Venom of the Annulated Sea Snake Hydrophis cyanocinctus: A Biochemically Simple but Genetically Complex Weapon

 , and
, and
Abstract
:1. Introduction
2. Results
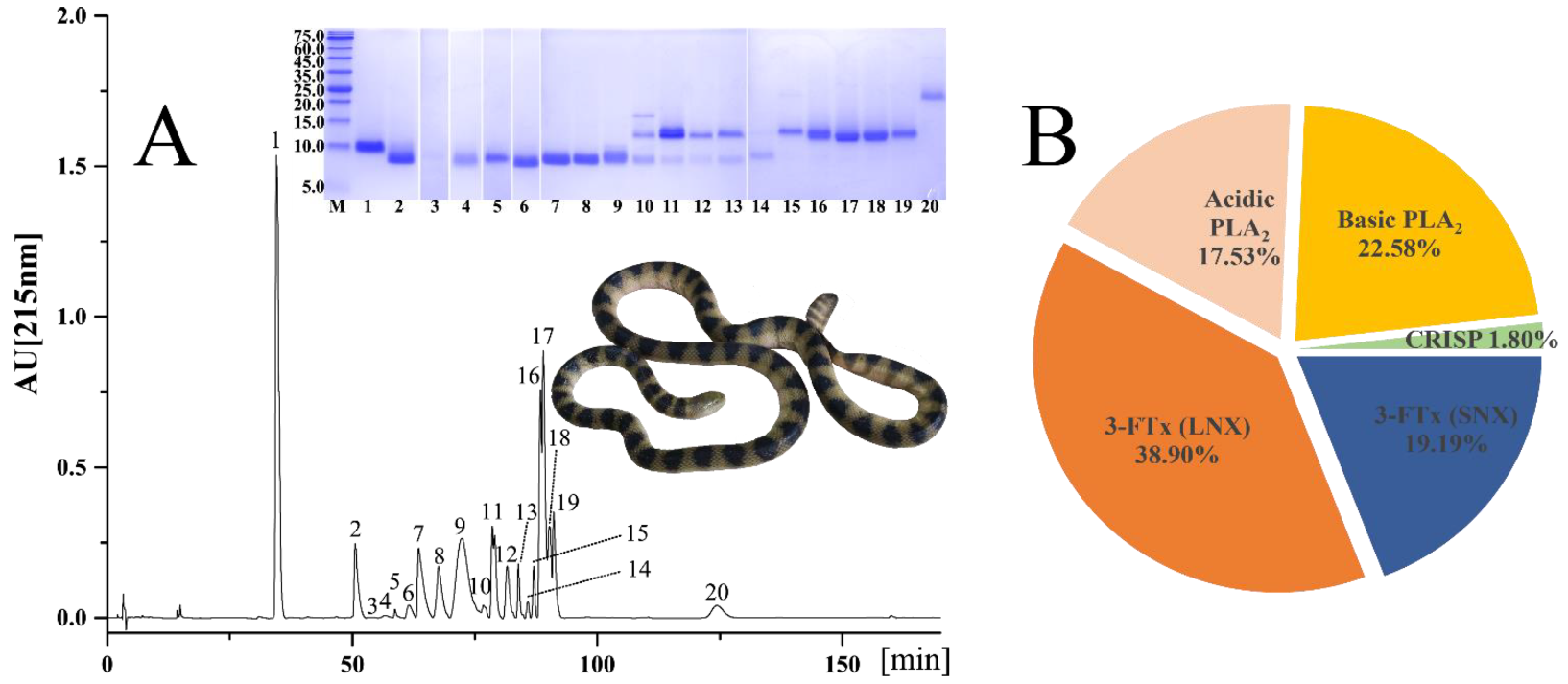
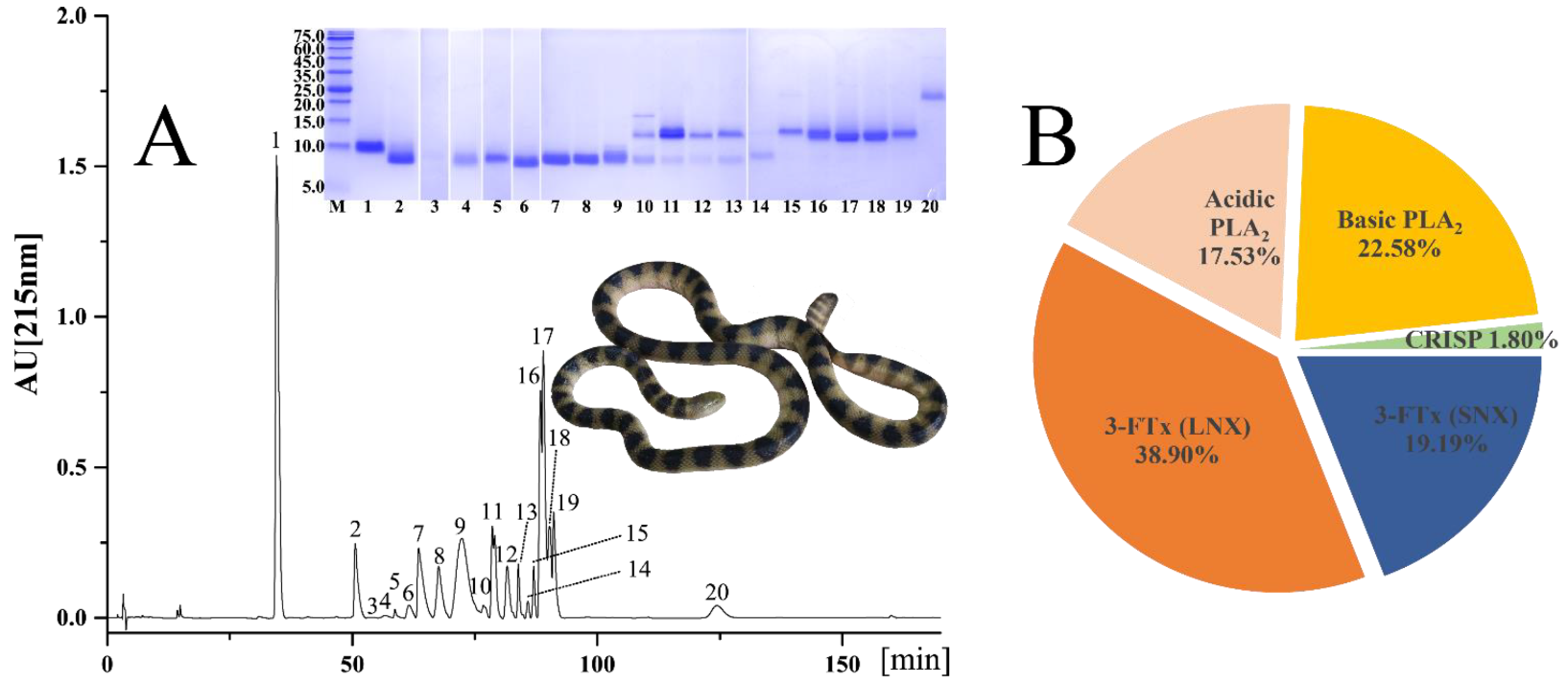
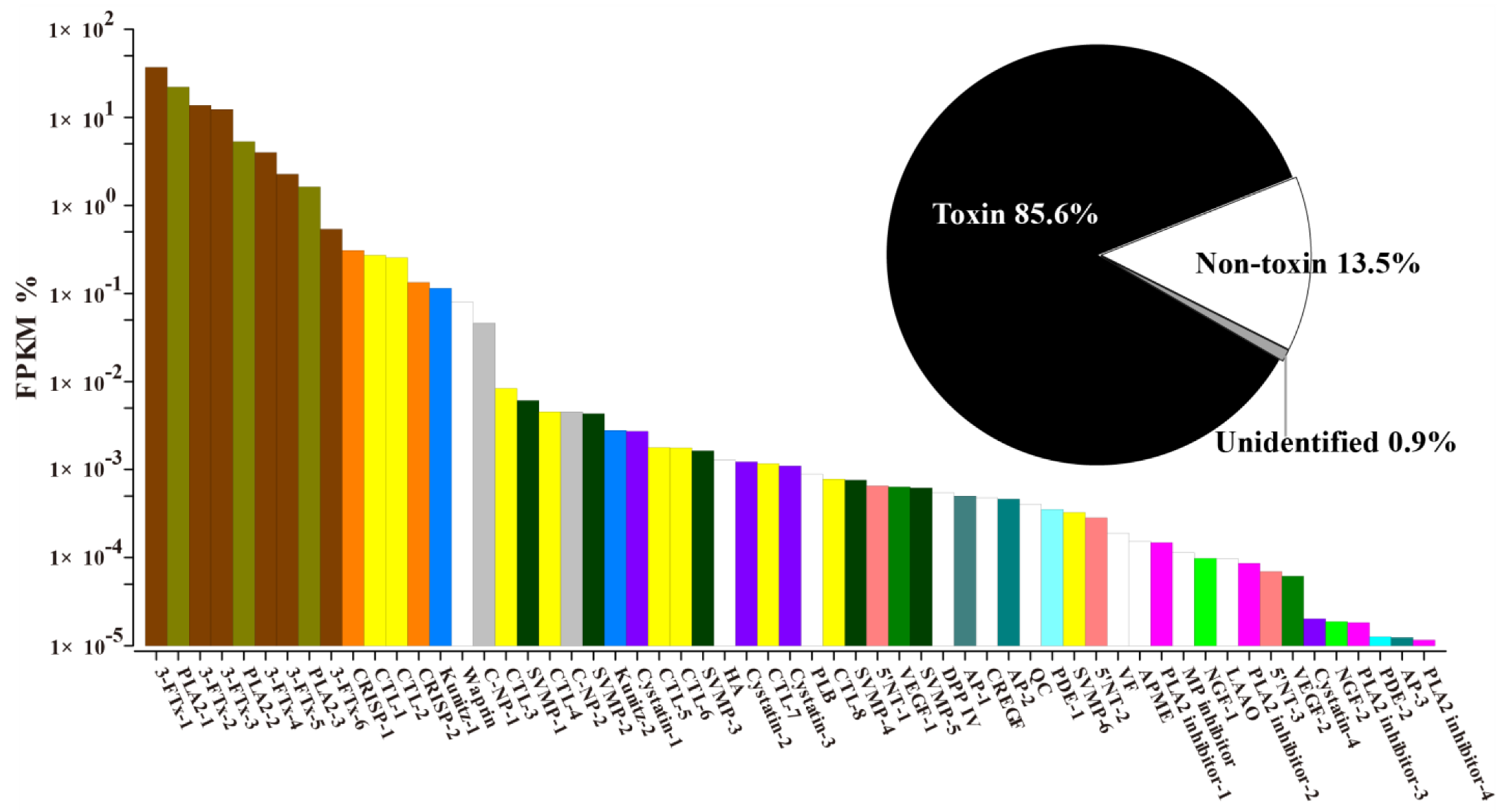
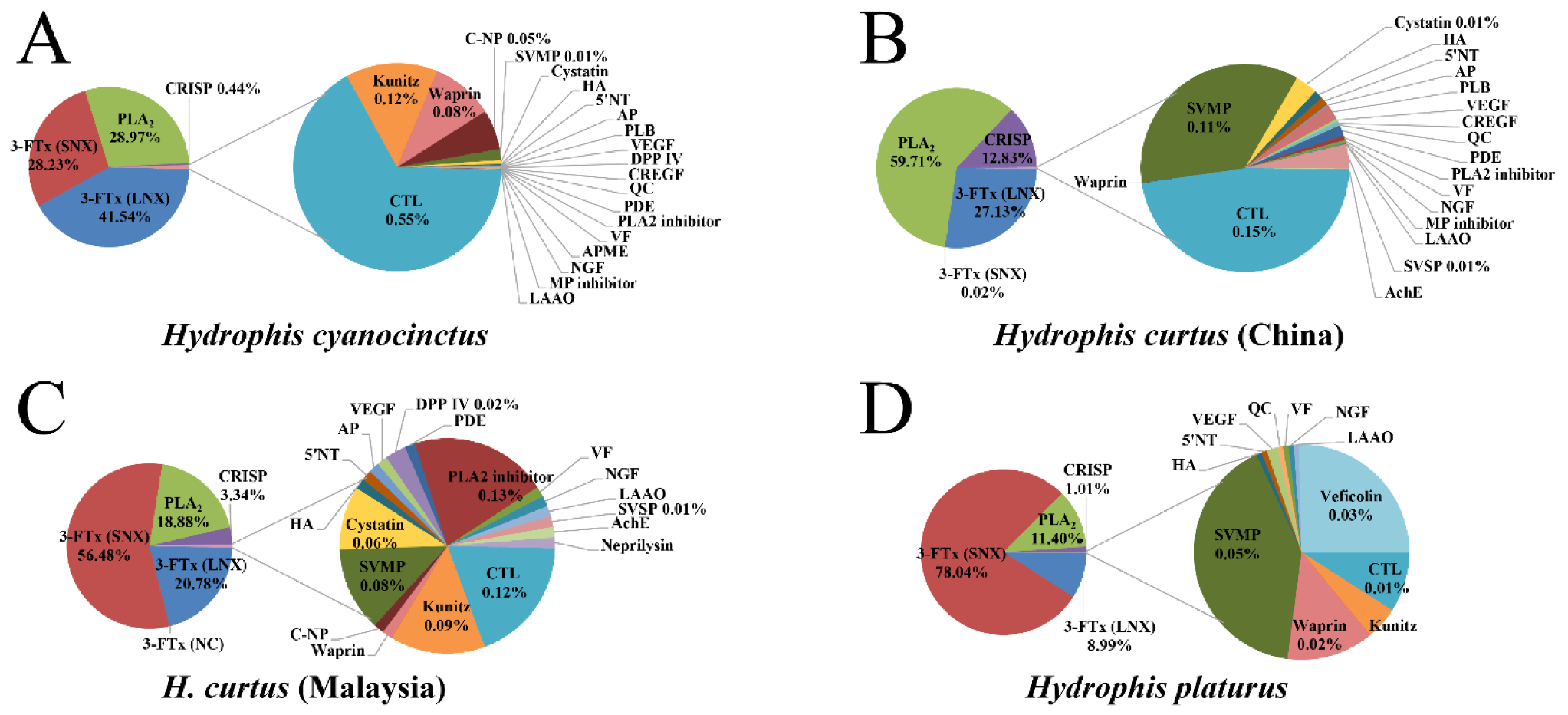
2.1. Venomic Profile
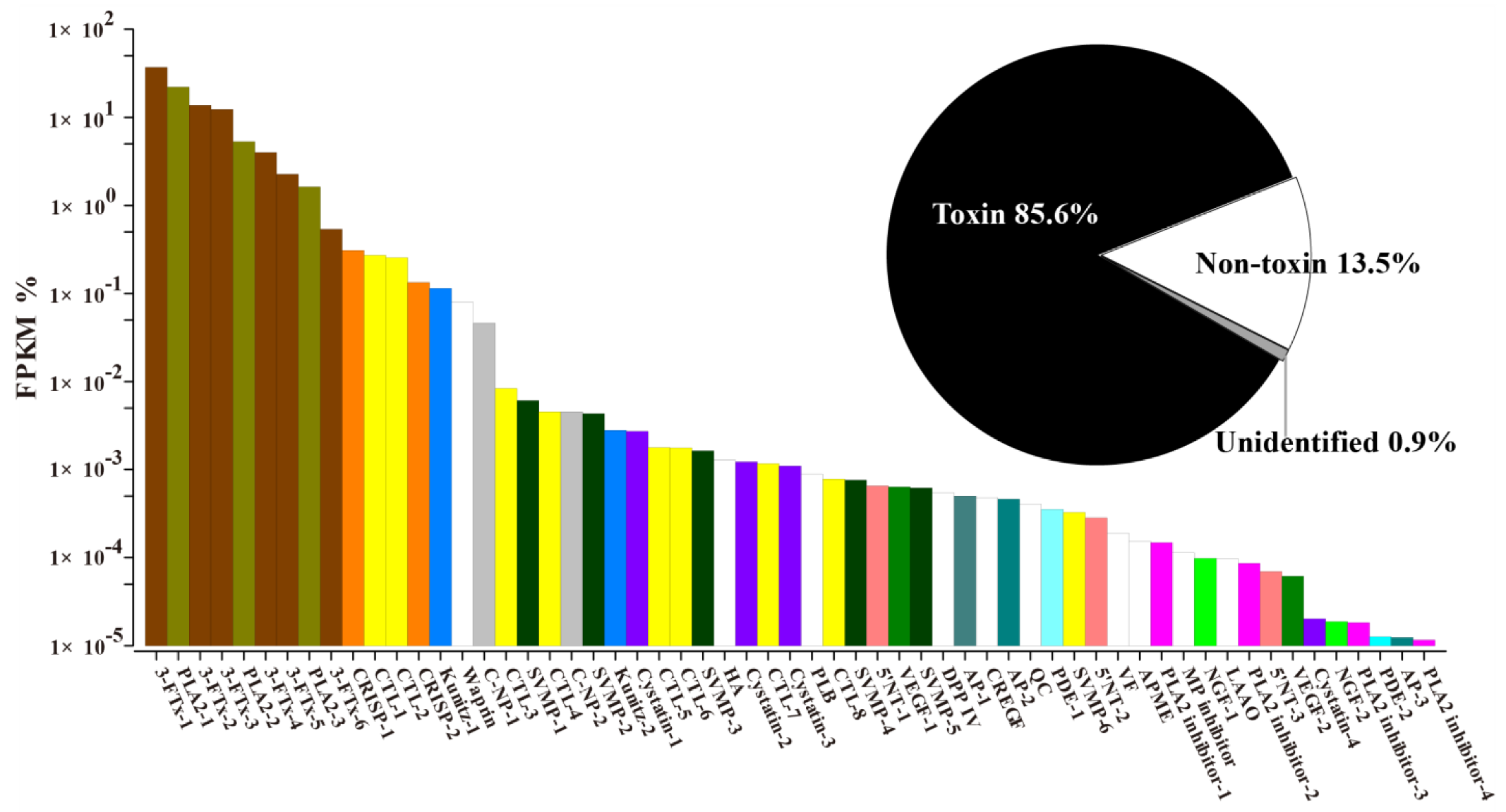
2.2. Transcriptome Assembly and Diversity of Toxin-Coding Unigenes
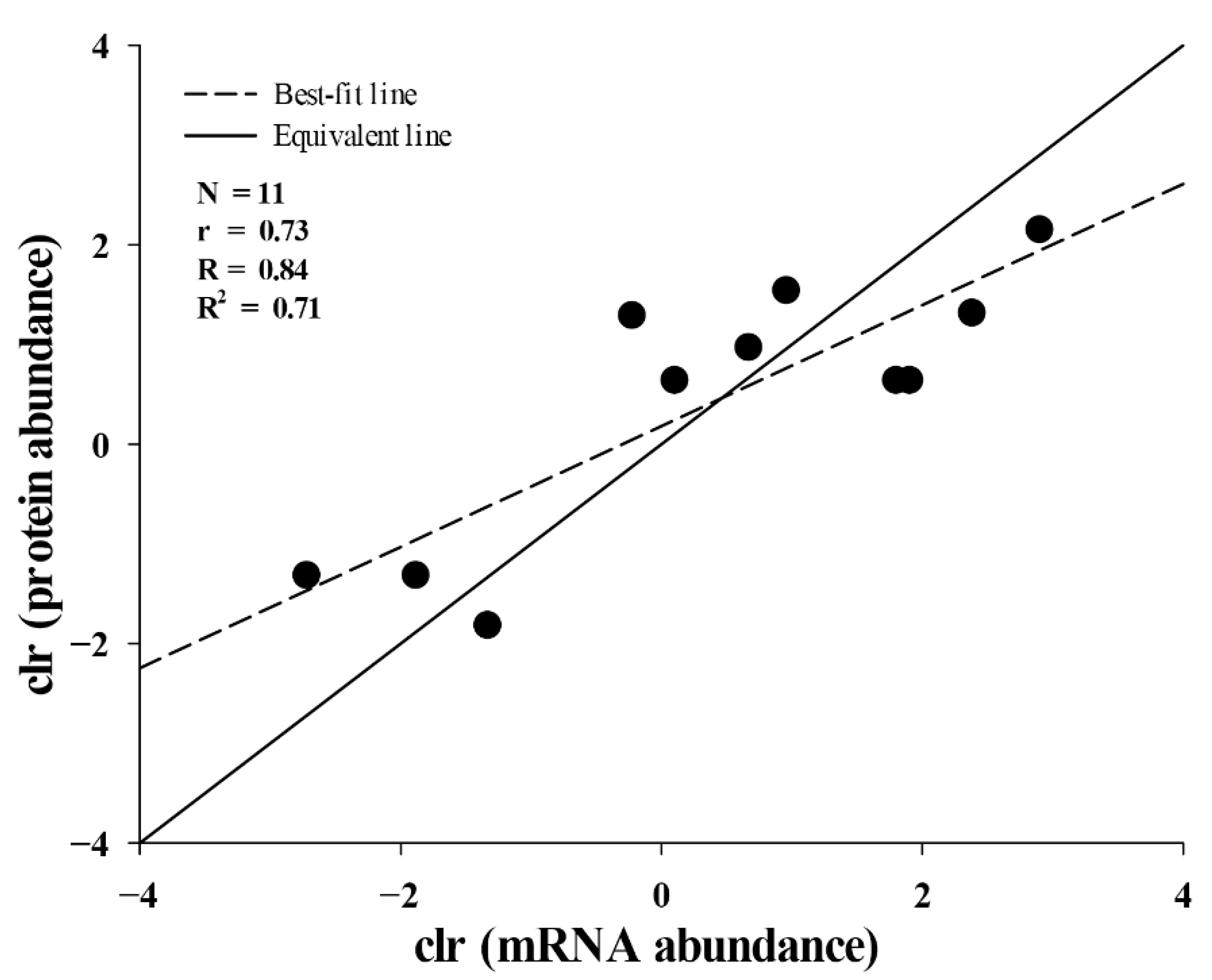
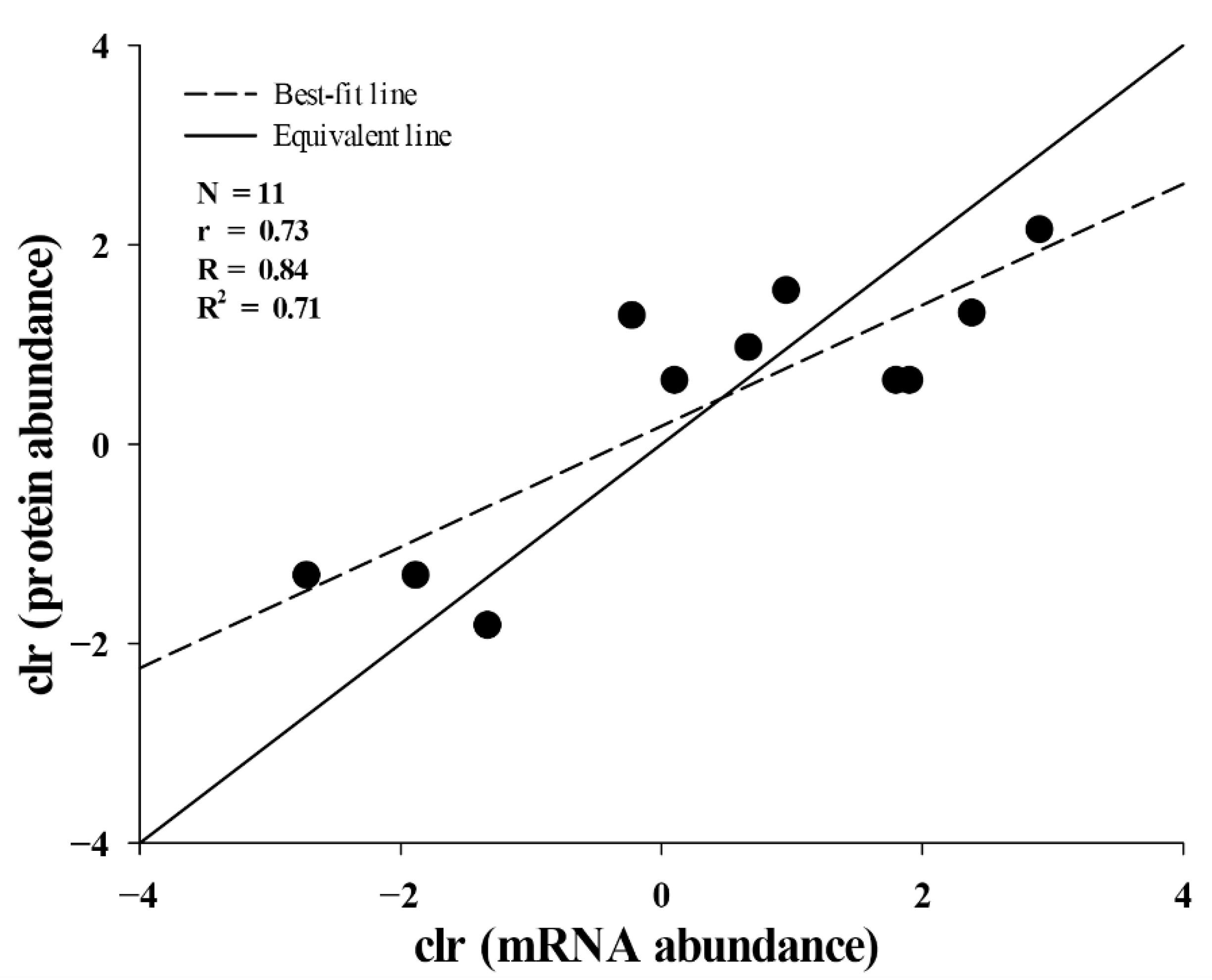
2.3. Correlation between Translational and Transcriptional Abundances of Toxins
2.4. Positive Selection in Evolutionary Adaption
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals and Ethics
5.2. Isolation of Venom Proteins by RP-HPLC and SDS-PAGE
5.3. Identification and Relative Abundance Estimation of Venom Proteins
5.4. Venom Gland cDNA Synthesis and Sequencing
5.5. Transcriptome Assembly, Annotation and Quantification
5.6. Detection of Positive Selection
5.7. Lethality
5.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gutiérrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17063. [Google Scholar] [CrossRef]
- Udyawer, V.; Goiran, C.; Shine, R.; Buijs, A. Peaceful coexistence between people and deadly wildlife: Why are recreational users of the ocean so rarely bitten by sea snakes? People Nat. 2021, 3, 335–346. [Google Scholar] [CrossRef]
- Wang, Y.-G.; Mao, J.-F.; Jin, L.-T.; Cao, C.-L.; Huang, Y.-X.; Wu, L.-Q.; Xie, J.; Pan, Q.-L. Epidemiology and prevention and treatment strategies of snake bite in coastal areas of Zhejiang. Chin. J. Crit. Care Med. 2011, 31, 541–544. [Google Scholar]
- Wang, N.; Li, Q.; Li, B.; Li, Z.; Li, H.; Tang, S.; Sawai, Y.; Kawanura, Y.; Toriba, M.; Kobayashi, T. An epidemiological study on the snakebites in Guangxi Province, China in 1990. J. Snake 1993, 5, 10–17. [Google Scholar]
- Karthikeyan, R.; Balasubramamian, T. Species diversity of sea snake (Hydrophiidae) distributed in the coramantal coast (East Coast of India). Int. J. Zool. Res. 2007, 3, 107–131. [Google Scholar]
- Rasmussen, A.R.; Murphy, J.C.; Ompi, M.; Gibbons, J.W.; Uetz, P. Marine Reptiles. PLoS ONE 2011, 6, e27373. [Google Scholar] [CrossRef] [Green Version]
- Cao, N.V.; Tao, N.T.; Moore, A.; Montoya, A.; Rasmussen, A.R.; Broad, K.; Voris, H.K.; Takacs, Z. Sea snake harvest in the gulf of Thailand. Conserv. Biol. 2014, 28, 1677–1687. [Google Scholar]
