Determination of Urinary Mycotoxin Biomarkers Using a Sensitive Online Solid Phase Extraction-UHPLC-MS/MS Method

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Online SPE-UHPLC-MS/MS Method Development
2.2. Method Performance
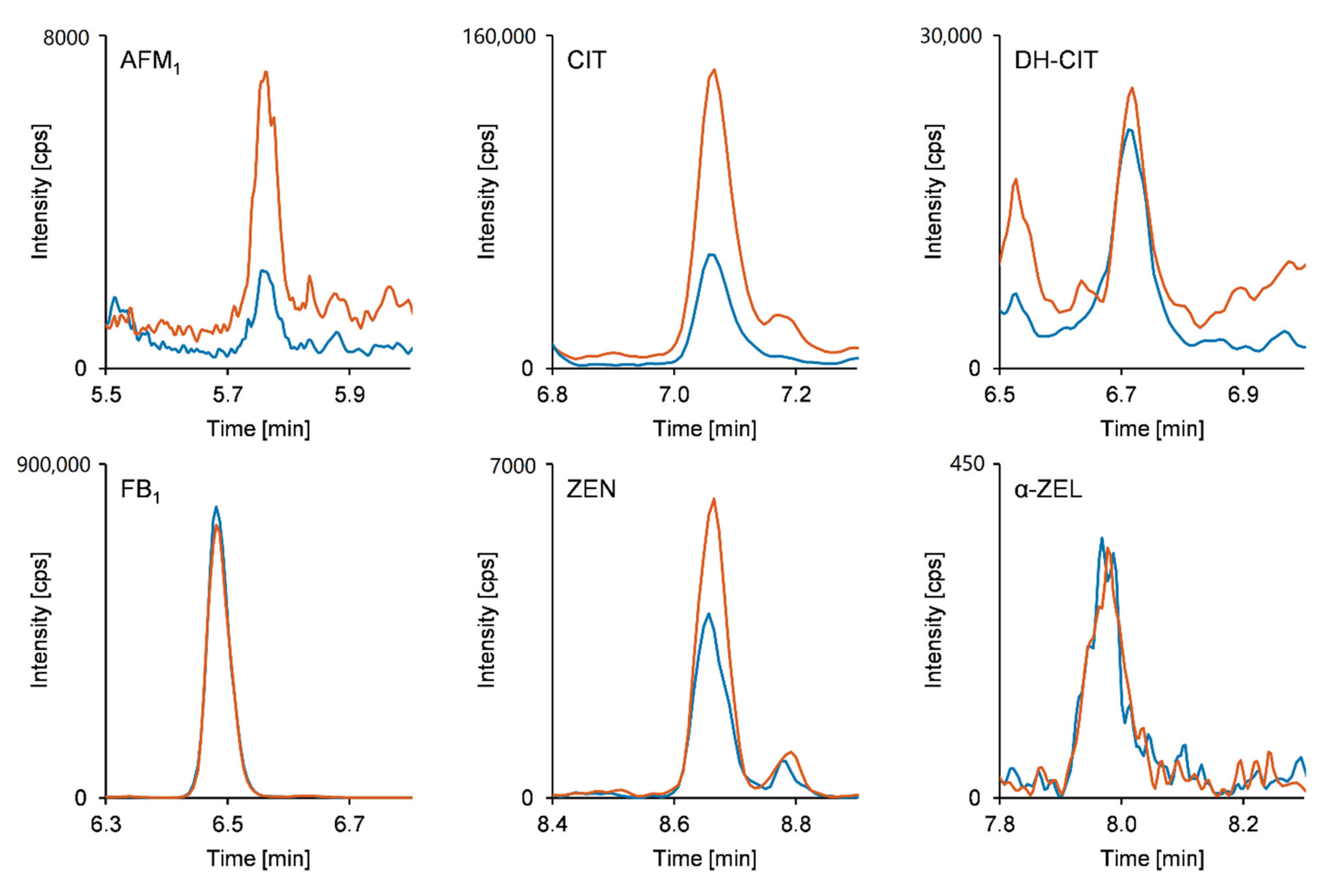
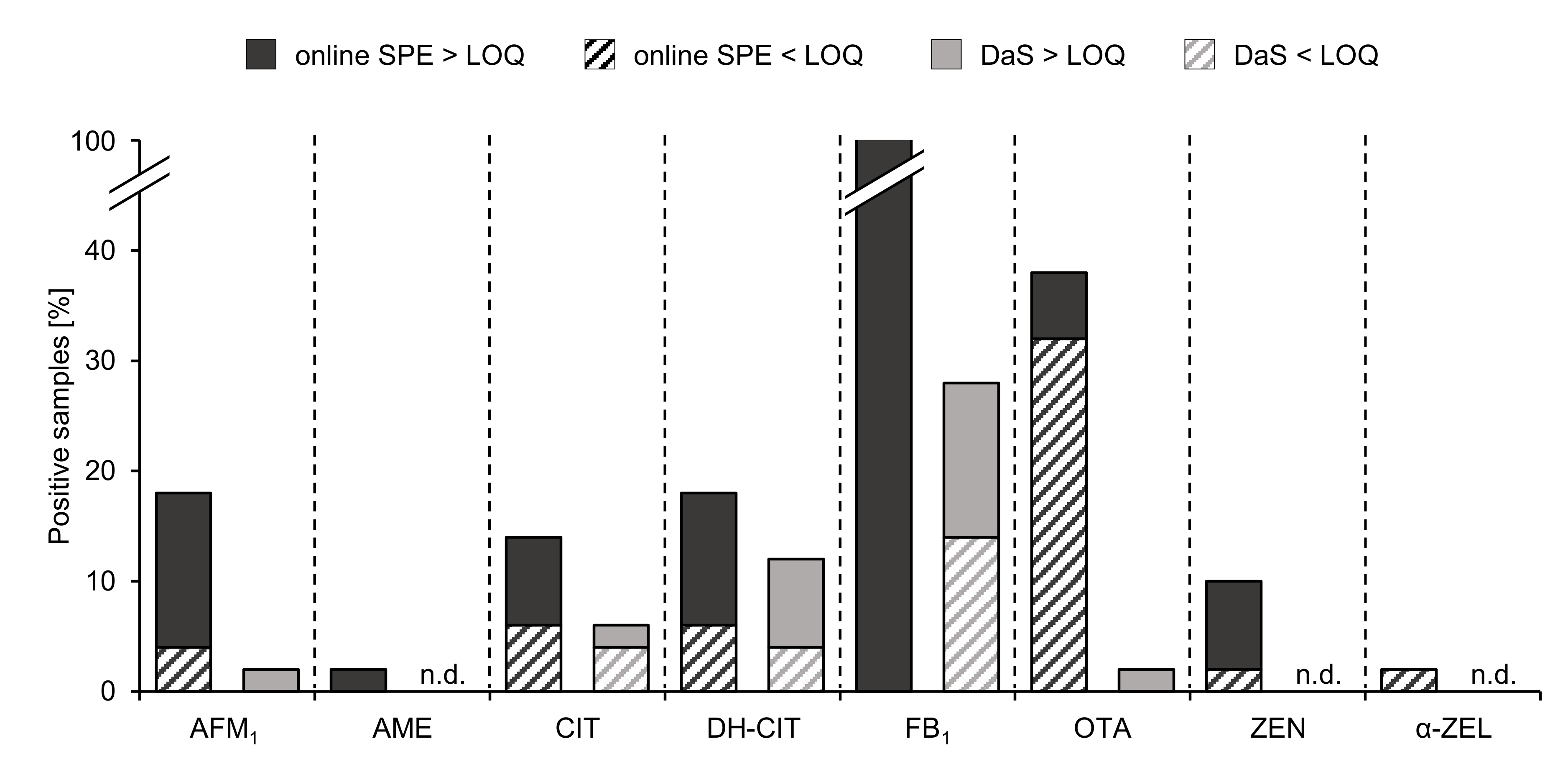
2.3. Analysis of Human Urine Samples
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Urine Samples
4.3. Sample Preparation
4.4. Online SPE-UHPLC-MS/MS Method
4.5. Method Validation
4.6. Analysis of Urine Samples Using a Dilute and Shoot Method
Supplementary Materials
Author Contributions
Funding
Institutional Review Board
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vidal, A.; Mengelers, M.; Yang, S.; de Saeger, S.; de Boevre, M. Mycotoxin biomarkers of exposure: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1127–1155. [Google Scholar] [CrossRef] [Green Version]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.C.; Flannery, B.; Isitt, C.; Ali, M.; Pestka, J. The role of biomarkers in evaluating human health concerns from fungal contaminants in food. Nutr. Res. Rev. 2012, 25, 162–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zöllner, P.; Mayer-Helm, B. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography—atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A 2006, 1136, 123–169. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [Green Version]
- Escrivá, L.; Font, G.; Manyes, L.; Berrada, H. Studies on the presence of mycotoxins in biological samples: An overview. Toxins 2017, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warth, B.; Sulyok, M.; Krska, R. LC-MS/MS-based multibiomarker approaches for the assessment of human exposure to mycotoxins. Anal. Bioanal. Chem. 2013, 405, 5687–5695. [Google Scholar] [CrossRef] [Green Version]
- Gerding, J.; Ali, N.; Schwartzbord, J.; Cramer, B.; Brown, D.L.; Degen, G.H.; Humpf, H.-U. A comparative study of the human urinary mycotoxin excretion patterns in Bangladesh, Germany, and Haiti using a rapid and sensitive LC-MS/MS approach. Mycotoxin Res. 2015, 31, 127–136. [Google Scholar] [CrossRef]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Mikula, H.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Abia, W.A.; Adam, G.; Fröhlich, J.; et al. Development and validation of a rapid multi-biomarker liquid chromatography/tandem mass spectrometry method to assess human exposure to mycotoxins. Rapid Commun. Mass Spectrom. 2012, 26, 1533–1540. [Google Scholar] [CrossRef]
