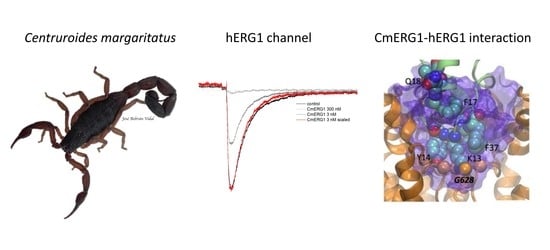
Colombian Scorpion Centruroides margaritatus: Purification and Characterization of a Gamma Potassium Toxin with Full-Block Activity on the hERG1 Channel

 , ,
, ,
Abstract
:
1. Introduction
2. Results
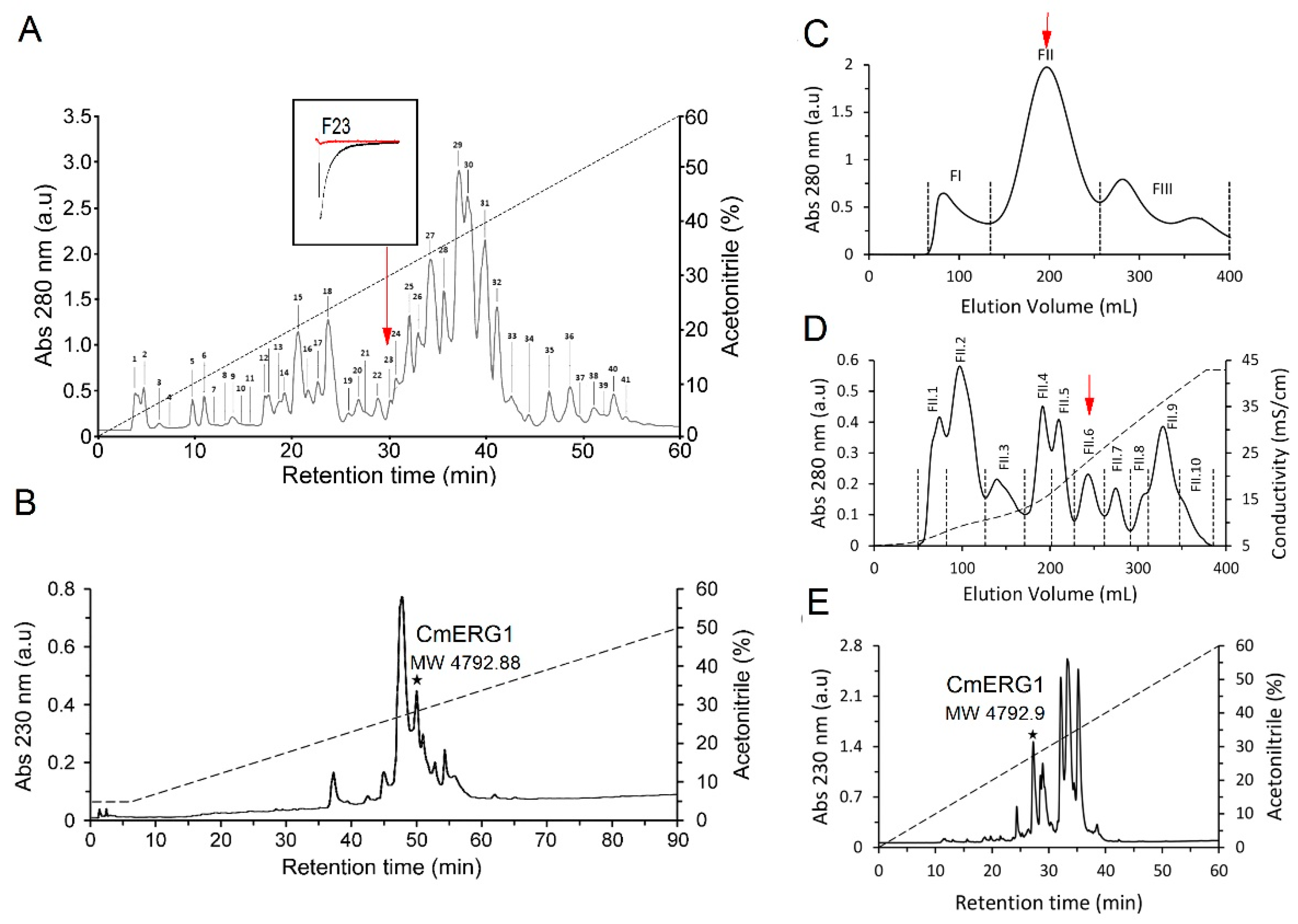
2.1. Peptide Isolation
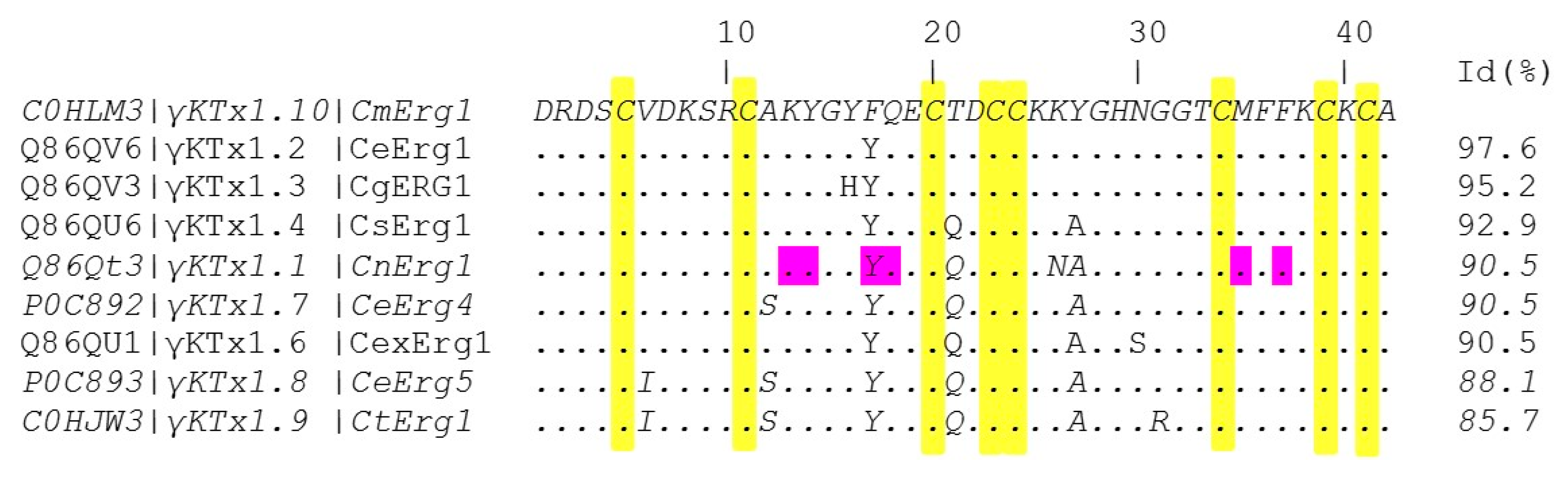
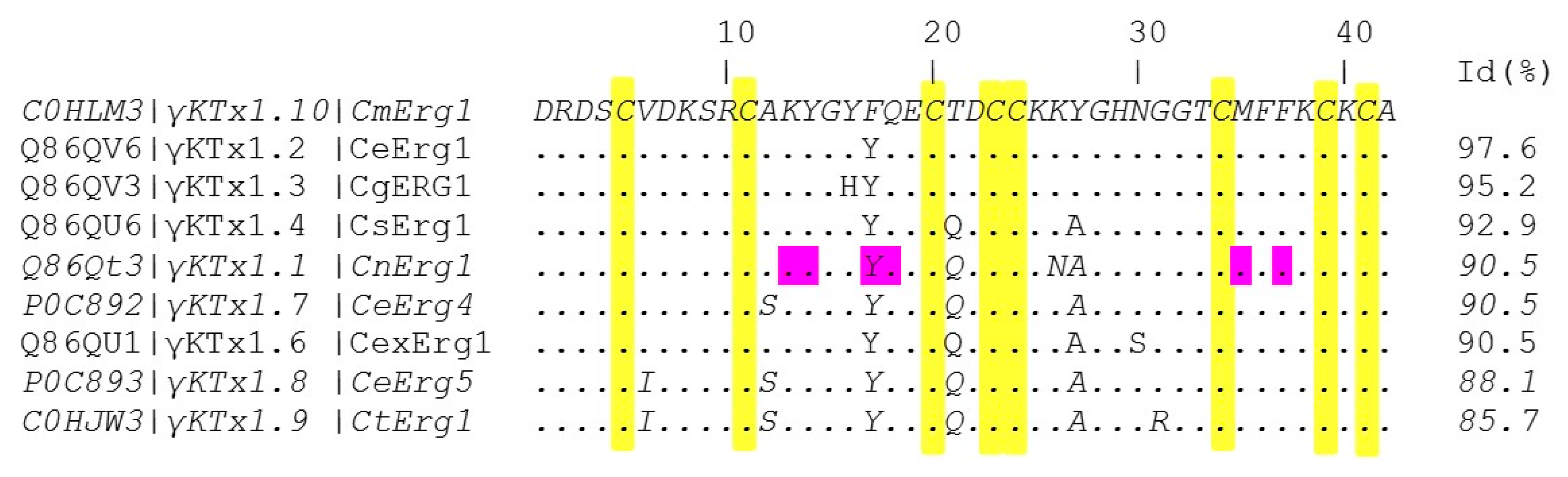
2.2. CmERG1 Primary Structure
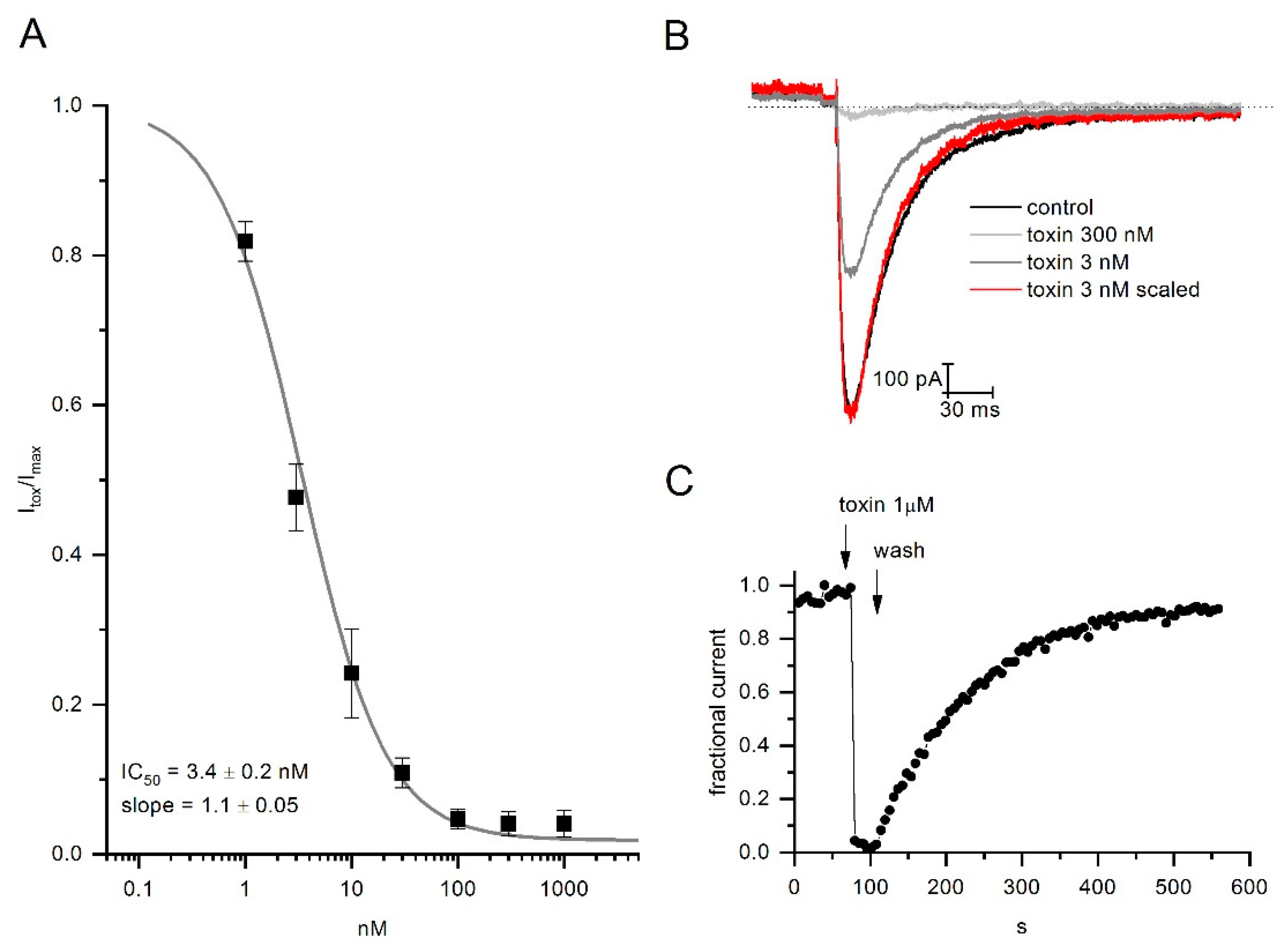
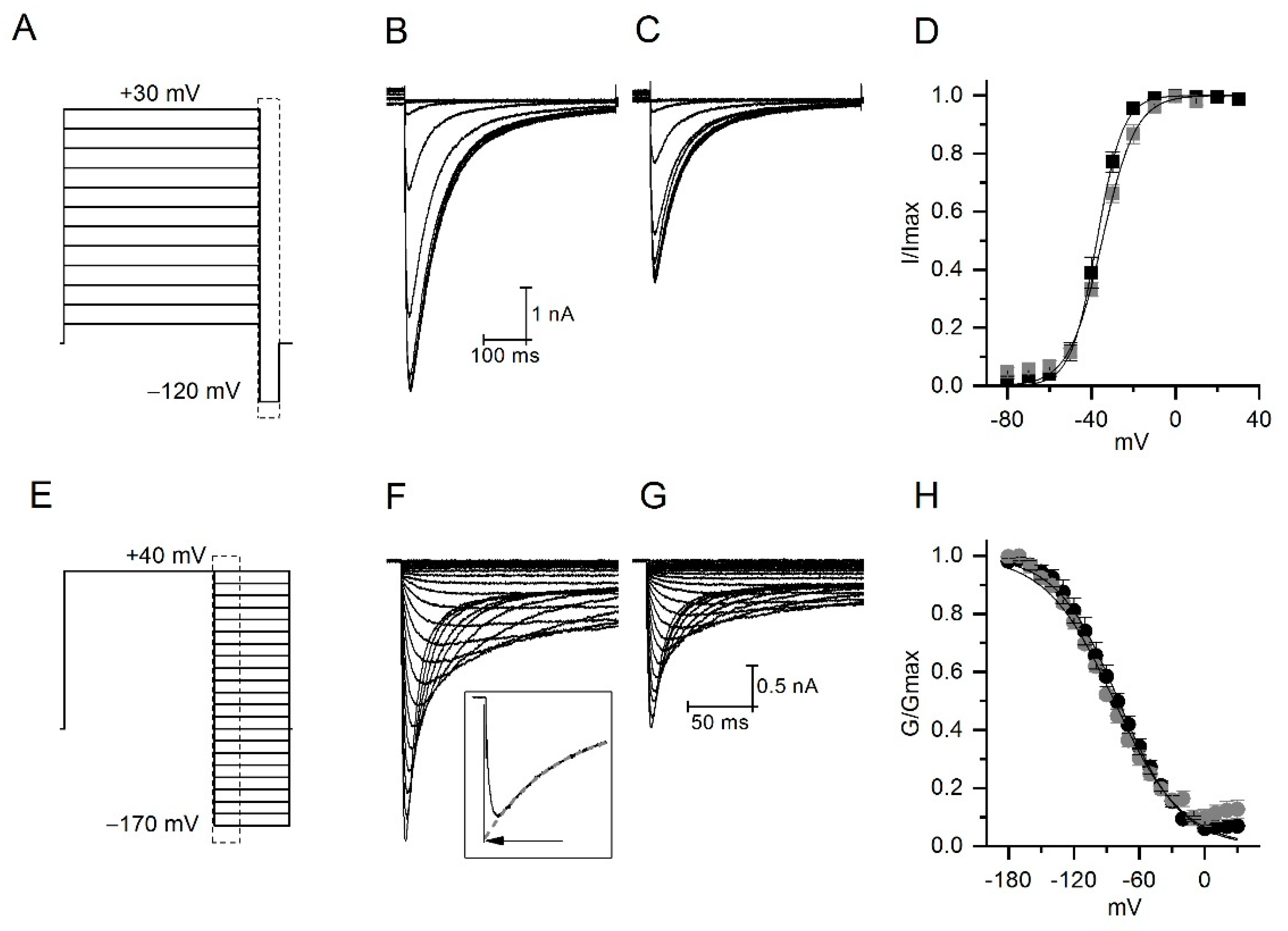
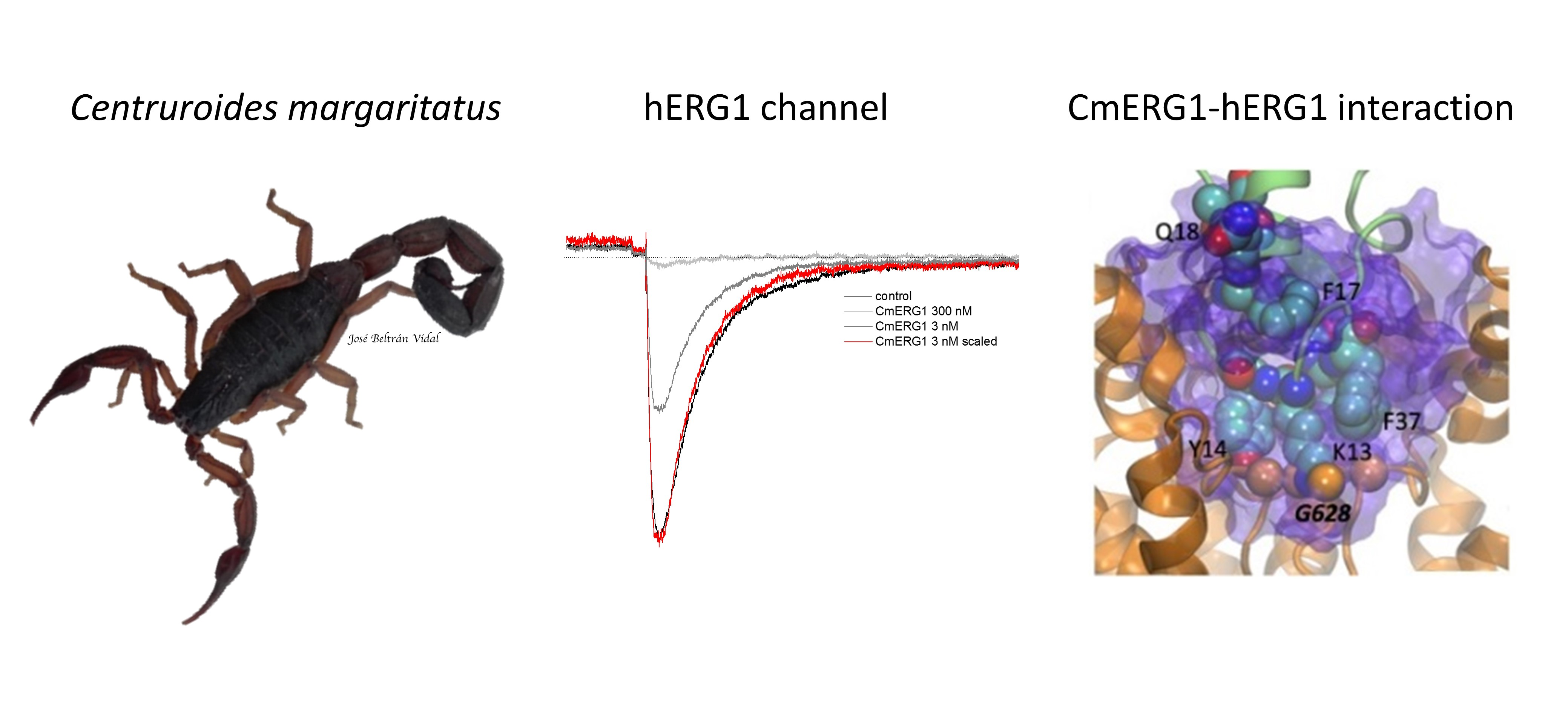
2.3. CmERG1 Block Action on hERG1 Channels
2.4. CmERG1 Does Not Modify the Activation and Inactivation Kinetics of hERG1 Channels
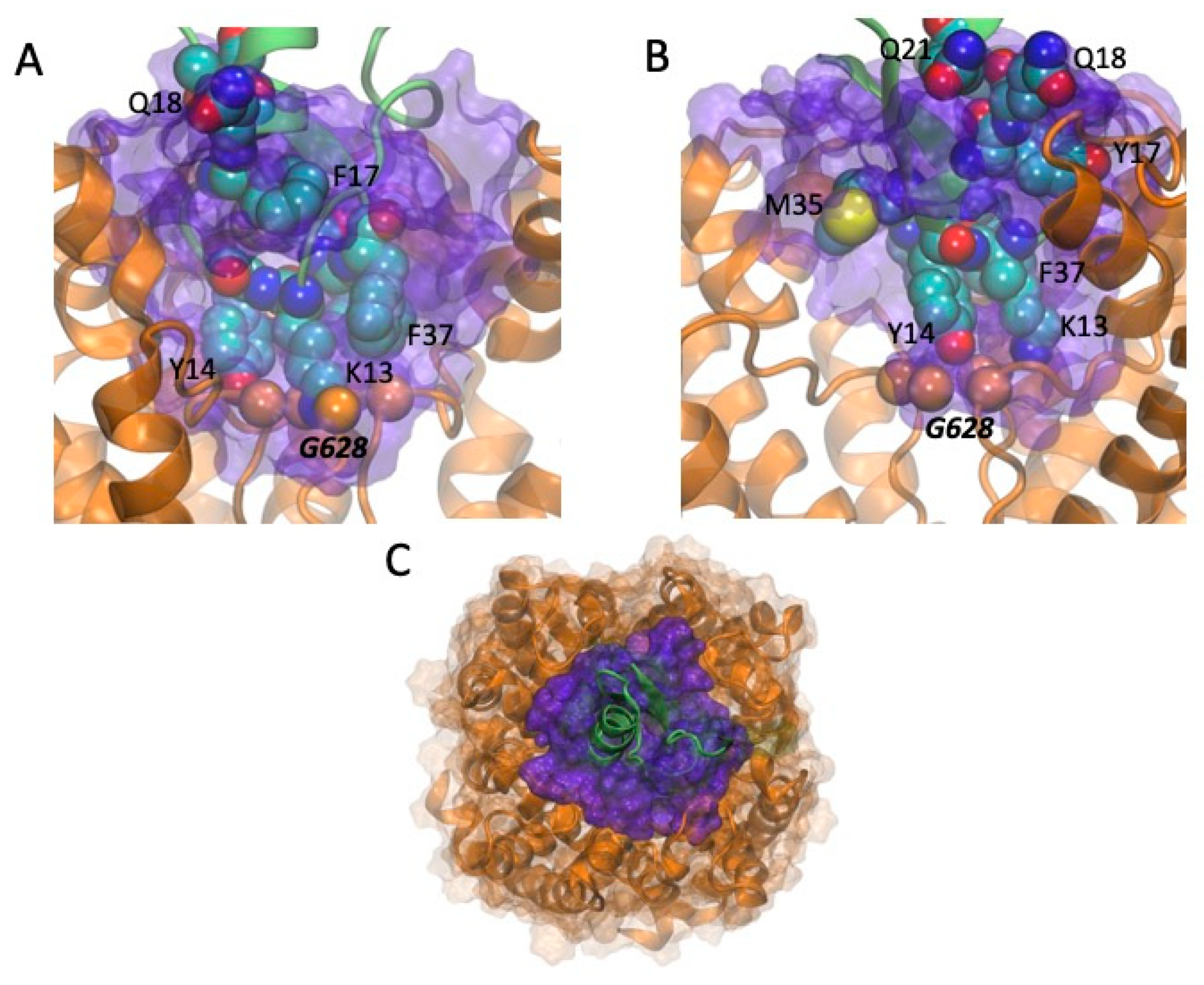
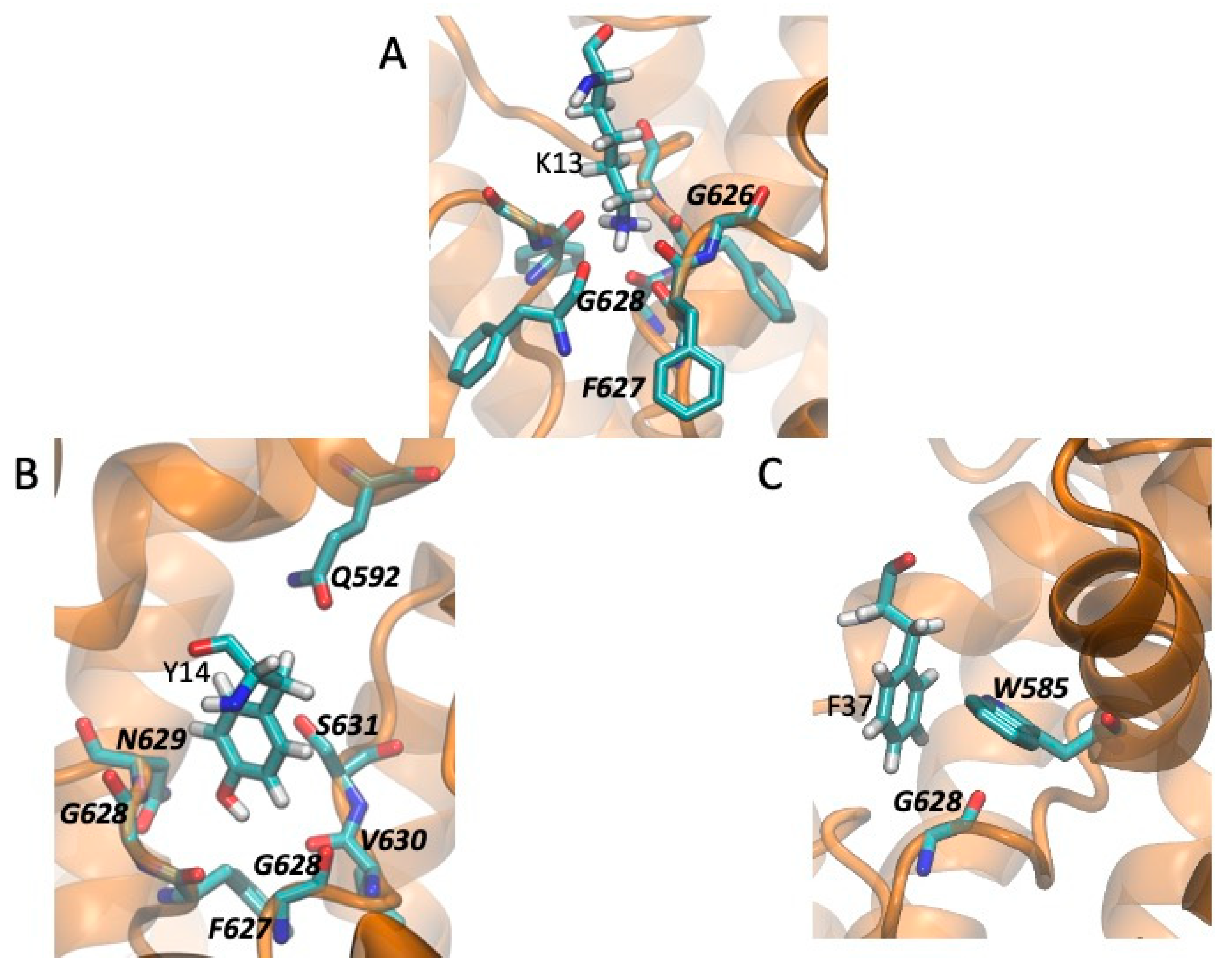
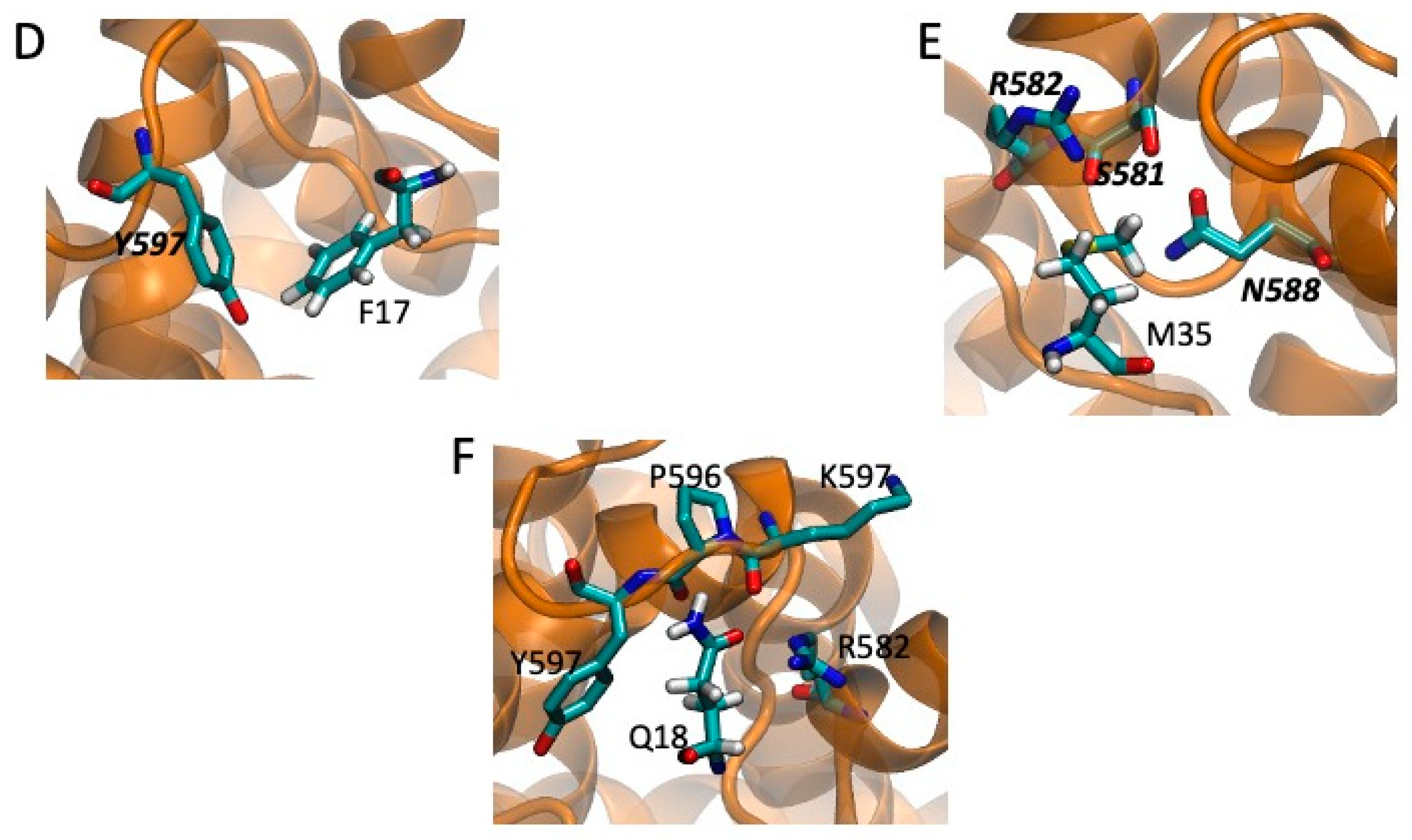
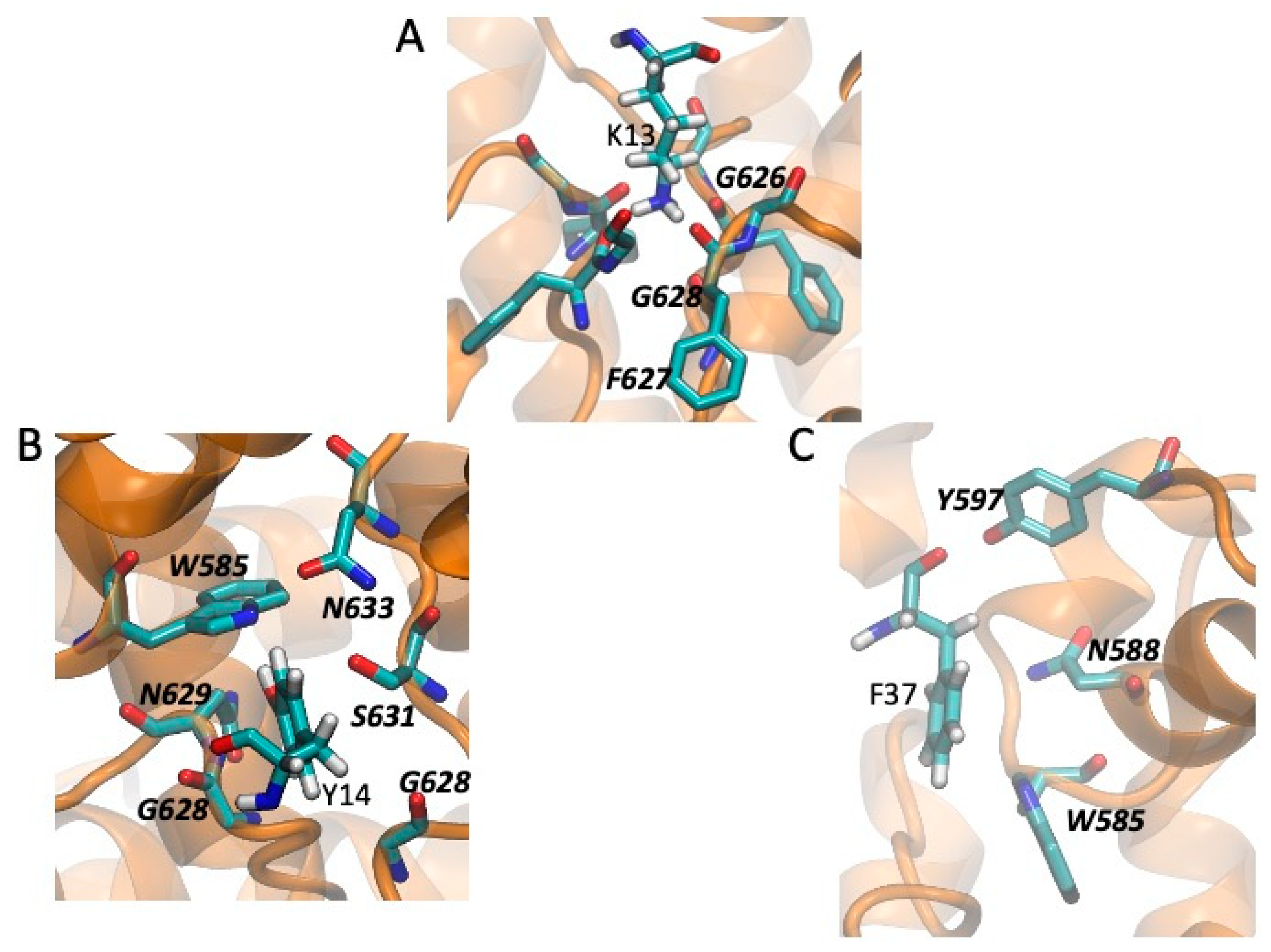
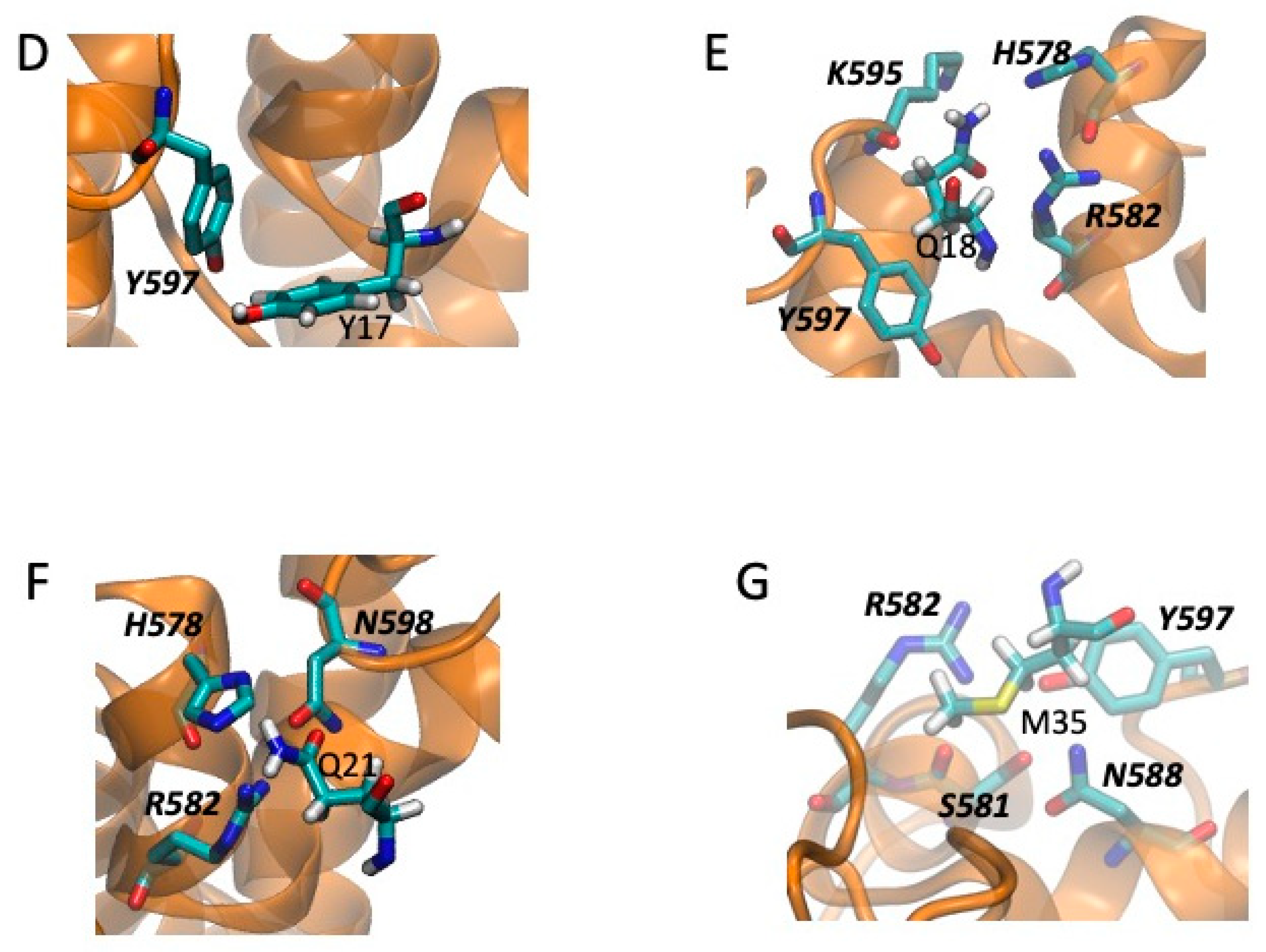
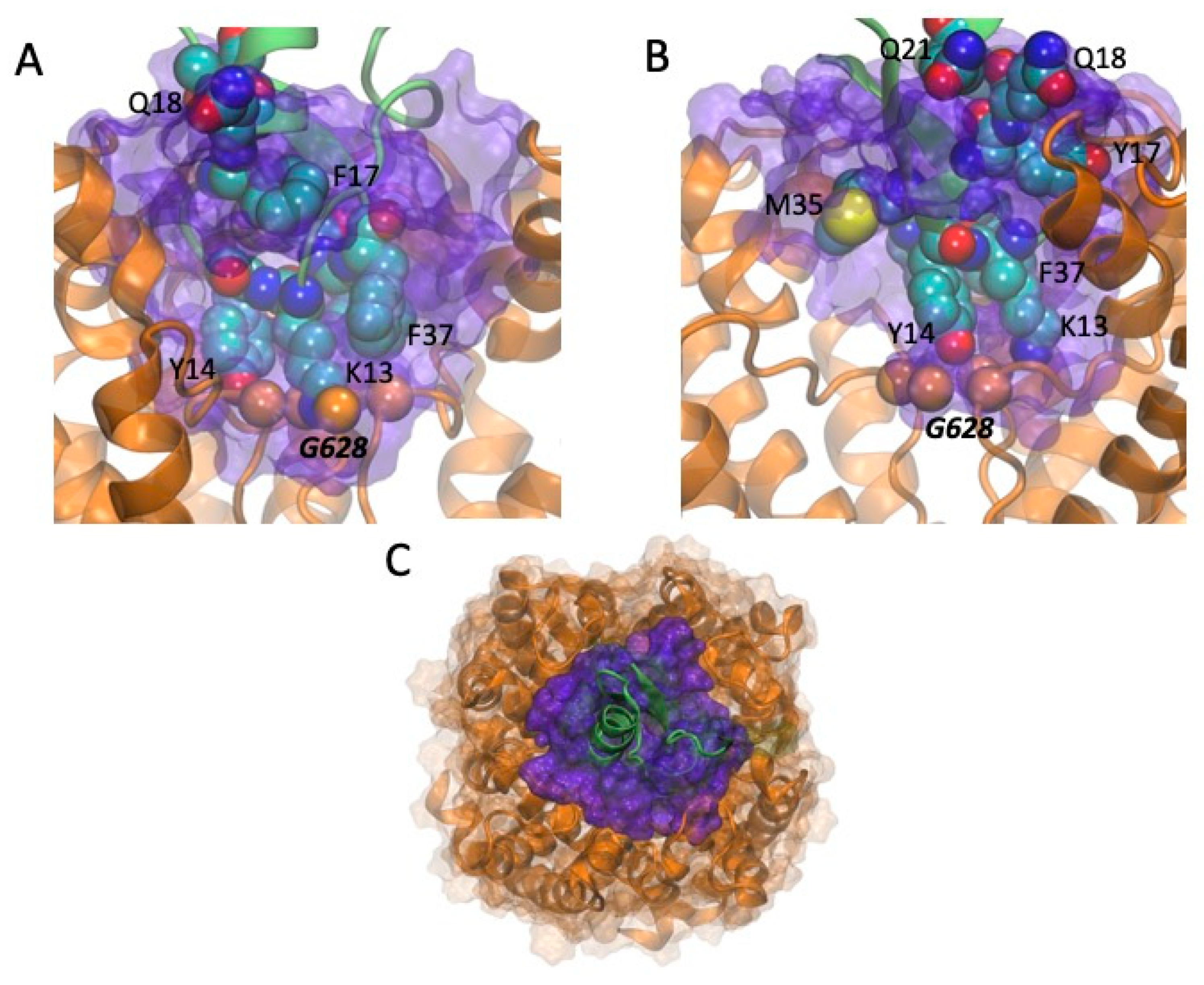
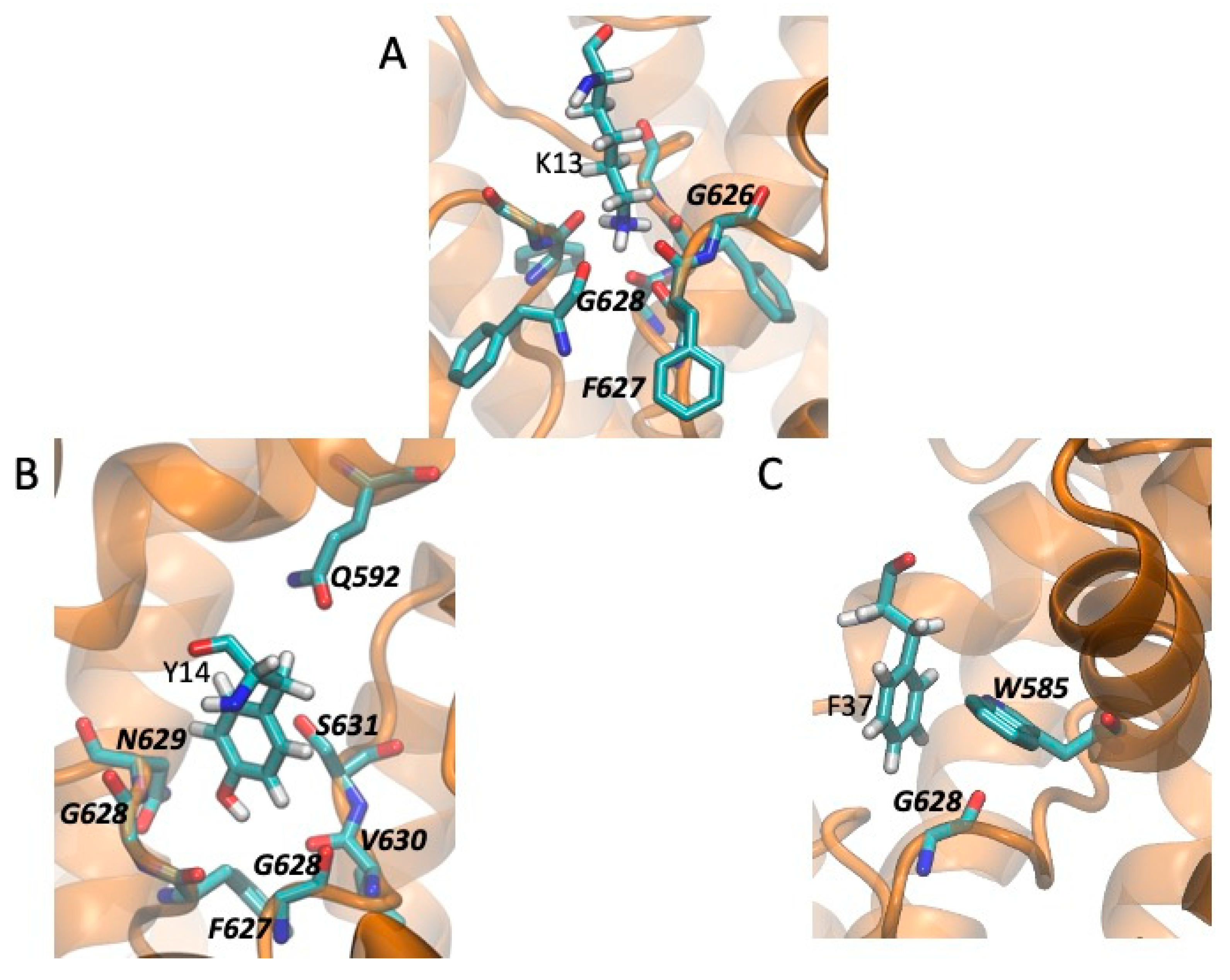
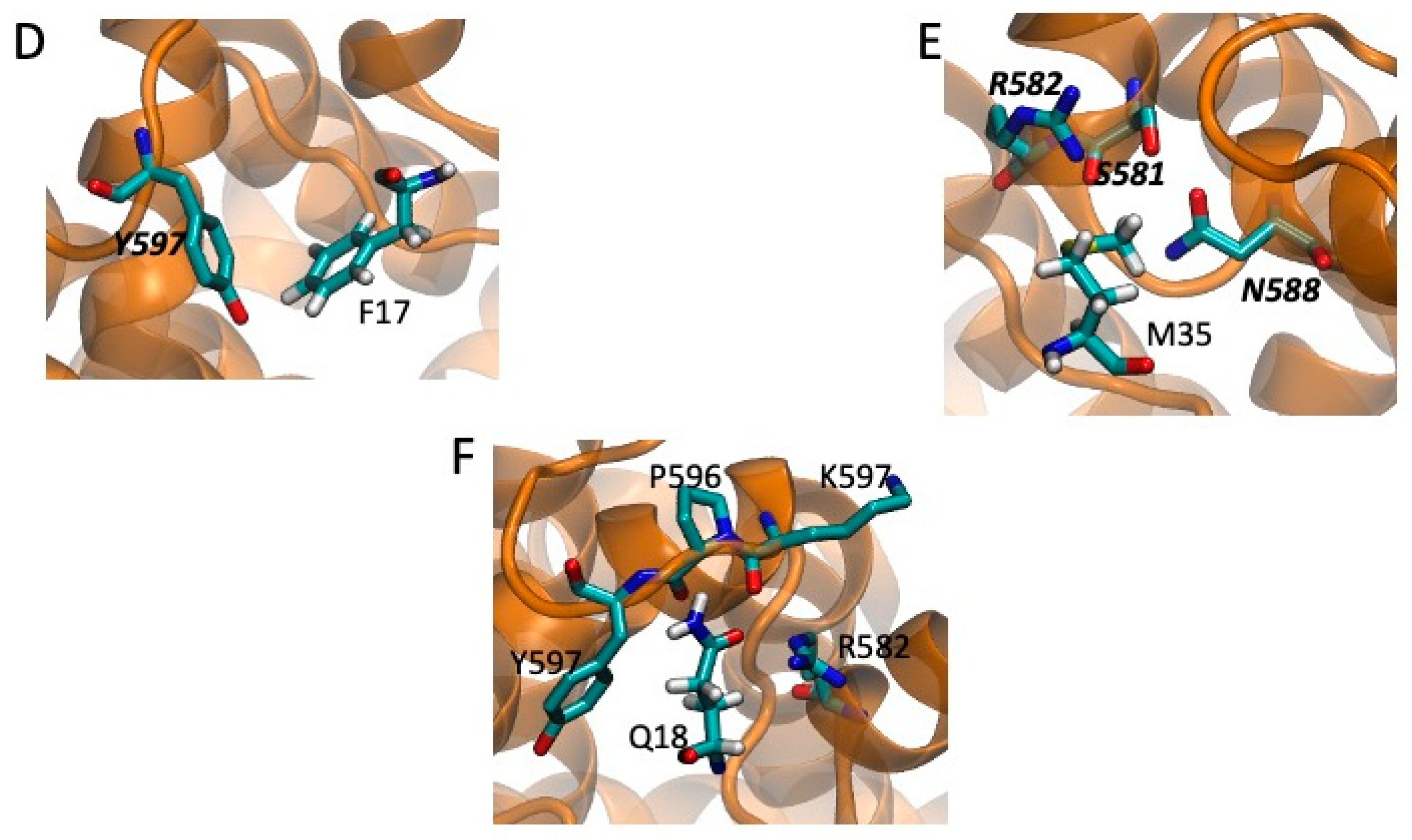
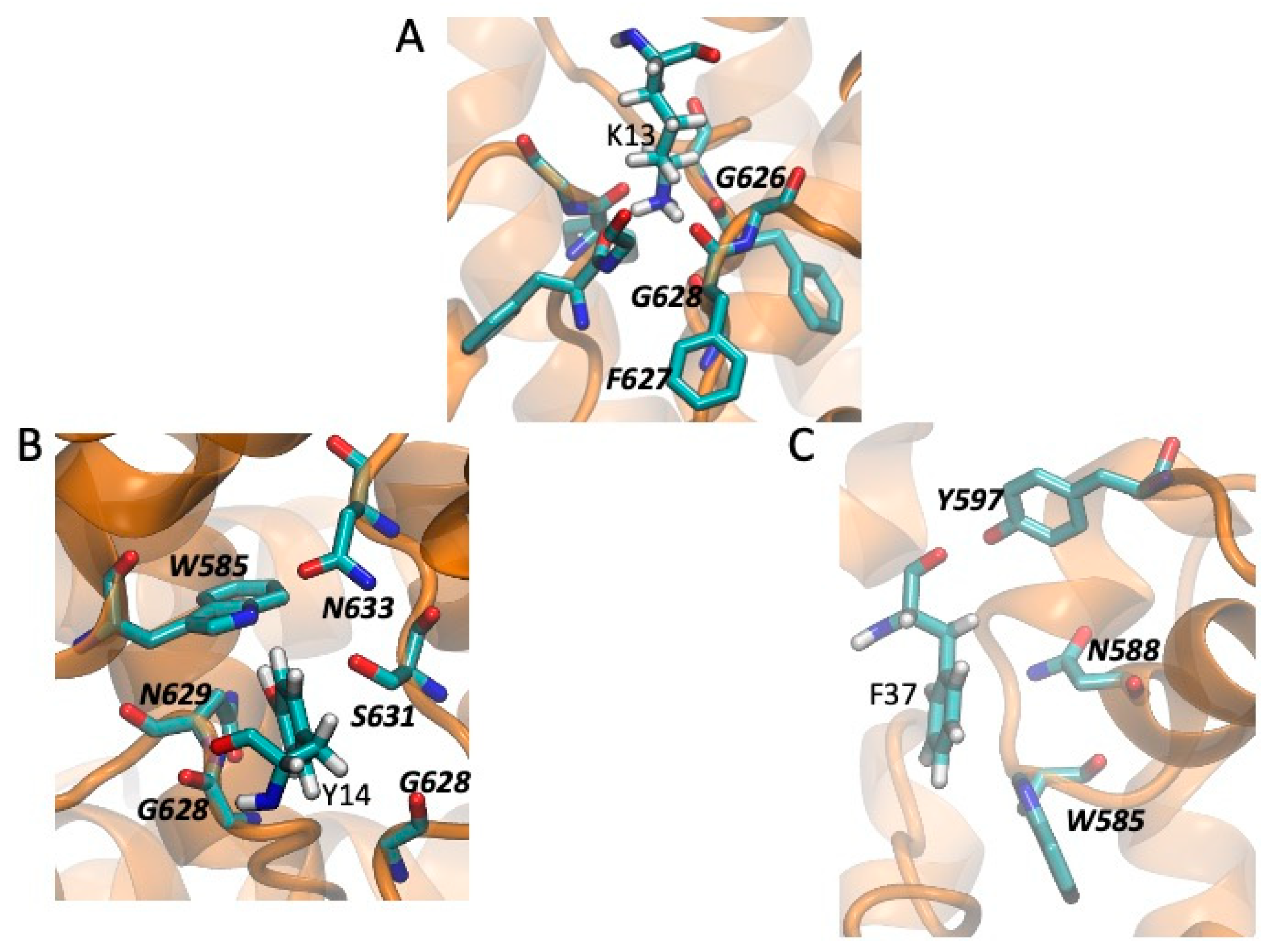
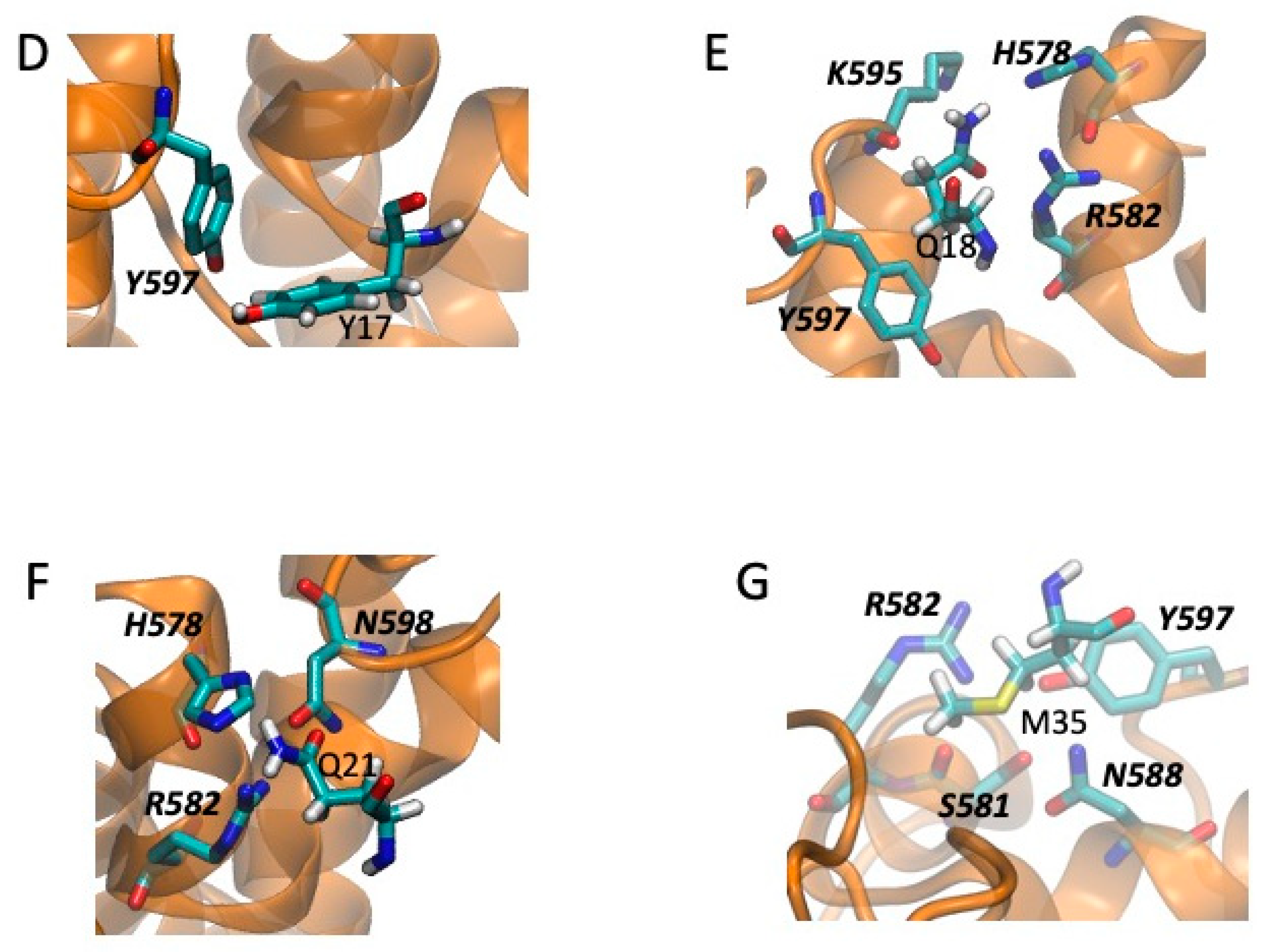
2.5. Models of Channel-Toxin Interactions
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Venom Source, Chemical and Reagents
5.2. Peptide Purification
5.3. Mass Spectrometry and Sequence by Edman Degradation
5.4. Reduction and Alkylation
5.5. Amino Acid Sequence Comparison of Peptide
5.6. Electrophysiology
5.6.1. Cells and Solutions
5.6.2. Patch-Clamp Recordings and Data Analysis
5.6.3. Statistical Analysis
5.7. Modeling of the Toxin-Channel Encounter Complexes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, M.J.; Ellsworth, S.A.; Nystrom, G.S. A global accounting of medically significant scorpions: Epidemiology, major toxins, and comparative resources in harmless counterparts. Toxicon 2018, 151, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Armas, L.F.; Sarmiento, D.L.; Flórez, E.L. Composición del género Centruroides marx, 1890 (Scorpiones buthidae) en Colombia, con la descripción de una nueva especie. Boletín Soc. Entomológica Aragonsa 2012, 50, 105–114. [Google Scholar]