- Udyawer, V.; Read, M.; Hamann, M.; Heupel, M.R.; Simpfendorfer, C.A. Importance of shallow tidal habitats as refugia from trawl fishing for sea snakes. J. Herpetol. 2016, 50, 527–533. [Google Scholar] [CrossRef]
- Karthikeyan, R.; Vijayalakshmi, S.; Balasubramamian, T. Feeding and parturition of female annulated sea snake Hydrophis cyanocinctus in captivity. Curr. Sci. 2008, 94, 660–664. [Google Scholar]
- Liu, L.; Chen, Z.; Liu, Y.; Shi, P. Studies on the sea snakes of the middle section of Fujian’s coastal waters and their feeding habits. Acta Herpetol. Sin. 1985, 4, 341–343. [Google Scholar]
- Zhang, L.-M.; Chen, Z.-L. Medicinal value of sea snakes. J. Tradit. Chin. Med. 2002, 30, 25–26. [Google Scholar]
- Suntrarachun, S.; Chanhome, L.; Sumontha, M. Identification of sea snake meat adulteration in meat products using PCR-RFLP of mitochondrial DNA. Food Sci. Hum. Wellness 2018, 7, 170–174. [Google Scholar] [CrossRef]
- Hao, E.-W.; Deng, J.-G.; Du, Z.-C.; Hou, X.-T. Study on conventional application and biological distribution of marine traditional Chinese medicine in Guangxi. J. Chin. Med. Mater. 2016, 39, 1262–1265. [Google Scholar]
- Phillips, C.M. Sea snake envenomation. Dermatol. Ther. 2002, 15, 58–61. [Google Scholar] [CrossRef]
- Milton, D.A.; Fry, G.C.; Dell, Q. Reducing impacts of trawling on protected sea snakes: By-catch reduction devices improve escapement and survival. Mar. Freshw. Res. 2009, 60, 824–832. [Google Scholar] [CrossRef]
- Tu, A.T.; Fulde, G. Sea snake bites. Clin. Dermatol. 1987, 5, 118–126. [Google Scholar] [CrossRef]
- Zhong, W.; Xie, G.-Q.; Zeng, G.-Q. Analysis of the secondary injury caused by marine organisms in drowning person in the South China Sea. Chin. J. Naut. Med. Hyperb. Med. 2008, 15, 175–177. [Google Scholar]
- Mackessy, S.P.; Tu, A.T. Biology of the Sea Snakes and Biochemistry of Their Venoms. In Toxin-Related Diseases: Poisons Originating from Plants, Animals and Spoilage; Tu, A.T., Ed.; Oxford & IBH Publishing Co.: New Delhi, India, 1993; pp. 305–351. [Google Scholar]
- Calvete, J.J.; Ghezellou, P.; Paiva, O.; Matainaho, T.; Ghassempour, A.; Goudarzi, H.; Kraus, F.; Sanz, L.; Williams, D.J. Snake venomics of two poorly known Hydrophiinae: Comparative proteomics of the venoms of terrestrial Toxicocalamus longissimus and marine Hydrophis cyanocinctus. J. Proteom. 2012, 75, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, K.Y.; Ng, T.S.; Sim, S.M.; Tan, N.H. Venom proteome of spine-bellied sea snake (Hydrophis curtus) from Penang, Malaysia: Toxicity correlation, immunoprofiling and cross-neutralization by sea snake antivenom. Toxins 2019, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, Q.-Q.; Wang, C.; Wang, B.-L.; Qiu, L.-L.; Zou, S.-J.; Zhang, F.-H.; Liu, G.-Y.; Zhang, L.-M. A comparative analysis of the proteomes and biological activities of the venoms from two sea snakes, Hydrophis curtus and Hydrophis cyanocinctus, from Hainan, China. Toxicon 2020, 187, 35–46. [Google Scholar] [CrossRef]