- Lemming, E.W.; Montes, A.M.; Schmidt, J.; Cramer, B.; Humpf, H.-U.; Moraeus, L.; Olsen, M. Mycotoxins in blood and urine of Swedish adolescents—possible associations to food intake and other background characteristics. Mycotoxin Res. 2019, 36, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Lindemann, V.; Olsen, M.; Cramer, B.; Humpf, H.-U. Dried urine spots as sampling technique for multi-mycotoxin analysis in human urine. Mycotoxin Res. 2021, 1–12. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, X.; Wu, L.; Zhou, S.; Gong, Z.; Zhao, Y.; Wu, Y. Development of a sensitive and reliable UHPLC-MS/MS method for the determination of multiple urinary biomarkers of mycotoxin exposure. Toxins 2020, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Šarkanj, B.; Ezekiel, C.N.; Turner, P.C.; Abia, W.A.; Rychlik, M.; Krska, R.; Sulyok, M.; Warth, B. Ultra-sensitive, stable isotope assisted quantification of multiple urinary mycotoxin exposure biomarkers. Anal. Chim. Acta 2018, 1019, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.T.; Powers, S.; Visconti, A. Simultaneous LC–MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebaut, A. Fast and sensitive LC–MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Vidal, A.; De Boevre, M.; De Saeger, S.; Nunes, C.; Torres, D.; Goios, A.; Lopes, C.; Assunção, R.; Alvito, P. Exposure assessment of Portuguese population to multiple mycotoxins: The human biomonitoring approach. Int. J. Hyg. Environ. Health 2019, 222, 913–925. [Google Scholar] [CrossRef]
- Rodriguez-Mozaz, S.; de Alda, M.J.L.; Barceló, D. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography—mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J. Chromatogr. A 2007, 1152, 97–115. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, C.; Zhang, Z.; Li, G. Review of online coupling of sample preparation techniques with liquid chromatography. Anal. Chim. Acta 2014, 815, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Pagano, I.; Russo, M.; Rastrelli, L. Rapid and automated analysis of aflatoxin M1 in milk and dairy products by online solid phase extraction coupled to ultra-high-pressure-liquid-chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1428, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Lhotská, I.; Gajdošová, B.; Solich, P.; Šatínský, D. Molecularly imprinted vs. reversed-phase extraction for the determination of zearalenone: A method development and critical comparison of sample clean-up efficiency achieved in an on-line coupled SPE chromatography system. Anal. Bioanal. Chem. 2018, 410, 3265–3273. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Russo, M.; Valdés, A.; Ibañez, C.; Rastrelli, L. A fully automated method for simultaneous determination of aflatoxins and ochratoxin A in dried fruits by pressurized liquid extraction and online solid-phase extraction cleanup coupled to ultra-high-pressure liquid chromatography—tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2899–2911. [Google Scholar] [CrossRef]
- Kholová, A.; Lhotská, I.; Uhrová, A.; Špánik, I.; Machyňáková, A.; Solich, P.; Švec, F.; Šatínský, D. Determination of ochratoxin A and ochratoxin B in archived Tokaj wines (vintage 1959–2017) using on-line solid phase extraction coupled to liquid chromatography. Toxins 2020, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Ates, E.; Mittendorf, K.; Stroka, J.; Senyuva, H. Determination of Fusarium mycotoxins in wheat, maize and animal feed using on-line clean-up with high resolution mass spectrometry. Food Addit. Contam. Part A 2013, 30, 156–165. [Google Scholar] [CrossRef]
- Rozentale, I.; Bogdanova, E.; Bartkevics, V. A rapid and sensitive method for the control of selected regulated and emerging mycotoxins in beer. World Mycotoxin J. 2018, 11, 503–517. [Google Scholar] [CrossRef]
- Ndaw, S.; Jargot, D.; Antoine, G.; Denis, F.; Melin, S.; Robert, A. Investigating multi-mycotoxin exposure in occupational settings: A biomonitoring and airborne measurement approach. Toxins 2021, 13, 54. [Google Scholar] [CrossRef]