- Guerrero-Vargas, J.A.; Rodríguez Buitrago, J.R.; Ayerbe, S.; Flórez Daza, E.; Beltrán Vidal, J.T. Scorpionism and Dangerous Species of ColombiaColombia. In Scorpion Venoms; Gopalakrishnakone, P., Possani, L.D., F. Schwartz, E., Rodríguez de la Vega, R.C., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 245–272. [Google Scholar] [CrossRef]
- Marinkelle, C.J.; Stahnke, H.L. Toxicological and clinical studies on Centruroides margaritatus (Gervais), a common scorpion in western Colombia. J. Med. Entomol. 1965, 2, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M.; Gordon, D.; Barzilai, M.G.; Kahn, R.; Cohen, L.; Moran, Y.; Zilberberg, N.; Froy, O.; Altman-Gueta, H.; Turkov, M.; et al. Molecular Description of Scorpion Toxin Interaction with Voltage-Gated Sodium Channels. In Scorpion Venoms; Gopalakrishnakone, P., Ferroni Schwartz, E., Possani, L.D., Rodríguez de la Vega, R.C., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 471–492. [Google Scholar] [CrossRef]
- Bartok, A.; Panyi, G.; Varga, Z. Potassium Channel-Blocking Peptide Toxins from Scorpion Venom. In Scorpion Venoms; Gopalakrishnakone, P., Ferroni Schwartz, E., Possani, L.D., Rodríguez de la Vega, R.C., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 493–521. [Google Scholar] [CrossRef]
- Rodríguez de la Vega, R.C.; Vidal, N.; Possani, L. Scorpion Peptides. In Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 423–429. [Google Scholar]
- Xiao, L.; Gurrola, G.B.; Zhang, J.; Valdivia, C.R.; SanMartin, M.; Zamudio, F.Z.; Zhang, L.; Possani, L.D.; Valdivia, H.H. Structure-function relationships of peptides forming the calcin family of ryanodine receptor ligands. J. Gen. Physiol. 2016, 147, 375–394. [Google Scholar] [CrossRef] [Green Version]
