The Protein-Independent Role of Phosphate in the Progression of Chronic Kidney Disease

 ,
,
Abstract
:1. Introduction
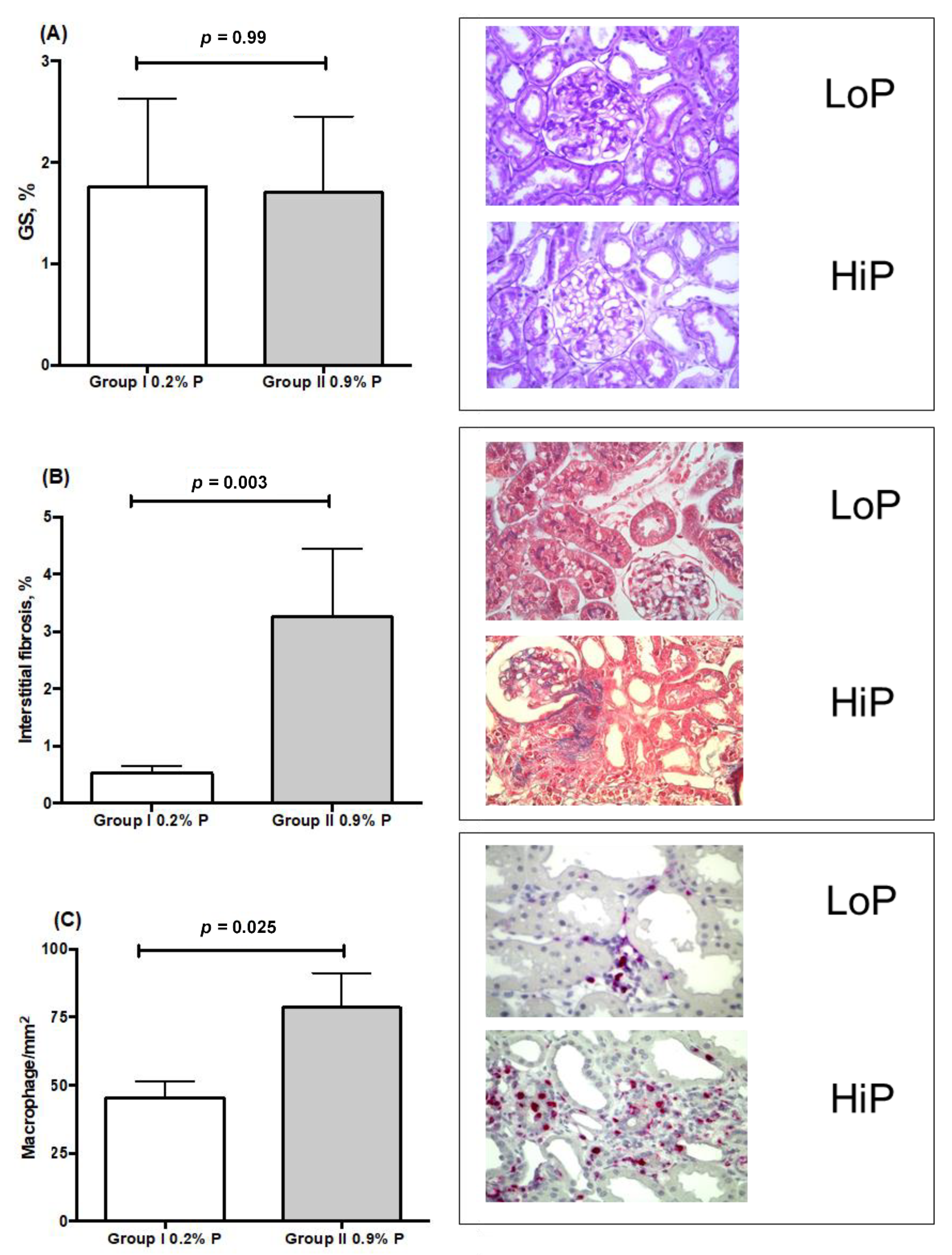
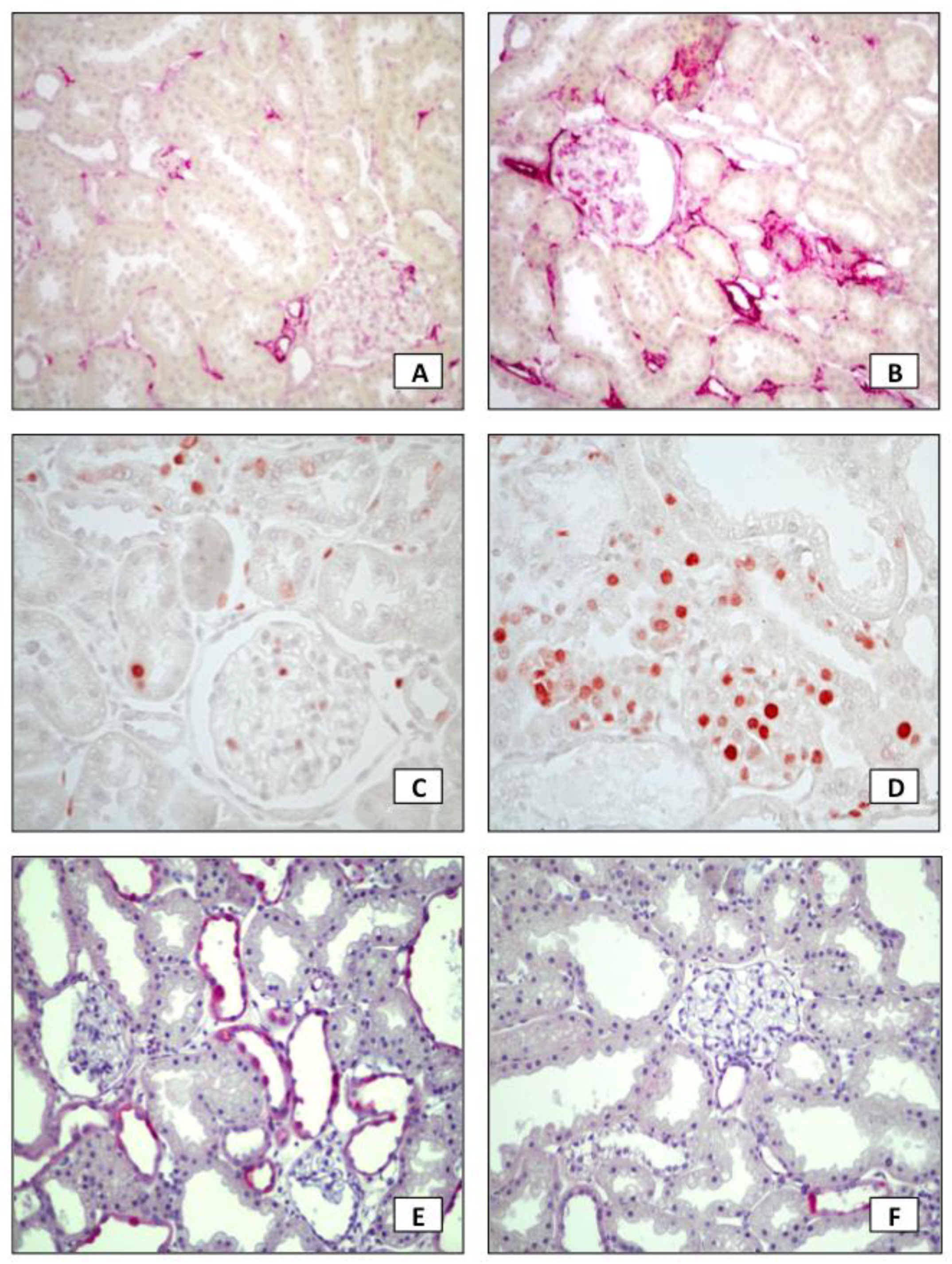
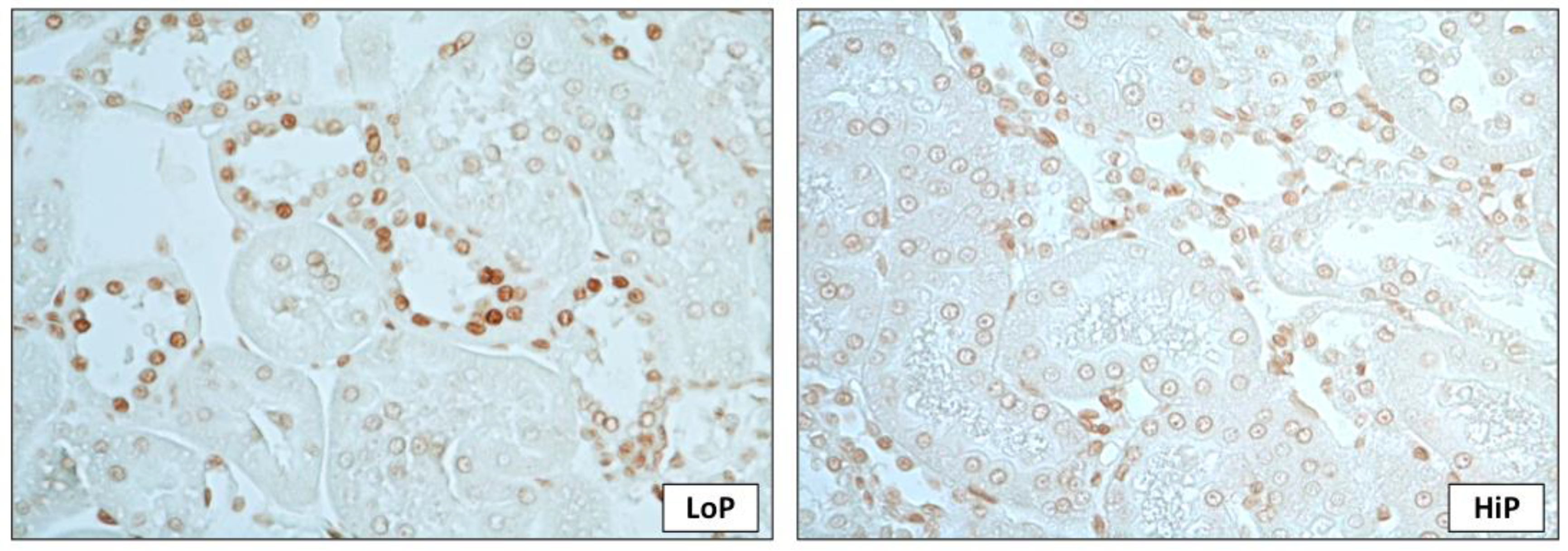
2. Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Analytic Techniques
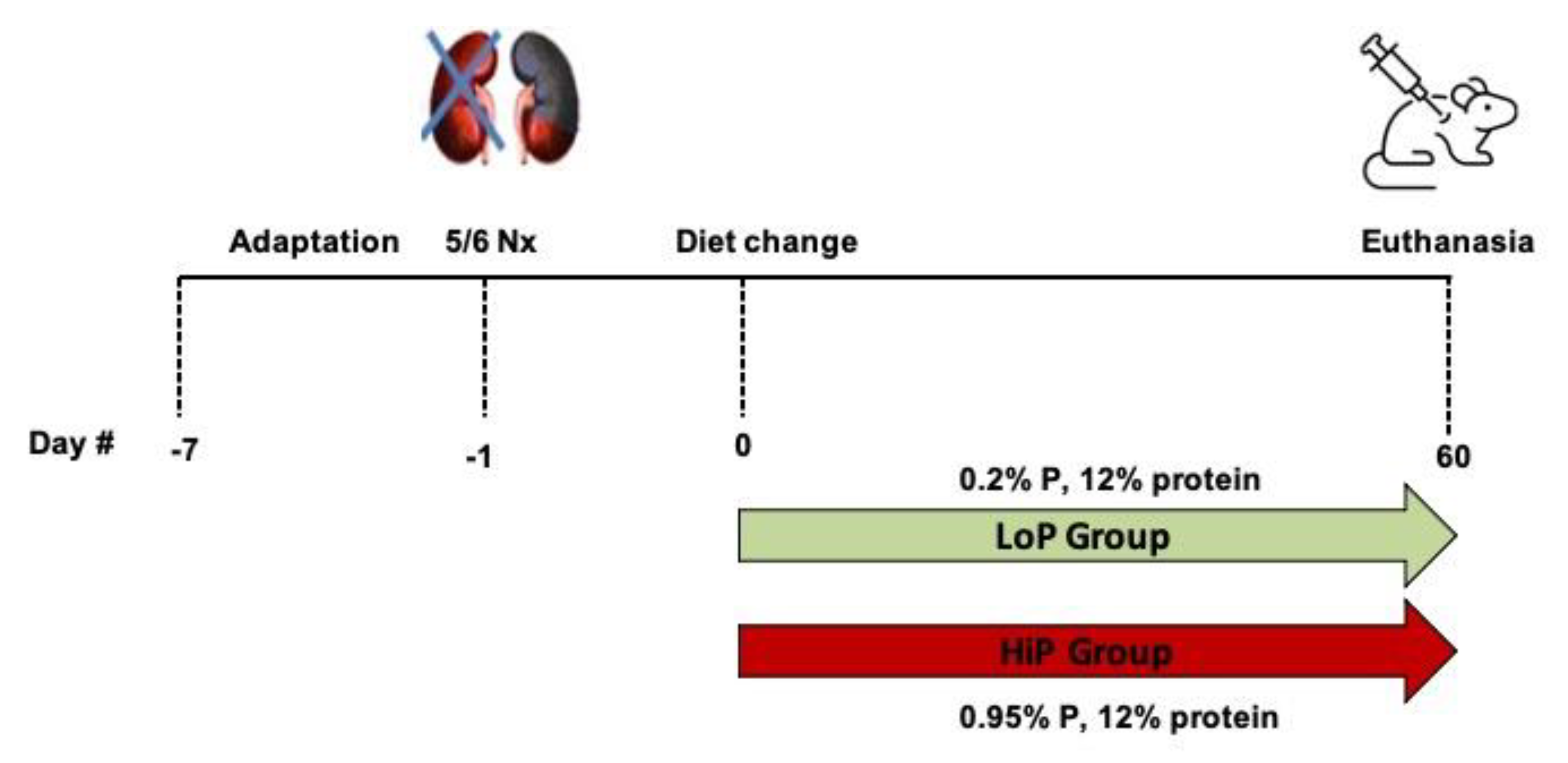
5.2. Experimental Groups
5.3. Histological Techniques
5.4. Immunohistochemistry
5.5. Apoptosis
5.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global prevalence of chronic kidney disease—A systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Wen, C.P.; Chang, C.H.; Tsai, M.K.; Lee, J.H.; Lu, P.J.; Tsai, S.P.; Wen, C.; Chen, C.H.; Kao, C.W.; Tsao, C.K.; et al. Diabetes with early kidney involvement may shorten life expectancy by 16 years. Kidney Int. 2017, 92, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Kawamura, T.; Bills, T.; Yoshioka, T.; Ichikawa, I.; Fogo, A. Blood pressure-independent effect of angiotensin inhibition on vascular lesions of chronic renal failure. Kidney Int. 1992, 42, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, L.F.; Orchard, T.J.; Kasiske, B.L. Effect of lipid reduction on the progression of renal disease: A meta-analysis. Kidney Int. 2001, 59, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishidoya, S.; Morrissey, J.; McCracken, R.; Reyes, A.; Klahr, S. Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int. 1995, 47, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldigier, J.C.; Kanjanbuch, T.; Ma, L.J.; Brown, N.J.; Fogo, A.B. Regression of existing glomerulosclerosis by inhibition of aldosterone. J. Am. Soc. Nephrol. 2005, 16, 3306–3314. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Kren, S.M.; Hostetter, T.H. Dietary protein restriction in established renal injury in the rat. Selective role of glomerular capillary pressure in progressive glomerular dysfunction. J. Clin. Investig. 