Identification and Characterization of Novel Proteins from Arizona Bark Scorpion Venom That Inhibit Nav1.8, a Voltage-Gated Sodium Channel Regulator of Pain Signaling

 ,
,  , , , and
, , , and
Abstract
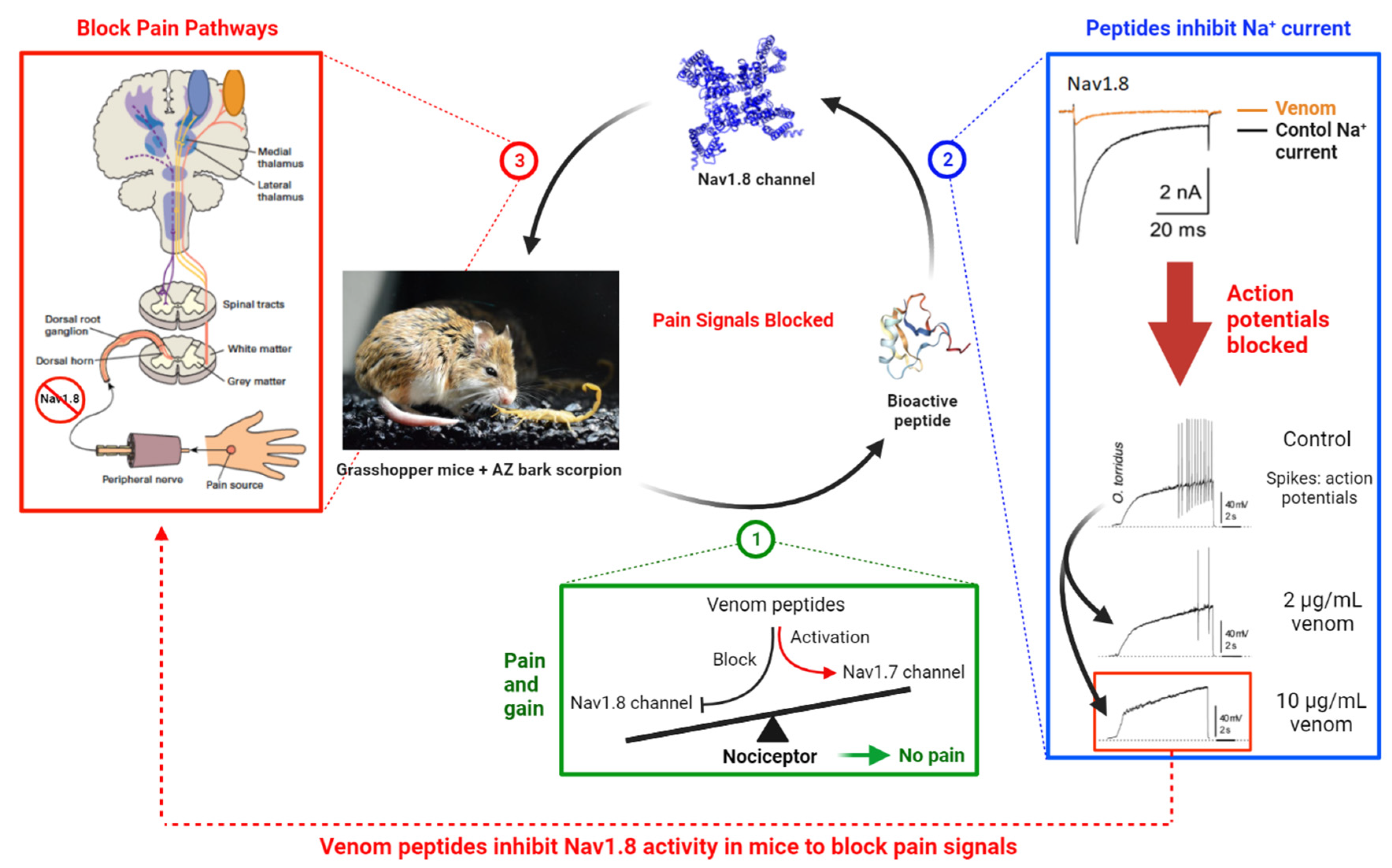
:1. Introduction
2. Results
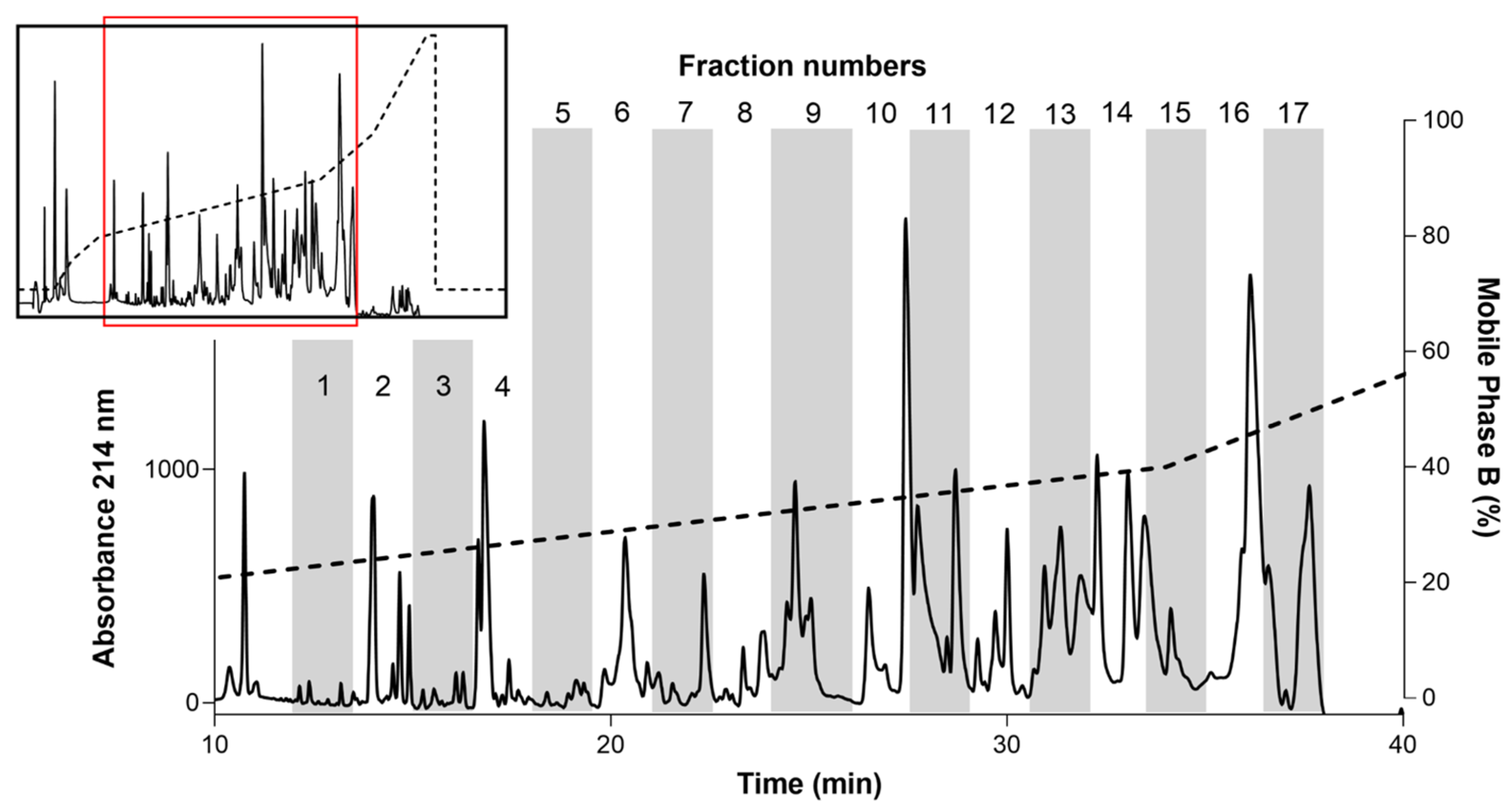
2.1. RPLC Fractionation of AZ Bark Scorpion Venom
2.2. Identification of Inhibitory Fractions from AZ Bark Scorpion Venom
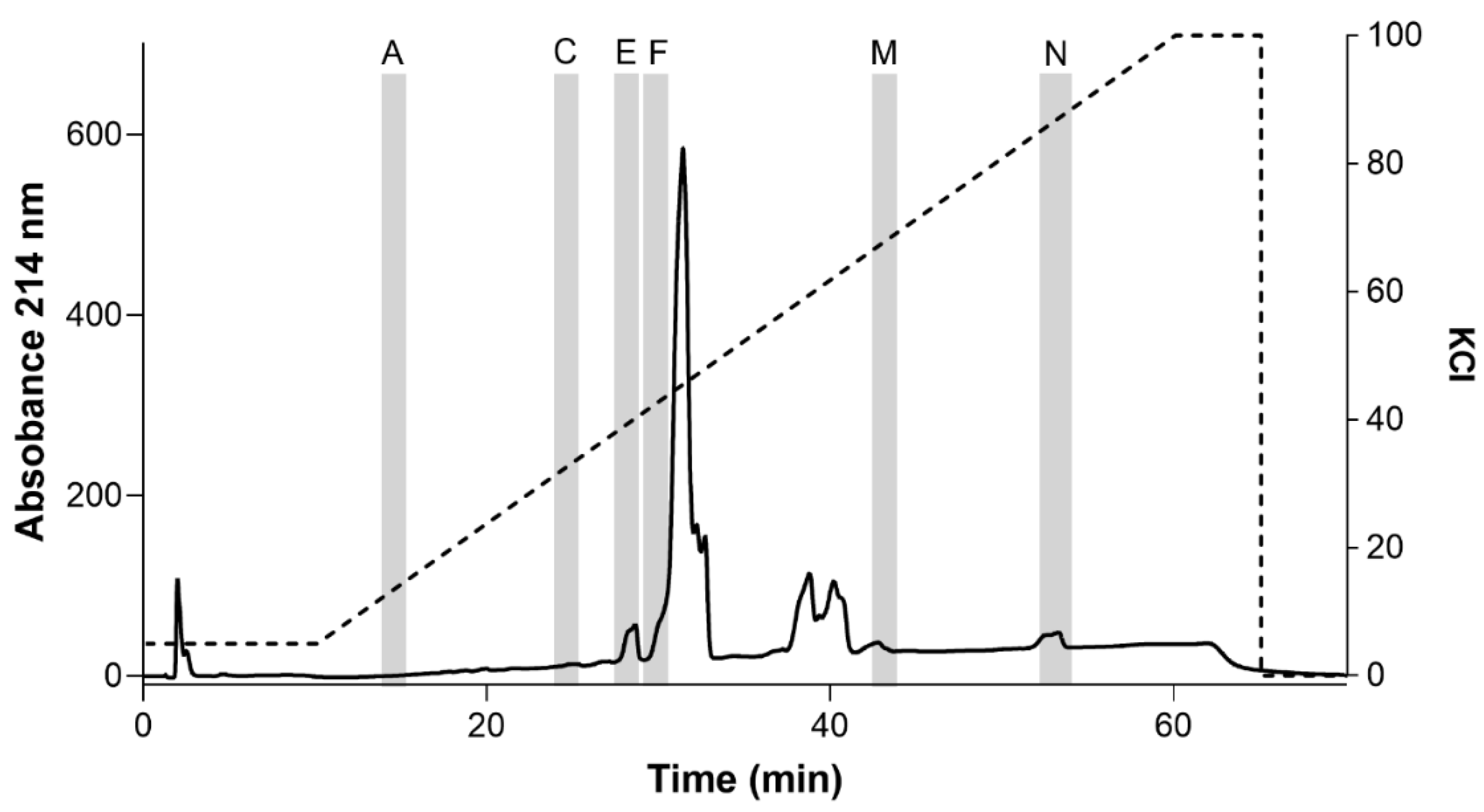
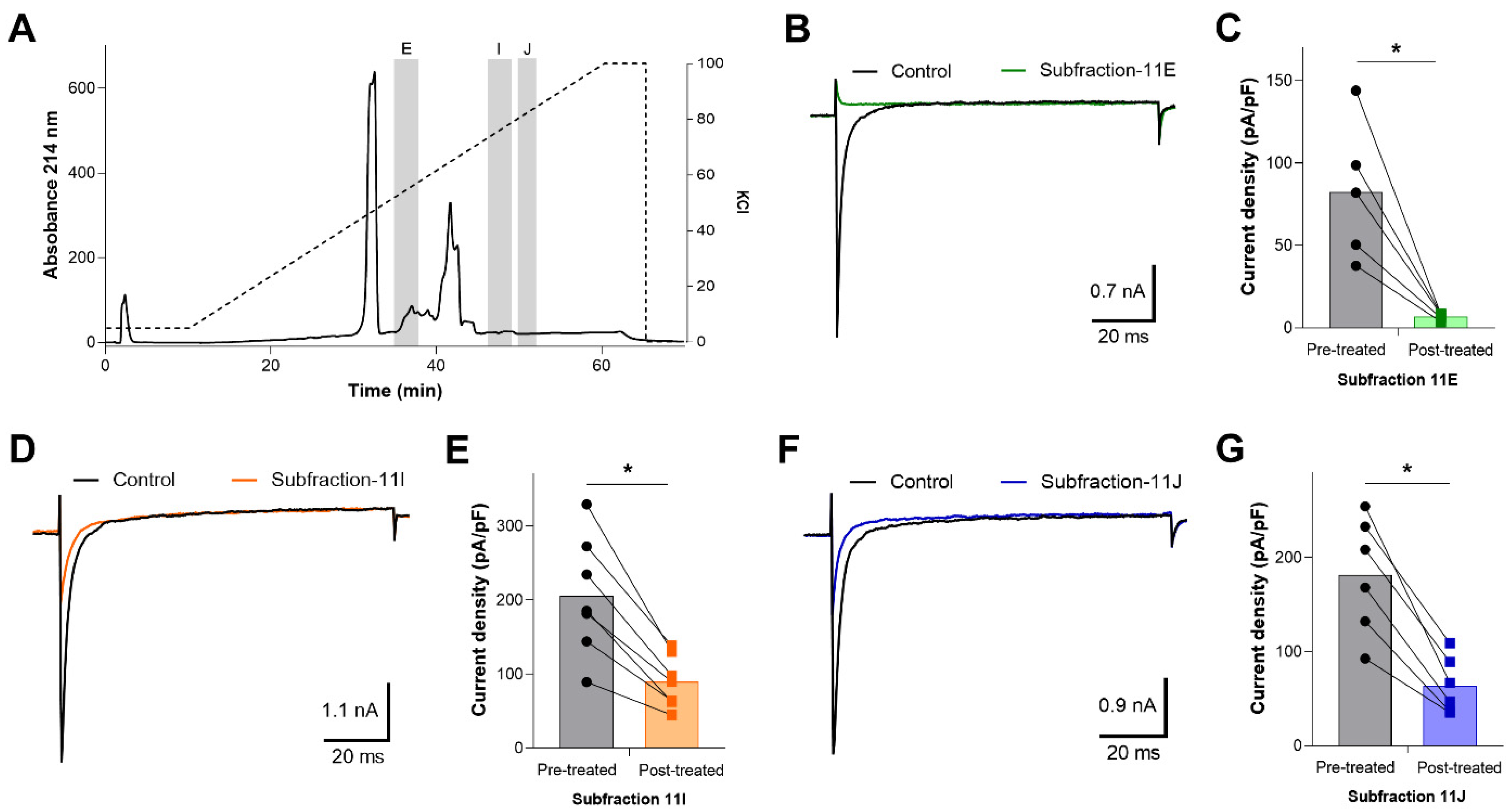
2.3. Ion Exchange Chromatography of Inhibitory Fractions 7, 11, 12, and 13 and Identification of Inhibitory Subfractions
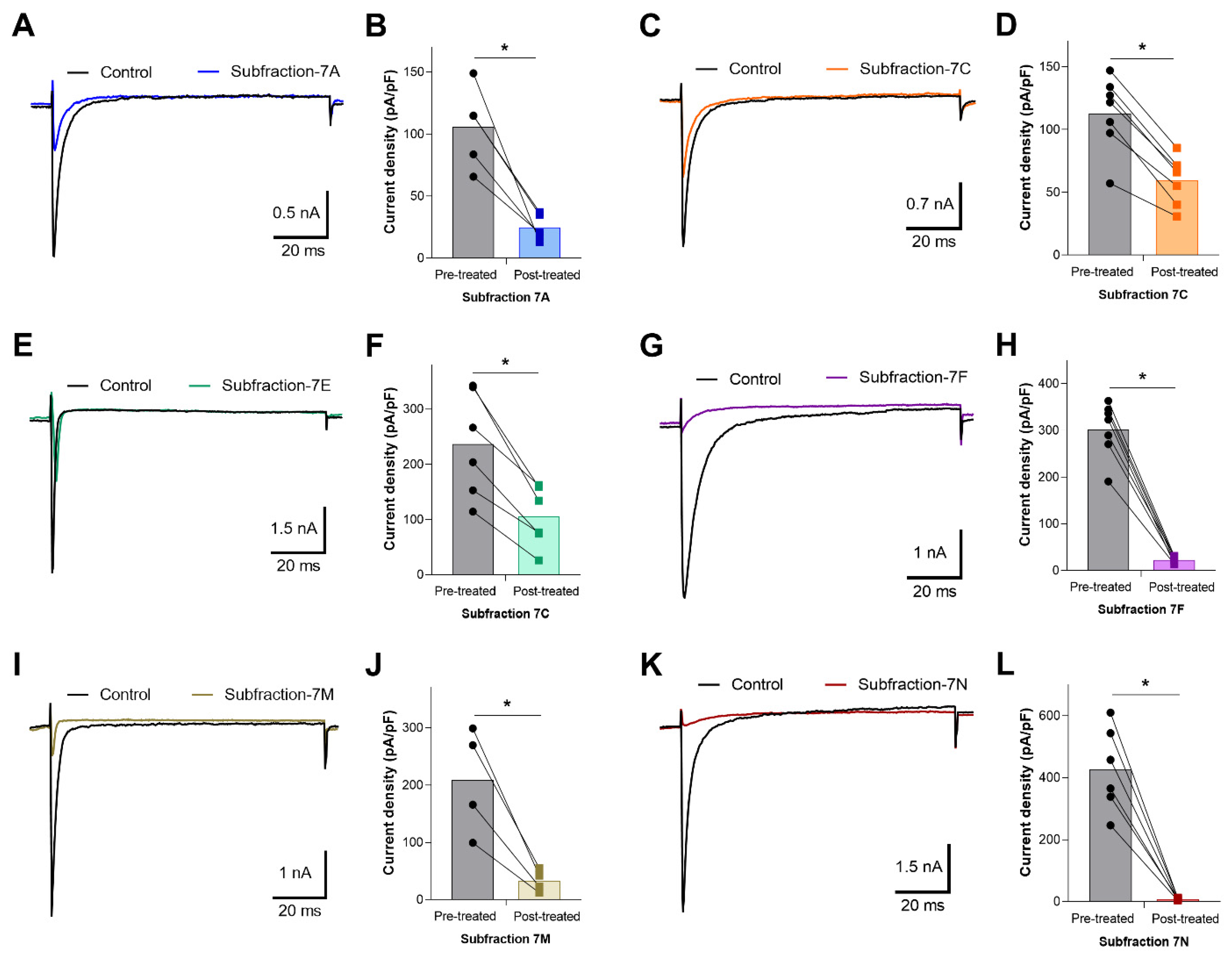
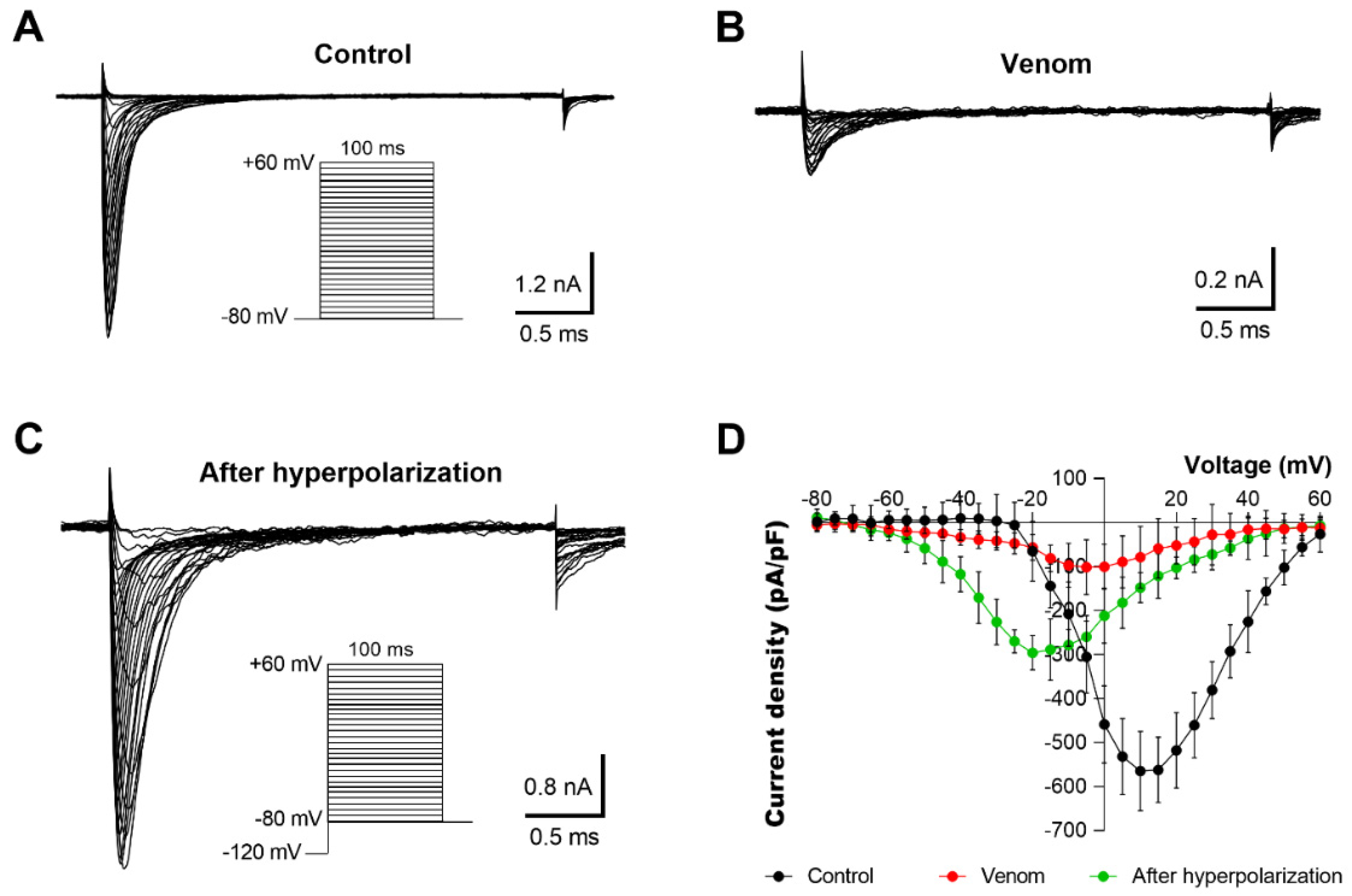
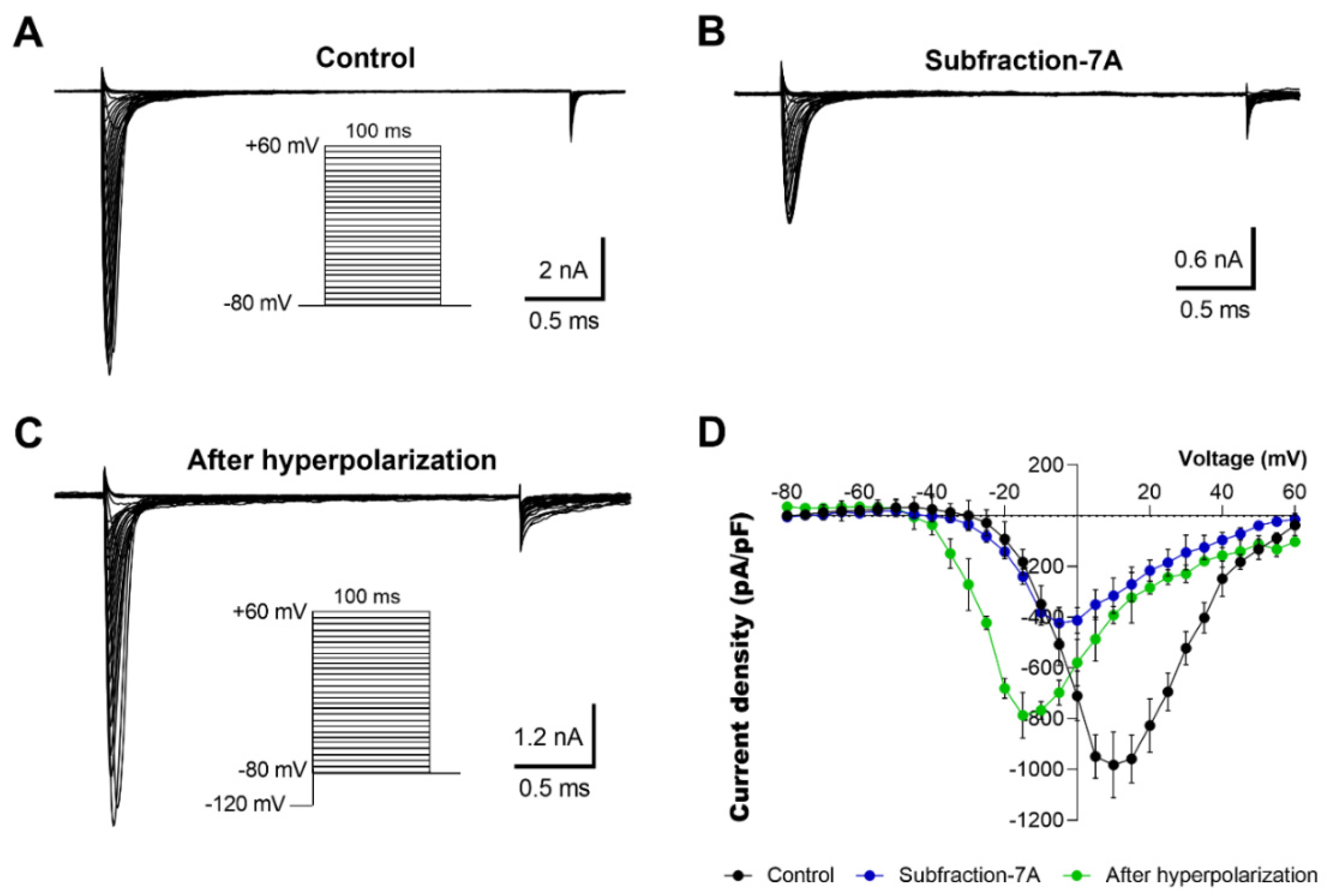
2.4. Electrophysiological Characterization of Voltage-Dependent Effects of AZ Bark Scorpion Venom and Inhibitory Subfractions
2.5. Proteomic Analyses of AZ Bark Scorpion Venom Subfractions That Inhibit OtNav1.8 Na+ Current
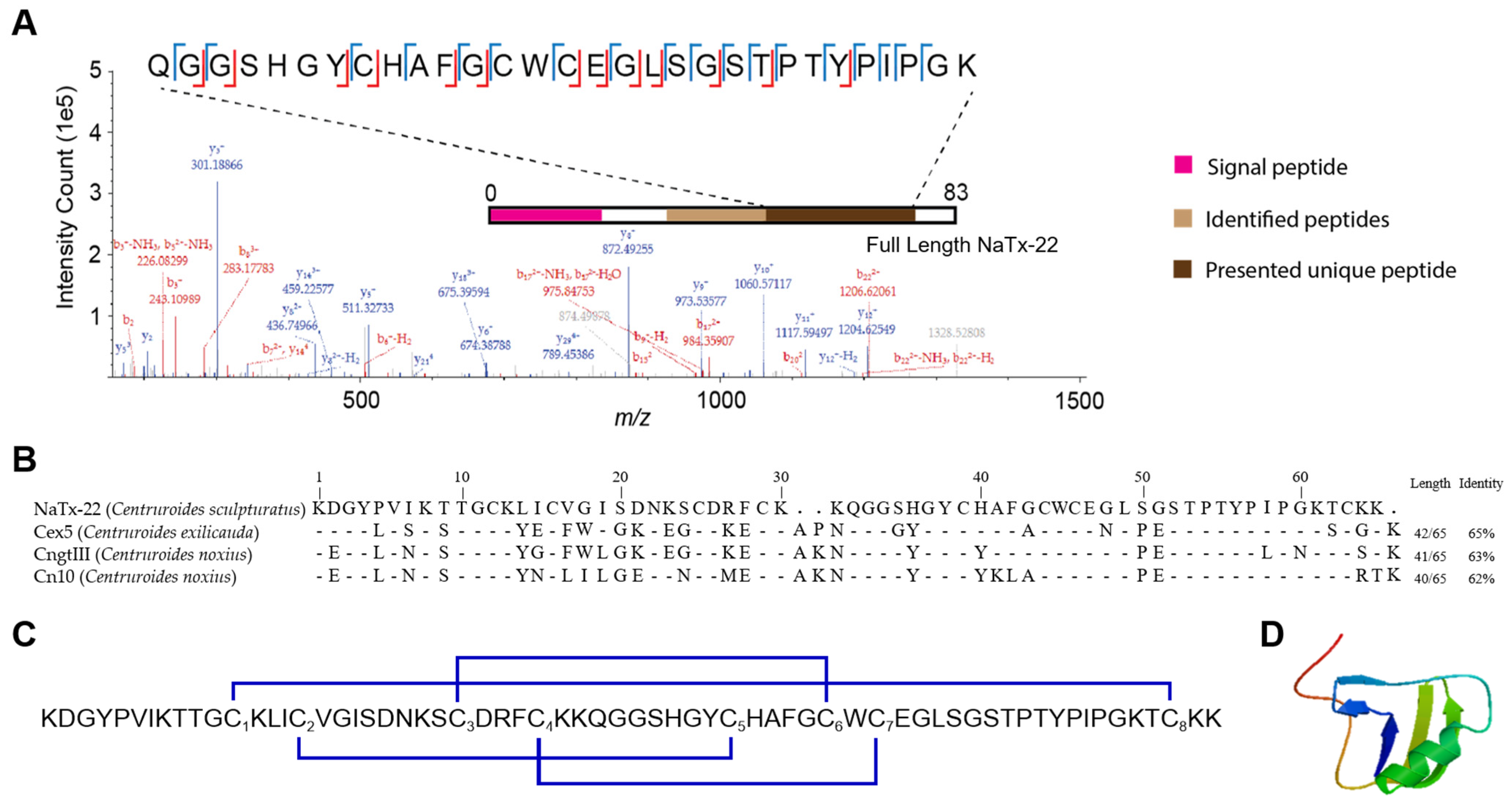
2.6. Structural Characterization of NaTx-22 from Subfraction 11E That Inhibits OtNav1.8
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Venom Extraction
5.2. Reversed-Phase LC Fractionation of the AZ Bark Scorpion Venom
5.3. Ion Exchange Liquid Chromatography to Isolate Individual Peptides
5.4. Culture and Transfection of ND7/23 Cells
5.5. Electrophysiology Recording
5.6. Sample Preparation for Proteomic Analyses
5.7. Proteomic Analyses (Liquid-Chromatography–Mass-Spectrometry)
5.8. Search of Peptides Analogs
5.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, D.J.; Lee, H.; Lo, L.; Shields, S.D.; Zylka, M.J.; Basbaum, A.I.; Anderson, D.J. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 9075–9080. [Google Scholar] [CrossRef] [Green Version]