- Neale, V.; Sotillo, J.; Seymour, J.; Wilson, D. The venom of the spine-bellied sea snake (Hydrophis curtus): Proteome, toxin diversity and intraspecific variation. Int. J. Mol. Sci. 2017, 18, 2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.H.; Wong, K.Y.; Tan, K.Y.; Tan, N.H. Venom proteome of the yellow-lipped sea krait, Laticauda colubrina from Bali: Insights into subvenomic diversity, venom antigenicity and cross-neutralization by antivenom. J. Proteom. 2017, 166, 48–58. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Wen, L.; Miao, Y.-F.; Du, Y.; Sun, Y.; Yin, Y.; Lin, C.-X.; Lin, L.-H.; Ji, X.; Gao, J.-F. Venom-gland transcriptomic, venomic, and antivenomic profiles of the spine-bellied sea snake (Hydrophis curtus) from the South China Sea. BMC Genom. 2021, 22, 520. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Lim, S.E.; Tan, N.H. Venomics of the beaked sea snake, Hydrophis schistosus: A minimalist toxin arsenal and its cross-neutralization by heterologous antivenoms. J. Proteom. 2015, 126, 121–130. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Gutiérrez, J.M.; Rasmussen, A.R.; Engmark, M.; Gravlund, P.; Sanders, K.L.; Lohse, B.; Lomonte, B. Danger in the reef: Proteome, toxicity, and neutralization of the venom of the olive sea snake, Aipysurus laevis. Toxicon 2015, 107 Pt B, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Durban, J.; Sasa, M.; Calvete, J.J. Venom gland transcriptomics and microRNA profiling of juvenile and adult yellow-bellied sea snake, Hydrophis platurus, from Playa del Coco (Guanacaste, Costa Rica). Toxicon 2018, 153, 96–105. [Google Scholar] [CrossRef]
- Peng, C.; Ren, J.-L.; Deng, C.; Jiang, D.; Wang, J.; Qu, J.; Chang, J.; Yan, C.; Jiang, K.; Murphy, R.W.; et al. The genome of shaw’s sea snake (Hydrophis curtus) reveals secondary adaptation to its marine environment. Mol. Biol. Evol. 2020, 37, 1744–1760. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y. De novo venom-gland transcriptomics of spine-bellied sea snake (Hydrophis curtus) from Penang, Malaysia-next-generation sequencing, functional annotation and toxinological correlation. Toxins 2021, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.L.; Rasmussen, A.R.; Elmberg, J.; De Silva, A.; Guinea, M.L.; Lee, M.S.Y. Recent rapid speciation and ecomorph divergence in Indo-Australian sea snakes. Mol. Ecol. 2013, 22, 2742–2759. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.-M. Snakes of China; Anhui Science and Technology Publishing House: Hefei, China, 2006. [Google Scholar]
- Zheng, J.; Wu, S.; Pang, F.; Mo, Y.; Xu, L.; Su, B.; Chang, M.; Zhuo, Z.; Lao, Z. Studies on the components of Lapemis hardwickii venom. Acta Sci. Nat. Univ. Sunyatseni 1986, 3, 122–127. [Google Scholar]
- Shi, F.; Zheng, W.-R. Determination of the venom yield and toxicity (LD50) in Hydrophis cyanocinctus from Dongshan, Fujian. J. Fujian Med. Coll. 1987, 21, 110–111. [Google Scholar]
- Calvete, J.J.; Juárez, P.; Sanz, L. Snake venomics. Strategy and applications. J. Mass Spectrom. 2007, 42, 1405–1414. [Google Scholar] [CrossRef]
- Pahari, S.; Bickford, D.; Fry, B.G.; Kini, R.M. Expression pattern of three-finger toxin and phospholipase A2 genes in the venom glands of two sea snakes, Lapemis curtus and Acalyptophis peronii: Comparison of evolution of these toxins in land snakes, sea kraits and sea snakes. BMC Evol. Biol. 2007, 7, 175. [Google Scholar] [CrossRef] [Green Version]
- Lomonte, B.; Pla, D.; Sasa, M.; Tsai, W.-C.; Solórzano, A.; Ureña-Díaz, J.M.; Fernández-Montes, M.L.; Mora-Obando, D.; Sanz, L.; Gutiérrez, J.M.; et al. Two color morphs of the pelagic yellow-bellied sea snake, Pelamis platura, from different locations of Costa Rica: Snake venomics, toxicity, and neutralization by antivenom. J. Proteom. 2014, 103, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casewell, N.R.; Wagstaff, S.C.; Wüster, W.; Cook, D.A.N.; Bolton, F.M.S.; King, S.I.; Pla, D.; Sanz, L.; Calvete, J.J.; Harrison, R.A. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 9205–9210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, R.H.; Guimarães, P.R.; Junqueira, M.; Neves-Ferreira, A.G.C.; Soares, M.R.; Chapeaurouge, A.; Trugilho, M.R.O.; León, I.R.; Rocha, S.L.G.; Oliveira-Carvalho, A.L.; et al. Bothrops insularis venomics: A proteomic analysis supported by transcriptomic-generated sequence data. J. Proteom. 2009, 72, 241–255. [Google Scholar] [CrossRef]
- Corrêa-Netto, C.; Junqueira-de-Azevedo, I.d.L.M.; Silva, D.A.; Ho, P.L.; Leitão-de-Araújo, M.; Alves, M.L.M.; Sanz, L.; Foguel, D.; Zingali, R.B.; Calvete, J.J. Snake venomics and venom gland transcriptomic analysis of Brazilian coral snakes, Micrurus altirostris and M. corallinus. J. Proteom. 2011, 74, 1795–1809. [Google Scholar] [CrossRef]