- Chuang, R.S.; Jaffe, H.; Cribbs, L.; Perez-Reyes, E.; Swartz, K.J. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat. Neurosci. 1998, 1, 668–674. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Z.; Cao, Z.; Li, W.; Wu, Y. Diverse Structural Features of Potassium Channels Characterized by Scorpion Toxins as Molecular Probes. Molecules 2019, 24, 2045. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Luo, F.; Feng, J.; Yang, W.; Zeng, D.; Zhao, R.; Cao, Z.; Liu, M.; Li, W.; Jiang, L.; et al. Genomic and structural characterization of Kunitz-type peptide LmKTT-1a highlights diversity and evolution of scorpion potassium channel toxins. PLoS ONE 2013, 8, e60201. [Google Scholar] [CrossRef] [Green Version]
- Mouhat, S.; Jouirou, B.; Mosbah, A.; de Waard, M.; Sabatier, J.M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 2004, 378, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kalium: Toxins Active on Potassium Channels. Available online: https://kaliumdb.org/ (accessed on 1 September 2019).
- Perrin, M.J.; Subbiah, R.N.; Vandenberg, J.I.; Hill, A.P. Human ether-a-go-go related gene (hERG) K+ channels: Function and dysfunction. Prog. Biophys. Mol. Biol. 2008, 98, 137–148. [Google Scholar] [CrossRef]
- Gurrola, G.B.; Rosati, B.; Rocchetti, M.; Pimienta, G.; Zaza, A.; Arcangeli, A.; Olivotto, M.; Possani, L.D.; Wanke, E. A toxin to nervous, cardiac, and endocrine ERG K+ channels isolated from Centruroides noxius scorpion venom. FASEB J. 1999, 13, 953–962. [Google Scholar] [CrossRef]
- Corona, M.; Gurrola, G.B.; Merino, E.; Cassulini, R.R.; Valdez-Cruz, N.A.; García, B.; Ramírez-Domínguez, M.E.; Coronas, F.I.V.; Zamudio, F.Z.; Wanke, E.; et al. A large number of novel Ergtoxin-like genes and ERG K+-channels blocking peptides from scorpions of the genus Centruroides. FEBS Lett. 2002, 532, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Gunay, B.C.; Yurtsever, M.; Durdagi, S. Elucidation of interaction mechanism of hERG1 potassium channel with scorpion toxins BeKm-1 and BmTx3b. J. Mol. Graph. Model. 2020, 96, 107504. [Google Scholar] [CrossRef] [PubMed]