- Le Pichon, C.E.; Chesler, A.T. The functional and anatomical dissection of somatosensory subpopulations using mouse genetics. Front. Neuroanat. 2014, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Peirs, C.; Seal, R.P. Neural Circuits for Pain: Recent Advances and Current Views. Science 2016, 354, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Renganathan, M.; Cummins, T.R.; Waxman, S.G. Contribution of Na+ 1.8 Sodium Channels to Action Potential Electrogenesis in DRG Neurons. J. Neurophysiol. 2001, 86, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Blair, N.T.; Bean, B.P. Roles of Tetrodotoxin (TTX)-Sensitive Na+ Current, TTX-Resistant Na+ Current, and Ca2+ Current in the Action Potentials of Nociceptive Sensory Neurons. J. Neurosci. 2002, 22, 10277–10290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, T.; Sheets, P.; Waxman, S. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 2007, 131, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.; Cummins, T.R. Unusual Voltage-Gated Sodium Currents as Targets for Pain. Curr. Top. Membr. 2016, 78, 599–638. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, J.; Waxman, S.G. Sodium channel Nav1.8: Emerging links to human disease. Neurology 2016, 86, 473–483. [Google Scholar] [CrossRef]
- Abrahamsen, B.; Zhao, J.; Asante, C.O.; Cendan, C.M.; Marsh, S.; Martinez-Barbera, J.P.; Nassar, M.A.; Dickenson, A.H.; Wood, J.N. The Cell and Molecular Basis of Mechanical, Cold, and Inflammatory Pain. Science 2008, 321, 702–705. [Google Scholar] [CrossRef] [Green Version]
- Cummins, T.R.; Waxman, S.G. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 1997, 17, 3503–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib-Hajj, S.D.; Fjell, J.; Cummins, T.R.; Zheng, Z.; Fried, K.; LaMotte, R.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar] [CrossRef]