- Rodrigues, R.S.; Boldrini-França, J.; Fonseca, F.P.P.; De la Torre, P.; Henrique-Silva, F.; Sanz, L.; Calvete, J.J.; Rodrigues, V.M. Combined snake venomics and venom gland transcriptomic analysis of Bothropoides pauloensis. J. Proteom. 2012, 75, 2707–2720. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; McGivern, J.J.; Wray, K.P.; Seavy, M.; Calvin, K.; Rokyta, D.R. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J. Proteom. 2014, 96, 145–158. [Google Scholar] [CrossRef]
- Durban, J.; Juárez, P.; Angulo, Y.; Lomonte, B.; Flores-Diaz, M.; Alape-Girón, A.; Sasa, M.; Sanz, L.; Gutiérrez, J.M.; Dopazo, J.; et al. Profiling the venom gland transcriptomes of Costa Rican snakes by 454 pyrosequencing. BMC Genom. 2011, 12, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durban, J.; Pérez, A.; Sanz, L.; Gómez, A.; Bonilla, F.; Rodríguez, S.; Chacón, D.; Sasa, M.; Angulo, Y.; Gutiérrez, J.M.; et al. Integrated “omics” profiling indicates that miRNAs are modulators of the ontogenetic venom composition shift in the Central American rattlesnake, Crotalus simus simus. BMC Genom. 2013, 14, 234. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genom. 2015, 16, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Aird, S.D.; Watanabe, Y.; Villar-Briones, A.; Roy, M.C.; Terada, K.; Mikheyev, A.S. Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genom. 2013, 14, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, E.P.; Rautsaw, R.M.; Strickland, J.L.; Holding, M.L.; Hogan, M.P.; Mason, A.J.; Rokyta, D.R.; Parkinson, C.L. Comparative venom-gland transcriptomics and venom proteomics of four Sidewinder Rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Sci. Rep. 2018, 8, 15534. [Google Scholar] [CrossRef] [PubMed]
- Lynch, V.J. Inventing an arsenal: Adaptive evolution and neofunctionalization of snake venom phospholipase A2 genes. BMC Evol. Biol. 2007, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, H.L.; Rossiter, W. Rapid evolution by positive selection and gene gain and loss: PLA2 venom genes in closely related Sistrurus rattlesnakes with divergent diets. J. Mol. Evol. 2008, 66, 151–166. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wüster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 2011, 28, 2637–2649. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Wray, K.P.; Lemmon, A.R.; Lemmon, E.M.; Caudle, S.B. A high-throughput venom-gland transcriptome for the eastern diamondback rattlesnake (Crotalus adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon 2011, 57, 657–671. [Google Scholar] [CrossRef]