- Korolkova, Y.V.; Kozlov, S.A.; Lipkin, A.V.; Pluzhnikov, K.A.; Hadley, J.K.; Filippov, A.K.; Brown, D.A.; Angelo, K.; Strøbaek, D.; Jespersen, T.; et al. An ERG channel inhibitor from the scorpion Buthus eupeus. J. Biol. Chem. 2001, 276, 9868–9876. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Vargas, J.M.; Restano-Cassulini, R.; Possani, L.D. Interacting sites of scorpion toxin ErgTx1 with hERG1 K+ channels. Toxicon 2012, 59, 633–641. [Google Scholar] [CrossRef]
- Wang, X.; Jimenez-Vargas, J.M.; Xu, C.; Possani, L.D.; Zhu, S. Positive selection-guided mutational analysis revealing two key functional sites of scorpion ERG K(+) channel toxins. Biochem. Biophys. Res. Commun. 2012, 429, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Tseng, G.-N.; Sonawane, K.D.; Korolkova, Y.V.; Zhang, M.; Liu, J.; Grishin, E.V.; Guy, H.R. Probing the Outer Mouth Structure of the hERG Channel with Peptide Toxin Footprinting and Molecular Modeling. Biophys. J. 2007, 92, 3524–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, A.P.; Sunde, M.; Campbell, T.J.; Vandenberg, J.I. Mechanism of block of the hERG K+ channel by the scorpion toxin CnErg1. Biophys. J. 2007, 92, 3915–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo-López, L.; García-Valdés, J.; Gurrola, G.B.; Robertson, G.A.; Possani, L.D. Mapping the receptor site for ergtoxin, a specific blocker of ERG channels. FEBS Lett. 2002, 510, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez de la Vega, R.C.; Merino, E.; Becerril, B.; Possani, L.D. Novel interactions between K+ channels and scorpion toxins. Trends Pharmacol. Sci. 2003, 24, 222–227. [Google Scholar] [CrossRef]
- Teruel, R. Primer registro de Centruroides margaritatus (Gervais, 1841) para Cuba (Scorpiones: Buthidae). Rev. Ibérica Aracnol. 2002, 5, 87–89. [Google Scholar]
- Khattabi, A.; Soulaymani-Bencheikh, R.; Achour, S.; Salmi, L.R.; Scorpion Consensus Expert, G. Classification of clinical consequences of scorpion stings: Consensus development. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, J.; Guerrero, J.; Nascimento, N.; Velasco, A.; Bezerra, P.; Farias, L. Respuesta hemodinámica de Rattus norvegicus a tres fracciones cromatográficas del veneno de Centruroides margaritatus (Gervais, 1841) (Scorpiones: Buthidae). Iatreia 2018, 31, S15. [Google Scholar]