- Göschl, L.; Gmeiner, G.; Enev, V.; Kratena, N.; Gärtner, P.; Forsdahl, G. Development and validation of a simple online-SPE method coupled to high-resolution mass spectrometry for the analysis of stanozolol-N-glucuronides in urine samples. Drug Test. Anal. 2020, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.E.; Serafim, A.B.; Morales-Agudelo, P.; Vidal, M.; Calafat, A.M.; Ospina, M. Quantification of DEET and neonicotinoid pesticide biomarkers in human urine by online solid-phase extraction high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2018, 411, 669–678. [Google Scholar] [CrossRef]
- De Wilde, L.; Roels, K.; Deventer, K.; Van Eenoo, P. Automated sample preparation for the detection and confirmation of hypoxia-inducible factor stabilizers in urine. Biomed. Chromatogr. 2021, 35, 4970. [Google Scholar] [CrossRef]
- Franco, L.T.; Khaneghah, A.M.; Lee, S.H.I.; Oliveira, C.A.F. Biomonitoring of mycotoxin exposure using urinary biomarker approaches: A review. Toxin Rev. 2019, 57, 1–21. [Google Scholar] [CrossRef]
- Ediage, E.N.; Di Mavungu, J.D.; Song, S.; Wu, A.; Van Peteghem, C.; De Saeger, S. A direct assessment of mycotoxin biomarkers in human urine samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2012, 741, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.H.; Mbuya, M.N.N.; Ntozini, R.; Moulton, L.H.; Stoltzfus, R.J.; Tavengwa, N.V.; Mutasa, K.; Majo, F.; Mutasa, B.; Mangwadu, G.; et al. Independent and combined effects of improved water, sanitation, and hygiene, and improved complementary feeding, on child stunting and anaemia in rural Zimbabwe: A cluster-randomised trial. Lancet Glob. Health 2019, 7, e132–e147. [Google Scholar] [CrossRef] [Green Version]
- Hickert, S.; Bergmann, M.; Ersen, S.; Cramer, B.; Humpf, H.-U. Survey of Alternaria toxin contamination in food from the German market, using a rapid HPLC-MS/MS approach. Mycotoxin Res. 2015, 32, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Hübner, F.; Harrer, H.; Fraske, A.; Kneifel, S.; Humpf, H.-U. Large scale purification of B-type fumonisins using centrifugal partition chromatography (CPC). Mycotoxin Res. 2012, 28, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cramer, B.; Harrer, H.; Nakamura, K.; Uemura, D.; Humpf, H.-U. Total synthesis and cytotoxicity evaluation of all ochratoxin A stereoisomers. Bioorganic Med. Chem. 2010, 18, 343–347. [Google Scholar] [CrossRef]
