dislocon will compete for mutant protein transit, and thereby allow a fraction of the mutant protein to avoid ERAD and traffic to its correct cellular address to rescue (at least in part) the deficiency disease phenotype.

Figure 2.
mCT restores F508delCFTR-dependent saliva production in the F508delCFTR mouse. Six Homozygous female F508delCFTR mice were injected i.v. with 400 ng (100 ng/mL blood) mCT every 48 h (from day 1) for two weeks. The dose was then increased to 1000 ng for a further three weeks, and CFTR dependent saliva secretion measured periodically (4 h post injection). Mice were left untreated for a further 11 days and saliva secretion measured. Three control mice were injected with saline and their saliva production did not change. No significant change in weight was observed for the treated mice, Saliva secretion in normal mice was measured as 6.7 µg/min/gm. After 40 days, the mice were left untreated for a further 10 days when the elevated saliva production returned to untreated levels. Importantly, no adverse effects were observed within the course of this study. These studies were performed under contract from ERAD Therapeutics by the Animal facilities at the Research Institute at the Hospital for Sick Children.
Figure 2.
mCT restores F508delCFTR-dependent saliva production in the F508delCFTR mouse. Six Homozygous female F508delCFTR mice were injected i.v. with 400 ng (100 ng/mL blood) mCT every 48 h (from day 1) for two weeks. The dose was then increased to 1000 ng for a further three weeks, and CFTR dependent saliva secretion measured periodically (4 h post injection). Mice were left untreated for a further 11 days and saliva secretion measured. Three control mice were injected with saline and their saliva production did not change. No significant change in weight was observed for the treated mice, Saliva secretion in normal mice was measured as 6.7 µg/min/gm. After 40 days, the mice were left untreated for a further 10 days when the elevated saliva production returned to untreated levels. Importantly, no adverse effects were observed within the course of this study. These studies were performed under contract from ERAD Therapeutics by the Animal facilities at the Research Institute at the Hospital for Sick Children.

Figure 3.
Effect of mCT on glucocerebrosidase A levels in N370 S Gaucher cells. N370S GBA Gaucher fibroblasts in 96 well microplate triplicates were treated with mCT as indicated and the expression level of GBA monitored by Western blot with MAB7410 using the quantitative Western Assay System (Protein Simple). These studies were carried out under contract by ERAD Therapeutics, in the laboratory of Dr Will Costain, NRC, Ottawa, ON, Canada. * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 3.
Effect of mCT on glucocerebrosidase A levels in N370 S Gaucher cells. N370S GBA Gaucher fibroblasts in 96 well microplate triplicates were treated with mCT as indicated and the expression level of GBA monitored by Western blot with MAB7410 using the quantitative Western Assay System (Protein Simple). These studies were carried out under contract by ERAD Therapeutics, in the laboratory of Dr Will Costain, NRC, Ottawa, ON, Canada. * p < 0.05, ** p < 0.01, *** p < 0.001.

Figure 4.
mCT reduces plasma glucosyl sphingosine in the N370S Gaucher mouse. N370S glucocerebrosidase Gaucher mice (25 gm) were injected with 400 ng mCT18L i.p. every 48 h for two weeks. Blood samples taken prior to (day 0) and after (day 14) the treatment period were then assayed for glucosyl sphingosine by mass spectrometry and the prior and after values compared. Thus, each mouse served as its own control. The mouse studies were performed under contract from ERAD Therapeutics, in the laboratory of Dr Lorne Clark, UBC and the (blinded) blood glucosyl ceramide mass spectrometric analyses were performed at the facilities at the Hospital for Sick Children.
Figure 4.
mCT reduces plasma glucosyl sphingosine in the N370S Gaucher mouse. N370S glucocerebrosidase Gaucher mice (25 gm) were injected with 400 ng mCT18L i.p. every 48 h for two weeks. Blood samples taken prior to (day 0) and after (day 14) the treatment period were then assayed for glucosyl sphingosine by mass spectrometry and the prior and after values compared. Thus, each mouse served as its own control. The mouse studies were performed under contract from ERAD Therapeutics, in the laboratory of Dr Lorne Clark, UBC and the (blinded) blood glucosyl ceramide mass spectrometric analyses were performed at the facilities at the Hospital for Sick Children.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lingwood, C. Therapeutic Uses of Bacterial Subunit Toxins. Toxins 2021, 13, 378. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060378
AMA Style
Lingwood C. Therapeutic Uses of Bacterial Subunit Toxins. Toxins. 2021; 13(6):378. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060378
Chicago/Turabian StyleLingwood, Clifford. 2021. "Therapeutic Uses of Bacterial Subunit Toxins" Toxins 13, no. 6: 378. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060378
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.