- Margres, M.J.; Aronow, K.; Loyacano, J.; Rokyta, D.R. The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms. BMC Genom. 2013, 14, 531. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Wray, K.P.; Margres, M.J. The genesis of an exceptionally lethal venom in the timber rattlesnake (Crotalus horridus) revealed through comparative venom-gland transcriptomics. BMC Genom. 2013, 14, 394. [Google Scholar] [CrossRef] [Green Version]
- McGivern, J.J.; Wray, K.P.; Margres, M.J.; Couch, M.E.; Mackessy, S.P.; Rokyta, D.R. RNA-seq and high-definition mass spectrometry reveal the complex and divergent venoms of two rear-fanged colubrid snakes. BMC Genom. 2014, 15, 1061. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Bielawski, J.P.; Yang, Z.-H. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 2001, 18, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Shan, L.-L.; Gao, J.-F.; Zhang, Y.-X.; Shen, S.-S.; He, Y.; Wang, J.; Ma, X.-M.; Ji, X. Proteomic characterization and comparison of venoms from two elapid snakes (Bungarus multicinctus and Naja atra) from China. J. Proteom. 2016, 138, 83–94. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novoassembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-Q.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Chanda, A.; Kalita, B.; Islam, T.; Patra, A.; Mukherjee, A.K. Proteomic analysis to unravel the complex venom proteome of eastern India Naja naja: Correlation of venom composition with its biochemical and pharmacological properties. J. Proteom. 2017, 156, 29–39. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.-F.; Wang, J.; He, Y.; Qu, Y.-F.; Lin, L.-H.; Ma, X.-M.; Ji, X. Proteomic and biochemical analyses of short-tailed pit viper (Gloydius brevicaudus) venom: Age-related variation and composition–activity correlation. J. Proteom. 2014, 105, 307–322. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Margres, M.J.; Calvin, K. Post-transcriptional mechanisms contribute little to phenotypic variation in snake venoms. G3 Genes Genomes Genet. 2015, 5, 2375–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | % | MW (kDa) | Peptide Ion | MS/MS-Derived Sequence | Protein Family/Species/Accession/Transcript ID | |
|---|---|---|---|---|---|---|
| m/z | z | |||||
| 1 | 19.19 | 9.3 | 775.3 | 2 | TTTNCAESSCYKK | 3-FTx (SNX); Hydrophis cyanocinctus; P25494; Hcy|29338, Hcy|29549, Hcy|28274 |