1986, 78, 1199–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.M.; Meyer, T.W.; Hostetter, T.H. Dietary protein intake and the progressive nature of kidney disease: The role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N. Engl. J. Med. 1982, 307, 652–659. [Google Scholar]
- Bellizzi, V. Low-protein diet or nutritional therapy in chronic kidney disease? Blood Purif. 2013, 36, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kusano, K.; Segawa, H.; Ohnishi, R.; Fukushima, N.; Miyamoto, K. Role of low protein and low phosphorus diet in the progression of chronic kidney disease in uremic rats. J. Nutr. Sci. Vitam. 2008, 54, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Neves, K.R.; Graciolli, F.G.; dos Reis, L.M.; Pasqualucci, C.A.; Moyses, R.M.; Jorgetti, V. Adverse effects of hyperphosphatemia on myocardial hypertrophy, renal function, and bone in rats with renal failure. Kidney Int. 2004, 66, 2237–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, J.L.; Lee, D.H.; Liapis, H.; Ritter, C.; Zhang, S.; Suarez, E.; Ferder, L.; Slatopolsky, E. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int. 2013, 84, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Cupisti, A.; Bolasco, P.; D’Alessandro, C.; Giannese, D.; Sabatino, A.; Fiaccadori, E. Protection of residual renal function and nutritional treatment: First step strategy for reduction of uremic toxins in end-stage kidney disease patients. Toxins 2021, 13, 289. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Su, X.; Xu, B.; Qiao, X.; Wang, L. Effect of diet protein restriction on progression of chronic kidney disease: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0206134. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of diet in renal disease study group. N. Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Shi, M.; Cho, H.J.; Zhang, J.; Pavlenco, A.; Liu, S.; Sidhu, S.; Huang, L.J.; Moe, O.W. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013, 84, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. alphaKlotho mitigates progression of AKI to CKD through activation of autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar] [CrossRef]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstadt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal. Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef] [PubMed]
- Meleti, Z.; Shapiro, I.M.; Adams, C.S. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone 2000, 27, 359–366. [Google Scholar] [CrossRef]
- Park, J.W.; Yook, J.M.; Ryu, H.M.; Choi, S.Y.; Morishita, M.; Do, J.Y.; Park, S.H.; Kim, C.D.; Choi, J.Y.; Chung, H.Y.; et al. Phosphate-induced apoptosis in human peritoneal mesothelial cells in vitro. Am. J. Nephrol. 2011, 34, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Priante, G.; Gianesello, L.; Ceol, M.; Del Prete, D.; Anglani, F. Cell death in the kidney. Int. J. Mol. Sci. 2019, 20, 3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da, J.; Xie, X.; Wolf, M.; Disthabanchong, S.; Wang, J.; Zha, Y.; Lv, J.; Zhang, L.; Wang, H. Serum phosphorus and progression of CKD and mortality: A meta-analysis of cohort studies. Am. J. Kidney Dis. 2015, 66, 258–265. [Google Scholar] [CrossRef]
- Lee, Y.J.; Okuda, Y.; Sy, J.; Obi, Y.; Kang, D.H.; Nguyen, S.; Hsiung, J.T.; Park, C.; Rhee, C.M.; Kovesdy, C.P.; et al. Association of mineral bone disorder with decline in residual kidney function in incident hemodialysis patients. J. Bone Miner. Res. 2019, 35, 317–325. [Google Scholar] [CrossRef]
- Ibels, L.S.; Alfrey, A.C.; Haut, L.; Huffer, W.E. Preservation of function in experimental renal disease by dietary restriction of phosphate. N. Engl. J. Med. 1978, 298, 122–126. [Google Scholar] [CrossRef]