- Gold, M.S.; Reichling, D.B.; Shuster, M.J.; Levine, J.D. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl. Acad. Sci. USA 1996, 93, 1108–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuse, K.; Chaplan, S.R.; McMahon, S.B.; Luo, Z.D.; Calcutt, N.A.; Scott, B.P.; Wood, J.N. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol. Cell. Neurosci. 1997, 10, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Cummins, T.R.; Ishikawa, K.; Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. SNS Na+ channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. Neuroreport 1998, 9, 967–972. [Google Scholar] [CrossRef]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Waxman, S.G. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Vasylyev, D.; Macala, L.J.; Gerrits, M.M.; Hoeijmakers, J.G.; Bekelaar, K.J.; Waxman, S.G. The G1662S Nav1.8 mutation in small fibre neuropathy: Impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 2014, 85, 499–505. [Google Scholar] [CrossRef]
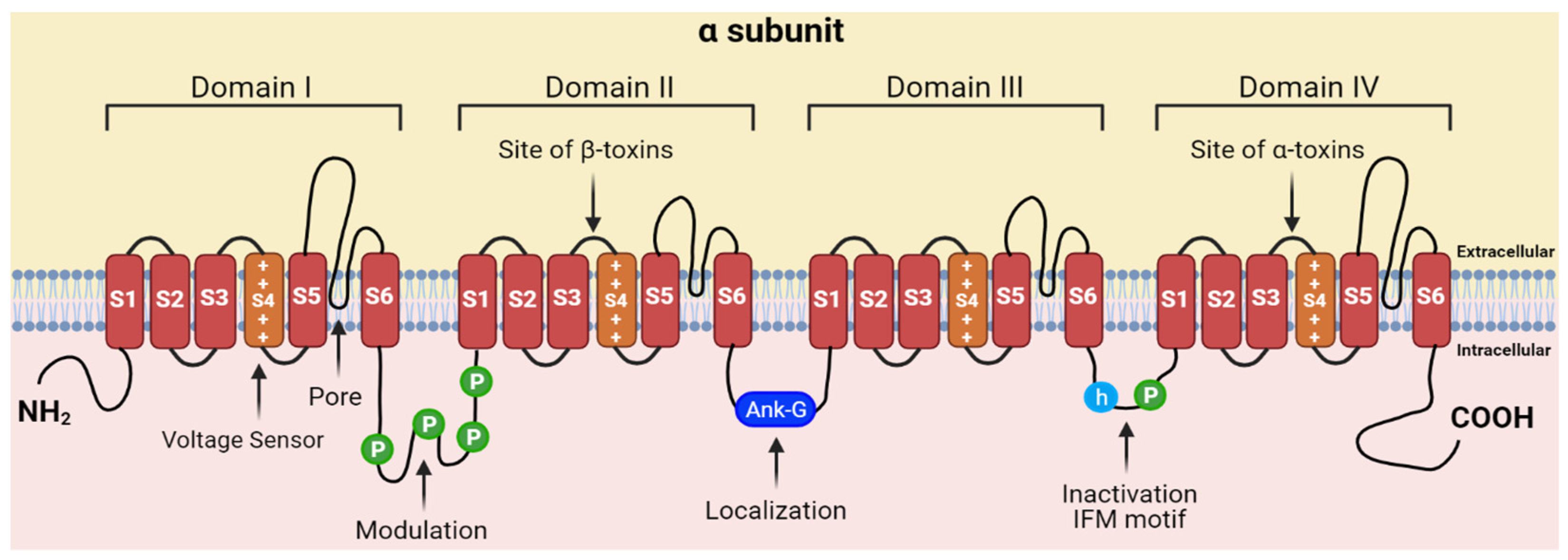
- Catterall, W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992, 72, S15–S48. [Google Scholar] [CrossRef]
- Catterall, W.A. From ion currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Vilin, Y.Y.; Ruben, P.C. Slow inactivation in voltage-gated sodium channels. Cell Biochem. Biophys. 2001, 35, 171–190. [Google Scholar] [CrossRef]
- Green, B.R.; Olivera, B.M. Venom Peptides From Cone Snails: Pharmacological Probes for Voltage-Gated Sodium Channels. Current Topics in Membranes, Robert, J.F., Sergei Yu, N., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 78, pp. 65–86. [Google Scholar] [CrossRef]