| 2 | 3.74 | 7.3 | 537.3 727.3 | 3 2 | XEFGCAATCPTVBR XEFGCAATCPTVB | 3-FTx (LNX); Laticauda colubrina; P0C8R8; Hcy|29273 |
| 3 | 0.27 | 7.3 | 537.3 727.3 | 3 2 | XEFGCAATCPTVBR XEFGCAATCPTVB | 3-FTx (LNX); L. colubrina; P0C8R8; Hcy|29273 |
| 4 | 0.22 | 7.3 | 805.4 727.3 | 2 2 | XEFGCAATCPTVBR XEFGCAATCPTVB | 3-FTx (LNX); L. colubrina; P0C8R8; Hcy|29273 |
| 5 | 0.23 | 7.4 | 805.4 727.3 | 2 2 | XEFGCAATCPTVBR XEFGCAATCPTVB | 3-FTx (LNX); L. colubrina; P0C8R8; Hcy|29273 |
| 6 | 1.48 | 7.0 | 537.3 727.3 | 3 2 | XEFGCAATCPTVBR XEFGCAATCPTVB | 3-FTx (LNX); L. colubrina; P0C8R8; Hcy|29273 |
| 7 | 6.17 | 7.3 | 531.9 479.9 623.2 | 3 3 2 | RXEMGCAATCPTVB XEMGCAATCPTVB SWCDAFCGSR | 3-FTx (LNX); Hydrophis hardwickii; Q8UW28; Hcy|29273 |
| 8 | 5.85 | 7.3 | 588.3 580.3 495.8 | 2 4 2 | GBVXEXGCTAB THPYBPETCPPGBNXCYBB VXEXGCTAB | 3-FTx (LNX); H. hardwickii; A3FM53; Hcy|29273 |
| 9 | 17.79 | 7.6 | 588.3 638.2 788.4 580.2 | 2 2 2 4 | GBVXEXGCTAB SWCDAFCSSR VXEXGCTABCPTVB THPYBPETCPPGBNXCYBB | 3-FTx (LNX); H. hardwickii; A3FM53; Hcy|29273, Hcy|29140 |
| 10 | 1.22 | 17.2 | 591.3 646.0 822.3 | 3 3 2 | BVCDCDVAAAECFAR NXVBFSYVXTCANHNR VCDCDVAAAECFAR | Basic PLA2; Hydrophis schistosus; P00610; Hcy|29208 |
| 11.8 | 591.3 646.0 548.6 | 3 3 3 | BVCDCDVAAAECFAR NXVBFSYVXTCANHNR VCDCDVAAAECFAR | Basic PLA2; H. schistosus; P00610; Hcy|29208 | ||
| 0.77 | 7.3 | 588.3 587.6 495.8 638.2 | 2 3 2 2 | GBVXEXGCTAB GBVXEXGCTABCPTVB VXEXGCTAB SWCDAFCSSR | 3-FTx (LNX); H. hardwickii; Q8UW29; Hcy|29273 | |
| 11 | 6.19 | 11.7 | 618.8 821.9 | 2 2 | BVCDCDVAAAB VCDCDVAAABCFAR | Basic PLA2; H. hardwickii; Q8UW30; Hcy|29208 |
| 7.3 | 591.3 548.6 646.0 | 3 3 3 | BVCDCDVAAAECFAR VCDCDVAAAECFAR NXVBFSYVXTCANHNR | Basic PLA2; H. schistosus; P00610; Hcy|29208 | ||
| 12 | 3.35 | 11.7 | 591.3 548.6 757.8 646.0 446.2 | 3 3 2 3 3 | BVCDCDVAAAECFAR VCDCDVAAAECFAR NAYNNANYNXDTB NXVBFSYVXTCANHNR XHDDCYGEAEB | Basic PLA2; H. schistosus; P00610; Hcy|29208 |
| 0.49 | 7.4 | 588.3 495.8 | 2 2 | GBVXEXGCTAB VXEXGCTAB | 3-FTx (LNX); H. hardwickii; Q8UW29; Hcy|29273 | |
| 13 | 1.51 | 12.0 | 822.3 591.3 | 2 3 | VCDCDVAAAECFAR BVCDCDVAAAECFAR | Basic PLA2; H. schistosus; P00610; Hcy|29208 |
| 0.80 | 7.3 | 588.3 638.2 587.6 | 2 2 3 | GBVXEXGCTAB SWCDAFCSSR GBVXEXGCTABCPTVB | 3-FTx (LNX); H. hardwickii; A3FM53; Hcy|29273 | |
| 14 | 0.29 | 12.0 | 591.3 548.6 | 3 3 | BVCDCDVAAAECFAR VCDCDVAAAECFAR | Basic PLA2; H. schistosus; P00610; Hcy|29123 |
| 1.10 | 7.7 | 773.4 588.3 587.6 495.8 638.2 | 3 2 3 2 2 | THPYBPETCPPGBNXCYBB GBVXEXGCTAB GBVXEXGCTABCPTVB VXEXGCTAB SWCDAFCSSR | 3-FTx (LNX); H. hardwickii; A3FM53; Hcy|29273, Hcy|29296 | |