- Selamet, U.; Tighiouart, H.; Sarnak, M.J.; Beck, G.; Levey, A.S.; Block, G.; Ix, J.H. Relationship of dietary phosphate intake with risk of end-stage renal disease and mortality in chronic kidney disease stages 3-5: The modification of diet in renal disease study. Kidney Int. 2016, 89, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Dhillon-Jhattu, S.; Sprague, S.M. Should phosphate management be limited to the KDIGO/ KDOQI guidelines? Semin. Dial. 2018, 31, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Russo, L.; Pota, A.; Mirenghi, F.; Russo, D. Acute effects of very-low-protein diet on FGF23 levels: A randomized study. Clin. J. Am. Soc. Nephrol. 2012, 7, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Nakai, K.; Kono, K.; Yonekura, Y.; Ito, J.; Fujii, H.; Nishi, S. Dietary phosphorus restriction by a standard low-protein diet decreased serum fibroblast growth factor 23 levels in patients with early and advanced stage chronic kidney disease. Clin. Exp. Nephrol. 2014, 18, 925–931. [Google Scholar] [CrossRef]
- Di Iorio, B.R.; Bellizzi, V.; Bellasi, A.; Torraca, S.; D’Arrigo, G.; Tripepi, G.; Zoccali, C. Phosphate attenuates the anti-proteinuric effect of very low-protein diet in CKD patients. Nephrol. Dial. Transpl. 2013, 28, 632–640. [Google Scholar] [CrossRef]
- Ix, J.H.; Anderson, C.A.; Smits, G.; Persky, M.S.; Block, G.A. Effect of dietary phosphate intake on the circadian rhythm of serum phosphate concentrations in chronic kidney disease: A crossover study. Am. J. Clin. Nutr. 2014, 100, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.J.; Hu, J.; Shiizaki, K.; Kuro-o, M.; Malter, J.S. Phosphate-induced renal fibrosis requires the prolyl isomerase Pin1. PLoS ONE 2016, 11, e0150093. [Google Scholar] [CrossRef] [Green Version]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.; Nagasu, H.; Morita, Y.; Yamaguchi, T.P.; Kanwar, Y.S.; Kashihara, N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am. J. Physiol. Renal. Physiol. 2012, 303, F1641–F1651. [Google Scholar] [CrossRef] [Green Version]