- Maertens, C.; Cuypers, E.; Amininasab, M.; Jalali, A.; Vatanour, H.; Tytgat, J. Potent modulation of the voltage-gated sodium channel Nav1.7 by OD1, a toxin from the scorpion Odonthobuthus doriae. Mol. Pharmacol. 2006, 70, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494J–499J. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, A.H.; Xiao, Y.; Scales, J.; Linse, K.D.; Rowe, M.P.; Cummins, T.R.; Zakon, H.H. Isolation and characterization of CvIV4: A pain inducing alpha-scorpion toxin. PLoS ONE 2011, 6, e23520. [Google Scholar] [CrossRef] [PubMed]
- Vandendriessche, T.; Olamendi-Portugal, T.; Zamudio, F.Z.; Possani, L.D.; Tytgat, J. Isolation and characterization of two novel scorpoin toxins: The α-toxin-like CeII8, specific for Nav1.7 channels and the classical anti-mammalian CeII9, specific for Nav1.4 channels. Toxicon 2010, 56, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bingham, J.-P.; Zhu, W.; Moczydlowski, E.; Liang, S.; Cummins, T.R. Tarantula Huwentoxin-IV Inhibits Neuronal Sodium Channels by Binding to Receptor Site 4 and Trapping the Domain II Voltage Sensor in the Closed Configuration. J. Biol. Chem. 2008, 283, 27300–27313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Blumenthal, K.; Jackson, J.O.; Liang, S.; Cummins, T.R. The Tarantula Toxins ProTx-II and Huwentoxin-IV Differentially Interact with Human Nav1.7 Voltage Sensors to Inhibit Channel Activation and Inactivation. Mol. Pharmacol. 2010, 78, 1124–1134. [Google Scholar] [CrossRef] [Green Version]
- Mohamed Abd El-Aziz, T.; Soares, A.G.; Stockand, J.D. Snake Venoms in Drug Discovery: Valuable Therapeutic Tools for Life Saving. Toxins 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Bordon, K.d.C.F.; Cologna, C.T.; Fornari-Baldo, E.C.; Pinheiro-Júnior, E.L.; Cerni, F.A.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; Cardoso, I.A.; et al. From Animal Poisons and Venoms to Medicines: Achievements, Challenges and Perspectives in Drug Discovery. Front. Pharmacol. 2020, 11, 1132. [Google Scholar] [CrossRef]
- Pennington, M.W.; Czerwinski, A.; Norton, R.S. Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef]