- Cramer, B.; Bretz, M.; Humpf, H.-U. Stable isotope dilution analysis of the Fusarium mycotoxin zearalenone. J. Agric. Food Chem. 2007, 55, 8353–8358. [Google Scholar] [CrossRef] [PubMed]
- Urry, W.; Wehrmeister, H.; Hodge, E.; Hidy, P. The structure of zearalenone. Tetrahedron Lett. 1966, 7, 3109–3114. [Google Scholar] [CrossRef]
- Bergmann, D.; Hübner, F.; Wibbeling, B.; Daniliuc, C.; Cramer, B.; Humpf, H.-U. Large-scale total synthesis of 13C3-labeled citrinin and its metabolite dihydrocitrinone. Mycotoxin Res. 2018, 34, 141–150. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC—MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Analyte | LOD a [ng/mL urine] | LOQ b [ng/mL urine] | Working Range [ng/mL urine] | R2 c | RA d [%] | Intraday f | Interday g | ||
|---|---|---|---|---|---|---|---|---|---|
| Accuracy [%] | RSD h [%] | Accuracy [%] | RSD h [%] | ||||||
| AFM1 | 0.0070 | 0.025 | 0.025–2.5 | 0.999 | 101 | 112/99 | 5/2 | 108/97 | 7/5 |
| ALT | 0.21 | 0.69 | 0.70–70 | 0.999 | 38 e | 116/97 | 4/1 | 107/94 | 8/4 |
| AME | 0.020 | 0.040 | 0.040–4.0 | 0.984 | 78 e | 179/133 | 9/7 | 172/117 | 11/14 |
| AOH | 0.27 | 0.91 | 0.90–90 | 0.993 | 42 e | 140/107 | 5/3 | 140/103 | 6/6 |
| CIT | 0.09 | 0.32 | 0.30–30 | 0.999 | 98 | 114/100 | 6/2 | 104/98 | 7/5 |
| DH-CIT | 0.07 | 0.23 | 0.20–20 | 0.998 | 96 | 110/98 | 10/3 | 109/97 | 6/6 |
| FB1 | 0.0042 | 0.014 | 0.014–1.4 | 0.999 | 147 | 119/114 | 13/10 | 134/128 | 18/11 |
| OTA | 0.0036 | 0.012 | 0.012–1.2 | 0.999 | 91 | 120/104 | 9/5 | 112/104 | 11/6 |
| ZEN | 0.010 | 0.035 | 0.035–3.5 | 0.992 | 103 | 119/117 | 16/6 | 101/116 | 25/8 |
| α-ZEL | 0.05 | 0.15 | 0.15–15 | 0.993 | 86 | 123/118 | 13/5 | 115/117 | 20/5 |
| β-ZEL | 0.05 | 0.15 | 0.15–15 | 0.997 | 80 | 95/104 | 10/4 | 94/110 | 27/8 |
| Sample Preparation | Online SPE | DaS a | DaS | DUS b | SPE b | SPE b | SPE + IAC b | Direct method/IAC | QuEChERS | LLE + SPE |
|---|---|---|---|---|---|---|---|---|---|---|
| LOD [ng/mL urine] | ||||||||||
| AFM1 | 0.0070 | 0.06 | 0.05 | — | — | 0.0003 | 0.06 | 0.002 c | — | 0.01 |
| ALT | 0.21 | 3.0 | — | — | 0.02 | — | — | — | — | — |
| AME | 0.020 | 0.3 | — | — | 0.001 | — | — | — | 0.5 | — |