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar] [CrossRef] [Green Version]
- Koh, N.; Fujimori, T.; Nishiguchi, S.; Tamori, A.; Shiomi, S.; Nakatani, T.; Sugimura, K.; Kishimoto, T.; Kinoshita, S.; Kuroki, T.; et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Morishita, K.; Shirai, A.; Kubota, M.; Katakura, Y.; Nabeshima, Y.; Takeshige, K.; Kamiya, T. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J. Nutr. 2001, 131, 3182–3188. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, S.; Wildey, G.M.; Howe, P.H. Transforming growth factor beta (TGFbeta)-induced apoptosis: The rise & fall of Bim. Cell Cycle 2009, 8, 11–17. [Google Scholar]
- August, P.; Suthanthiran, M. Transforming growth factor beta and progression of renal disease. Kidney Int. 2003, 64, S99–S104. [Google Scholar] [CrossRef] [Green Version]
- Custodio, M.R.; Koike, M.K.; Neves, K.R.; dos Reis, L.M.; Graciolli, F.G.; Neves, C.L.; Batista, D.G.; Magalhaes, A.O.; Hawlitschek, P.; Oliveira, I.B.; et al. Parathyroid hormone and phosphorus overload in uremia: Impact on cardiovascular system. Nephrol. Dial. Transpl. 2012, 27, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 2010, 12, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Chung, A.C.; Lan, H.Y. Role TGF-Beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.C.; Zhang, H.; Kong, Y.Z.; Tan, J.J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, E.; Carlini, R.G.; Clesca, P.; Arminio, A.; Suniaga, O.; De Elguezabal, K.; Weisinger, J.R.; Hruska, K.A.; Bellorin-Font, E. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodeling. Kidney Int. 2003, 63, 1915–1923. [Google Scholar] [CrossRef] [Green Version]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 1999, 104, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, F.; Afrin, S.; Forbes-Hernandez, T.Y.; Gasparrini, M.; Cianciosi, D.; Reboredo-Rodriguez, P.; Varela-Lopez, A.; Quiles, J.L.; Battino, M. Autophagy in human health and disease: Novel therapeutic opportunities. Antioxid. Redox Signal. 