- Guerrero-Vargas, J.; Ayerbe, S.; Rada-Mendoza, M.; Vélez, P.; Beltran, J.D.G. Preliminary toxinological characterization of the venom of the scorpion Centruroides margaritatus (Buthidae, Gervais, 1841) of the valle of the Patía, Colombia. J. Venom. Anim. Toxins Incl. Trop. Dis. 2007, 13, 228. [Google Scholar]
- Garcia-Calvo, M.; Leonard, R.J.; Novick, J.; Stevens, S.P.; Schmalhofer, W.; Kaczorowski, G.J.; Garcia, M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993, 268, 18866–18874. [Google Scholar] [CrossRef]
- Bartok, A.; Toth, A.; Somodi, S.; Szanto, T.G.; Hajdu, P.; Panyi, G.; Varga, Z. Margatoxin is a non-selective inhibitor of human Kv1.3 K+ channels. Toxicon 2014, 87, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Vargas, J. Análise Proteômica Parcial da Peçonha do Escorpião Colombiano Centruroides margaritatus. Master’s Thesis, University of Brasilia, Brasilia, Brazil, 2008. [Google Scholar]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Pardo-Lopez, L.; Zhang, M.; Liu, J.; Jiang, M.; Possani, L.D.; Tseng, G.-N. Mapping the Binding Site of a Humanether-a-go-go-related Gene-specific Peptide Toxin (ErgTx) to the Channel’s Outer Vestibule. J. Biol. Chem. 2002, 277, 16403–16411. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.M.; Bansal, P.; Alewood, P.F.; Bursill, J.A.; Kuchel, P.W.; Vandenberg, J.I. Solution structure of CnErg1 (Ergtoxin), a HERG specific scorpion toxin. FEBS Lett. 2003, 539, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Gwee, M.C.; Nirthanan, S.; Khoo, H.-E.; Gopalakrishnakone, P.; Kini, R.M.; Cheah, L.-S. Autonomic effects of some scorpion venoms and toxins. Clin. Exp. Pharmacol. Physiol. 2002, 29, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.L.A.; Neto, E.B.; de Freitas, L.A.; Dorce, V.A.C. Effects of Brazilian scorpion venoms on the central nervous system. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, A.; Turkdogan, K.A.; Akoz, A.; Avcil, M.; Dagli, B.; Canakci, S.E. A rare complication of scorpion venom: Atrial fibrillation. Am. J. Emerg. Med. 2016, 34, 938.e931–938.e933. [Google Scholar] [CrossRef]
- Cordeiro, F.F.; Sakate, M.; Fernandes, V.; Cuyumjian, P.R. Clinical and cardiovascular alterations produced by scorpion envenomation in dogs. J. Venom. Anim. Toxins Incl. Trop. Dis. 2006, 12, 19–43. [Google Scholar] [CrossRef] [Green Version]
- Romero-Imbachi, M.R.; Cupitra, N.; Angel, K.; Gonzalez, B.; Estrada, O.; Calderon, J.C.; Guerrero-Vargas, J.; Beltran, J.; Narvaez-Sanchez, R. Centruroides margaritatus scorpion complete venom exerts cardiovascular effects through alpha-1 adrenergic receptors. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2021, 240, 108939. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Tytgat, J.; Chandy, K.G.; Garcia, M.L.; Gutman, G.A.; Martin-Eauclaire, M.-F.; van der Walt, J.J.; Possani, L.D. A unified nomenclature for short-chain peptides isolated from scorpion venoms: α-KTx molecular subfamilies. Trends Pharmacol. Sci. 1999, 20, 444–447. [Google Scholar] [CrossRef]
- Torres, A.M.; Bansal, P.S.; Sunde, M.; Clarke, C.E.; Bursill, J.A.; Smith, D.J.; Bauskin, A.; Breit, S.N.; Campbell, T.J.; Alewood, P.F.; et al. Structure of the HERG K+ channel S5P extracellular linker: Role of an amphipathic alpha-helix in C-type inactivation. J. Biol. Chem. 2003, 278, 42136–42148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenal, K.; Xu, C.Q.; Wolff, N.; Wecker, K.; Gurrola, G.B.; Zhu, S.Y.; Chi, C.W.; Possani, L.D.; Tytgat, J.; Delepierre, M. Exploring structural features of the interaction between the scorpion toxinCnErg1 and ERG K+ channels. Proteins 2004, 56, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Cao, Z.; Yin, S.; Dai, C.; Wu, Y.; Li, W. Interaction Simulation of hERG K+ Channel with Its Specific BeKm-1 Peptide: Insights into the Selectivity of Molecular Recognition. J. Proteome Res. 2007, 6, 611–620. [Google Scholar] [CrossRef]