- Lazarovici, P. Snake- and Spider-Venom-Derived Toxins as Lead Compounds for Drug Development. Methods Mol. Biol. 2020, 2068, 3–26. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [Green Version]
- McDermott, A. News Feature: Venom back in vogue as a wellspring for drug candidates. Proc. Natl. Acad. Sci. USA 2020, 117, 10100. [Google Scholar] [CrossRef] [Green Version]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep. 2017, 7, 39662. [Google Scholar] [CrossRef]
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.B.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective Nav1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; Deuis, J.R.; Dekan, Z.; Jin, A.-H.; Alewood, P.F.; King, G.F.; Herzig, V.; Vetter, I. Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of Nav1.7. Biomedicines 2020, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, U.M.; Zuurbier, K.R.; Whoolery, C.W.; Plautz, E.J.; Chambliss, K.L.; Kong, X.; Zhang, S.; Kim, S.H.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; et al. Selective Nonnuclear Estrogen Receptor Activation Decreases Stroke Severity and Promotes Functional Recovery in Female Mice. Endocrinology 2018, 159, 3848–3859. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Structure-Function Map of the Receptor Site for beta-Scorpion Toxins in Domain II of Voltage-gated Sodium Channels. J. Biol. Chem. 2011, 286, 33641–33651. [Google Scholar] [CrossRef] [Green Version]
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef] [Green Version]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 2007, 49, 142–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clairfeuille, T.; Cloake, A.; Infield, D.T.; Llongueras, J.P.; Arthur, C.P.; Li, Z.R.; Jian, Y.W.; Martin-Eauclaire, M.F.; Bougis, P.E.; Ciferri, C.; et al. Structural basis of alpha-scorpion toxin action on Na-v channels. Science 2019, 363, eaav8573. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, J.; Bosmans, F. Using voltage-sensor toxins and their molecular targets to investigate NaV1.8 gating. J. Physiol. 2018, 596, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, A.H.; Xiao, Y.; Rowe, M.P.; Cummins, T.R.; Zakon, H.H. Voltage-gated sodium channel in grasshopper mice defends against bark scorpion toxin. Science 2013, 342, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Couraud, F.; Jover, E. Mechanism of Action of Scorpion Toxins. In Handbook of Natural Toxins; Tu, A.T., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1984; Volume 2, pp. 659–678. [Google Scholar]
- Corona, M.; Valdez-Cruz, N.A.; Merino, E.; Zurita, M.; Possani, L.D. Genes and peptides from the scorpion Centruroides sculpturatus Ewing, that recognize Na+-channels. Toxicon 2001, 39, 1893–1898. [Google Scholar] [CrossRef]
- Possani, L.D.; Becerril, B.; Delepierre, M.; Tytgat, J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 1999, 264, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possani, L.D.; Merino, E.; Corona, M.; Bolivar, F.; Becerril, B. Peptides and genes coding for scorpion toxins that affect ion-channels. Biochimie 2000, 82, 861–868. [Google Scholar] [CrossRef]
- Rodriguez de la Vega, R.C.; Possani, L.D. Current views on scorpion toxins specific for K+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef]
- Rodriguez de la Vega, R.; Possani, L.D. Overview of scorpion toxins specific for Na+ channels and related peptides: Biodiversity, structure-function relationships and evolution. Toxicon 2005, 46, 831–844. [Google Scholar] [CrossRef]
- Simard, J.M.; Meves, H.; Watt, D.D. Neurotoxins in venom from the North American scorpion, Centruroides sculpturatus Ewing. In Natural Toxins: Toxicolgy, Chemistry and Safety; Keeler, R.F., Mandava, N.B., Tu, A.T., Eds.; Alaken, Inc.: Fort Collins, CO, USA, 1992; Volume 1, pp. 236–263. [Google Scholar]