2019, 30, 577–634. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef]
- Fujimura, R.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba-Hamano, T.; Minami, S.; Sakai, S.; Matsuda, J.; Hesaka, A.; Yonishi, H.; et al. Autophagy protects kidney from phosphate-induced mitochondrial injury. Biochem. Biophys. Res. Commun. 2020, 524, 636–642. [Google Scholar] [CrossRef]
- Arias, S.C.; Valente, C.P.; Machado, F.G.; Fanelli, C.; Origassa, C.S.; de Brito, T.; Camara, N.O.; Malheiros, D.M.; Zatz, R.; Fujihara, C.K. Regression of albuminuria and hypertension and arrest of severe renal injury by a losartan-hydrochlorothiazide association in a model of very advanced nephropathy. PLoS ONE 2013, 8, e56215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teles, F.; Machado, F.G.; Ventura, B.H.; Malheiros, D.M.; Fujihara, C.K.; Silva, L.F.; Zatz, R. Regression of glomerular injury by losartan in experimental diabetic nephropathy. Kidney Int. 2009, 75, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, F.L.; Mortensen, P.B. Interstitial fibrosis of the renal cortex in minimal change lesion and its correlation with renal function. A quantitative study. Virchows Arch. A Pathol. Anat. Histol. 1979, 383, 265–270. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LoP (0.2% P) n = 20 | HiP (0.95% P) n = 19 | p Value | |
|---|---|---|---|
| Initial weight, g | 240 ± 2 | 241 ± 3 | 0.746 |
| Final weight, g | 315 ± 4 | 287 ± 8 | 0.002 |
| Caudal pressure, mmHg | 194 ± 4 | 185 ± 6 | 0.231 |
| Urinary albumine, mg/24 h | 17 ± 4 | 19 ± 4 | 0.849 |
| Creatinine clearance, mL/min | 0.65 ± 0.04 | 0.49 ± 0.05 | 0.019 |
| FE of Phosphate, % | 0.7 ± 0.3 | 84 ± 7.8 | <0.0001 |