- Rodríguez de la Vega, R.C.; Barraza, O.; Restano-Cassulini, R.; Possani, L. Toxins Active on hERG Channel: Structure and Function. In Animal Toxins: State of the Art—Perspectives in Health and Biotechnology; Lima, M.E.d., Pimenta, A.M.d.C., Martin-Eauclaire, M.F., Zingali, R.B., Rochat, H., Eds.; EDITORA UFMG: Belo Horizonte, Brazil, 2009; pp. 193–204. [Google Scholar]
- Smith, P.L.; Baukrowitz, T.; Yellen, G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 1996, 379, 833–836. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 2017, 169, 422.e410–430.e410. [Google Scholar] [CrossRef] [Green Version]
- Dickson, C.J.; Velez-Vega, C.; Duca, J.S. Revealing Molecular Determinants of hERG Blocker and Activator Binding. J. Chem. Inf. Modeling 2020, 60, 192–203. [Google Scholar] [CrossRef]
- Khan, H.M.; Guo, J.; Duff, H.J.; Tieleman, D.P.; Noskov, S.Y. Refinement of a cryo-EM structure of hERG: Bridging structure and function. Biophys. J. 2021, 120, 738–748. [Google Scholar] [CrossRef]
- Miranda, W.E.; DeMarco, K.R.; Guo, J.; Duff, H.J.; Vorobyov, I.; Clancy, C.E.; Noskov, S.Y. Selectivity filter modalities and rapid inactivation of the hERG1 channel. Proc. Natl. Acad. Sci. USA 2020, 117, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Perissinotti, L.L.; de Biase, P.M.; Guo, J.; Yang, P.C.; Lee, M.C.; Clancy, C.E.; Duff, H.J.; Noskov, S.Y. Determinants of Isoform-Specific Gating Kinetics of hERG1 Channel: Combined Experimental and Simulation Study. Front. Physiol. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Molecular Mass (Da) |
|---|---|
| F13 | 3704.97 |
| F14 | 3718.19 |
| F15 | 3543.74/4178.82/3228.7 |
| F16 | 3571.8/4191.71 |
| F17 | 3572.21 |
| F18 | 4177.69 |
| F19 | 4176.97 |
| F20 | 2820.1/3980.01 |
| F21 | 2819.8/3979.7 |
| F22 | 4916.12/6898.7/7942.6 |
| * F23 | 6838.9/4792.1 |
| F31 | 6793.37/3376.05 |