- Rowe, A.H.; Rowe, M.P. Risk assessment by grasshopper mice (Onychomys spp.) feeding on neurotoxic prey (Centruroides spp.). Anim. Behav. 2006, 71, 725–734. [Google Scholar] [CrossRef]
- Rowe, A.H.; Rowe, M.P. Physiological resistance of grasshopper mice (Onychomys spp.) to Arizona bark scorpion (Centruroides exilicauda) venom. Toxicon 2008, 52, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, C.; Xu, X.; Zhang, Y.; Gong, X.; Yang, Z.; Zhang, H.; Tang, D.; Liang, S.; Liu, Z. Naja atra venom peptide reduces pain by selectively blocking the voltage-gated sodium channel Nav1.8. J. Biol. Chem. 2019, 294, 7324–7334. [Google Scholar] [CrossRef]
- Valdez-Cruz, N.A.; Dávila, S.; Licea, A.; Corona, M.; Zamudio, F.Z.; García-Valdes, J.; Boyer, L.; Possani, L.D. Biochemical, genetic and physiological characterization of venom components from two species of scorpions: Centruroides exilicauda Wood and Centruroides sculpturatus Ewing. Biochimie 2004, 86, 387–396. [Google Scholar] [CrossRef]
- Yaksh, T.L.; Woller, S.A.; Ramachandran, R.; Sorkin, L.S. The search for novel analgesics: Targets and mechanisms. F1000Prime Rep. 2015, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Tikhonov, D.B. Computational Structural Pharmacology and Toxicology of Voltage-Gated Sodium Channels. Curr. Top. Membr. 2016, 78, 117–144. [Google Scholar] [CrossRef]
- French, R.J.; Yoshikami, D.; Sheets, M.F.; Olivera, B.M. The Tetrodotoxin Receptor of Voltage-Gated Sodium Channels—Perspectives from Interactions with μ-Conotoxins. Mar. Drugs 2010, 8, 2153–2161. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.C.; Qu, Y.; Tanada, T.N.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 1996, 271, 15950–15962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, M.J.; Ellsworth, S.A.; Rokyta Darin, R. Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 2018, 142, 14–29. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin ID | Full Sequence |