| AOH | 0.27 | 1.5 | — | — | 0.01 | 0.01 | — | — | 0.4 | — |
| CIT | 0.09 | 1.5 | — | — | — | 0.003 | — | 0.001 c | 0.5 | 2.88 |
| DH-CIT | 0.07 | 0.2 | — | — | — | 0.003 | — | 0.01 | — | — |
| FB1 | 0.0042 | 1.0 | 0.5 | 0.16 | 0.02 | 0.001 | 0.05 | 0.05 | 0.21 | 0.05 |
| OTA | 0.0036 | 0.01 | 0.05 | 0.004 | 0.01 | 0.0003 | 0.01 | 0.001 c | 0.01 | 0.03 |
| ZEN | 0.010 | 0.3 | 0.4 | 0.09 | — | 0.001 | — | 0.02 | 0.20 | 1.24 |
| α-ZEL | 0.05 | 0.5 | 0.5 | 0.17 | — | 0.003 | 0.8 | 0.05 | 0.61 | 0.61 |
| β-ZEL | 0.05 | 0.6 | 0.5 | 0.26 | — | 0.001 | 2.2 | 0.05 | 0.91 | 1.1 |
| Reference | This method | [10] | [9] | [11] | [12] | [13] | [14] | [15] | [16] | [30] |
| Concentration [ng/mL] | ||||||
|---|---|---|---|---|---|---|
| Analyte | Positive Samples % (n) | Quantitated Samples % (n) | Mean a | Median a | Maximum | SD b Quantitated Samples |
| AFM1 | 18 (9) | 14 (7) | 0.18 | 0.064 | 0.87 | 0.31 |
| AME | 2 (1) | 2 (1) | — | — | 0.11 | — |
| CIT | 14 (7) | 8 (4) | 1.5 | 1.4 | 2.3 | 0.57 |
| DH-CIT | 18 (9) | 12 (6) | 0.85 | 0.86 | 1.3 | 0.42 |
| FB1 | 100 (50) | 100 (50) | 0.68 | 0.29 | 4.6 c, d | 0.95 |
| OTA | 38 (19) | 6 (3) | 0.060 | 0.044 | 0.11 | 0.046 |
| ZEN | 10 (5) | 8 (4) | 0.23 | 0.11 | 0.40 | 0.14 |
| α-ZEL | 2 (1) | <LOQ | — | — | — | — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, J.; Cramer, B.; Turner, P.C.; Stoltzfus, R.J.; Humphrey, J.H.; Smith, L.E.; Humpf, H.-U. Determination of Urinary Mycotoxin Biomarkers Using a Sensitive Online Solid Phase Extraction-UHPLC-MS/MS Method. Toxins 2021, 13, 418. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060418
Schmidt J, Cramer B, Turner PC, Stoltzfus RJ, Humphrey JH, Smith LE, Humpf H-U. Determination of Urinary Mycotoxin Biomarkers Using a Sensitive Online Solid Phase Extraction-UHPLC-MS/MS Method. Toxins. 2021; 13(6):418. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060418
Chicago/Turabian StyleSchmidt, Jessica, Benedikt Cramer, Paul C. Turner, Rebecca J. Stoltzfus, Jean H. Humphrey, Laura E. Smith, and Hans-Ulrich Humpf. 2021. "Determination of Urinary Mycotoxin Biomarkers Using a Sensitive Online Solid Phase Extraction-UHPLC-MS/MS Method" Toxins 13, no. 6: 418. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060418