| Fraction | Retention Time (min) | Molecular Mass (Da) |
|---|---|---|
| FII-6 | 24.5 | 2820.5 |
| FII-6 | 27.4 | 4792.8 |
| FII-6 | 28.7 | 4045.7 |
| FII-6 | 29.1 | 4309.5; 3994 |
| FII-6 | 32.3 | 6992 |
| FII-6 | 33.4 | 4177; 6470 |
| FII-6 | 34.6 | 6613.5; 6868.1 |
| FII-6 | 35.4 | 6724 |
| FII-7 | 20.66 | 3229 |
| FII-7 | 28.4 | 4045.6 |
| FII-7 | 29.2 | 6838.8 |
| FII-8 | 23.2 | 4177.5 |
| FII-8 | 25.2 | 3980.2 |
| FII-8 | 29.2 | 8475.1; 4133.5 |
| FII-8 | 37.9 | 7547.6; 7998.1 |
| FII-9 | 21.6 | 3704.2; 3815.1 |
| FII-9 | 26.1 | 4178.4 |
| FII-9 | 26.4 | 8475.8; 4178 |
| FII-9 | 27 | 7498.0; 7620.7 |
| FII-9 | 27.2 | 7497.4; 7618 |
| FII-10 | 18.7 | 3703.7; 3718.2 |
| FII-10 | 23.2 | 8441; 5027.8; 4915 |
| FII-10 | 28.6 | 7497; 7620 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltrán-Vidal, J.; Carcamo-Noriega, E.; Pastor, N.; Zamudio-Zuñiga, F.; Guerrero-Vargas, J.A.; Castaño, S.; Possani, L.D.; Restano-Cassulini, R. Colombian Scorpion Centruroides margaritatus: Purification and Characterization of a Gamma Potassium Toxin with Full-Block Activity on the hERG1 Channel. Toxins 2021, 13, 407. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060407
Beltrán-Vidal J, Carcamo-Noriega E, Pastor N, Zamudio-Zuñiga F, Guerrero-Vargas JA, Castaño S, Possani LD, Restano-Cassulini R. Colombian Scorpion Centruroides margaritatus: Purification and Characterization of a Gamma Potassium Toxin with Full-Block Activity on the hERG1 Channel. Toxins. 2021; 13(6):407. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060407
Chicago/Turabian StyleBeltrán-Vidal, José, Edson Carcamo-Noriega, Nina Pastor, Fernando Zamudio-Zuñiga, Jimmy Alexander Guerrero-Vargas, Santiago Castaño, Lourival Domingos Possani, and Rita Restano-Cassulini. 2021. "Colombian Scorpion Centruroides margaritatus: Purification and Characterization of a Gamma Potassium Toxin with Full-Block Activity on the hERG1 Channel" Toxins 13, no. 6: 407. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060407