|---|---|
| NaTx-22 | KDGYPVIKTTGCKLICVGISDNKSCDRFCKKQGGSHGYCHAFGCWCEGLSGSTPTYPIPGKTCKK |
| NaTx-4 | KEGYLVNKETGCKLACVTTGENKNCKLDCKNQGGSKGYCLLFRCFCEGLSESTPTFPIPGKTCSGK |
| NaTx-36 | KDGYPMRSDGCTIACLFDNDFCNRKCVEQKGKSGYCYFWKQSCYCEGLPDDKVYDSATSKCRA |
| NaTx-13 | SKKEIPGGYPVNQFKCTYECAHADTDHIRCKNLCKKLGGSWGYCYWNTCYCEYLPDSVPQKNSIEVFSCGATIVGVPDTEQQ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El-Aziz, T.M.; Xiao, Y.; Kline, J.; Gridley, H.; Heaston, A.; Linse, K.D.; Ward, M.J.; Rokyta, D.R.; Stockand, J.D.; Cummins, T.R.; et al. Identification and Characterization of Novel Proteins from Arizona Bark Scorpion Venom That Inhibit Nav1.8, a Voltage-Gated Sodium Channel Regulator of Pain Signaling. Toxins 2021, 13, 501. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070501
Abd El-Aziz TM, Xiao Y, Kline J, Gridley H, Heaston A, Linse KD, Ward MJ, Rokyta DR, Stockand JD, Cummins TR, et al. Identification and Characterization of Novel Proteins from Arizona Bark Scorpion Venom That Inhibit Nav1.8, a Voltage-Gated Sodium Channel Regulator of Pain Signaling. Toxins. 2021; 13(7):501. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070501
Chicago/Turabian StyleAbd El-Aziz, Tarek Mohamed, Yucheng Xiao, Jake Kline, Harold Gridley, Alyse Heaston, Klaus D. Linse, Micaiah J. Ward, Darin R. Rokyta, James D. Stockand, Theodore R. Cummins, and et al. 2021. "Identification and Characterization of Novel Proteins from Arizona Bark Scorpion Venom That Inhibit Nav1.8, a Voltage-Gated Sodium Channel Regulator of Pain Signaling" Toxins 13, no. 7: 501. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070501