| Hematocrit, % | 44 ± 1 | 40 ± 1 | 0.0002 |
| Serum creatinine, mg/dL | 0.9 ± 0.03 | 1.1 ± 0.06 | 0.026 |
| Serum urea, mg/dL | 89 ± 3 | 83 ± 4 | 0.302 |
| Serum phosphate, mg/dL | 5.8 ± 0.4 | 9.5 ± 0.7 | <0.0001 |
| Ionized calcium, mmol/L | 1.24 ± 0.06 | 1.08 ± 0.05 | 0.072 |
| FGF-23, pg/mL | 306 ± 37 | 4435 ± 610 | <0.0001 |
| PTH, pg/mL | 560 ± 178 | 3644 ± 70 | <0.0001 |
| LoP (0.2% P) n = 13 | HiP (0.95% P) n = 12 | p | |
|---|---|---|---|
| α-SMA, % | 0.48 (0.2; 0.8) | 2.92 (2; 3.8) | <0.01 |
| ED-1, cell/mm2 | 45 (27; 67) | 74 (42; 93) | <0.01 |
| p-SMAD, cell/mm2 | 75.8 (18; 128) | 86.7 (19; 149) | 0.71 |
| PCNA, cell/mm2 | 67.7 ± 22 | 167.3 ± 50 | <0.01 |
| Apoptosis, cell/mm2 | 267.3 ± 179 | 173.3 ± 59 | 0.09 |
| Beclin, cell/mm2 | 10.4 ± 0.7 | 8.3 ± 0.5 | 0.024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duayer, I.F.; Duque, E.J.; Fujihara, C.K.; de Oliveira, I.B.; dos Reis, L.M.; Machado, F.G.; Graciolli, F.G.; Jorgetti, V.; Zatz, R.; Elias, R.M.; et al. The Protein-Independent Role of Phosphate in the Progression of Chronic Kidney Disease. Toxins 2021, 13, 503. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070503
Duayer IF, Duque EJ, Fujihara CK, de Oliveira IB, dos Reis LM, Machado FG, Graciolli FG, Jorgetti V, Zatz R, Elias RM, et al. The Protein-Independent Role of Phosphate in the Progression of Chronic Kidney Disease. Toxins. 2021; 13(7):503. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070503
Chicago/Turabian StyleDuayer, Irene Faria, Eduardo Jorge Duque, Clarice Kazue Fujihara, Ivone Braga de Oliveira, Luciene Machado dos Reis, Flavia Gomes Machado, Fabiana Giorgetti Graciolli, Vanda Jorgetti, Roberto Zatz, Rosilene Motta Elias, and et al. 2021. "The Protein-Independent Role of Phosphate in the Progression of Chronic Kidney Disease" Toxins 13, no. 7: 503. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070503