| 15 | 2.05 | 12.3 | 455.2 | 2 | TAAXCFAR | Acidic PLA2; Tropidechis carinatus; Q45Z26; Hcy|29123 |
| 16 | 7.15 | 11.4 | 455.2 644.3 | 2 3 | TAAXCFAR DNNDECBAFXCNCDR | Acidic PLA2; T. carinatus; Q45Z28; Hcy|29123 |
| 17 | 10.02 | 10.4 | 455.8 687.7 | 2 3 | XTXYSWB CFABAPYNNBNYNXDTB | Basic PLA2; Austrelaps superbus; Q9PUH5; Hcy|29057 |
| 18 | 3.21 | 10.7 | 644.3 455.2 | 3 2 | DNNDECBAFXCNCDR AFXCNCDR | Acidic PLA2; Notechis scutatus scutatus; Q9PSN5; Hcy|29123 |
| 19 | 5.11 | 11.4 | 455.3 601.8 528.2 | 2 2 2 | TAAXCFAR GGSGTPVDEXDR AFXCNCDR | Acidic PLA2; N. s. scutatus; Q9PSN5; Hcy|29057, Hcy|29123 |
| 20 | 1.80 | 23.6 | 448.2 | 3 | CTFAHSPEHTR | CRISP; H. hardwickii; Q8UW11; Hcy|29512, Hcy|29717 |
| Fraction | Toxin | Intraperitoneal LD50 (μg/g) * |
|---|---|---|
| - | Crude venom | 0.26 (0.23–0.30) |
| - | Whole venom protein ** | 0.16 (0.12–0.20) |
| 1 | Short neurotoxin | 0.09 (0.07–0.12) |
| 7 | Long neurotoxin | 0.14 (0.09–0.21) |
| 11 | Basic PLA2 | >0.6 |
| 18 | Acidic PLA2 | >2.0 |
| Toxins (No.) | M1: Nearly Neutral | −lnL | M2: Positive Selection | −lnL | M0: ω | Δ a | p-Value b | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3-FTx (1, 2, 3) | p: | 0.56 | 0.44 | 608.01 | p: | 0.45 | 0.27 | 0.28 | 588.51 | 2.79 | 39.00 | 3.40 × 10−9 * |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 9.84 | ||||||
| 5NT (1, 2) | p: | 0.60 | 0.40 | 3831.74 | p: | 0.90 | 0.00 | 0.10 | 3829.66 | 0.40 | 4.16 | 0.12 |
| ω: | 0.00 | 1.00 | ω: | 0.21 | 1.00 | 2.57 | ||||||
| AP (1) | p: | 0.63 | 0.37 | 5224.95 | p: | 0.93 | 0.00 | 0.07 | 5221.58 | 0.40 | 6.74 | 0.03 |
| ω: | 0.00 | 1.00 | ω: | 0.22 | 1.00 | 3.95 | ||||||
| CRISP (1, 2) | p: | 0.47 | 0.53 | 2499.04 | p: | 0.40 | 0.39 | 0.22 | 2458.11 | 1.38 | 81.87 | 0.00* |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 4.74 | ||||||
| CTL (1) | p: | 0.56 | 0.44 | 1046.59 | p: | 0.82 | 0.00 | 0.18 | 1045.29 | 0.52 | 2.60 | 0.27 |
| ω: | 0.00 | 1.00 | ω: | 0.19 | 1.00 | 2.49 | ||||||
| CTL (2) | p: | 0.60 | 0.40 | 1177.31 | p: | 0.80 | 0.07 | 0.13 | 1172.58 | 0.56 | 9.46 | 8.83 × 10−3 |
| ω: | 0.06 | 1.00 | ω: | 0.22 | 1.00 | 3.07 | ||||||
| CTL (3) | p: | 0.55 | 0.45 | 1186.50 | p: | 0.60 | 0.00 | 0.40 | 1184.23 | 0.62 | 4.55 | 0.10 |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 1.65 | ||||||
| Cystatin (1) | p: | 0.29 | 0.71 | 979.80 | p: | 0.96 | 0.00 | 0.04 | 969.73 | 1.15 | 20.13 | 4.25 × 10−5 * |
| ω: | 0.04 | 1.00 | ω: | 0.84 | 1.00 | 11.02 | ||||||
| DPP IV | p: | 0.64 | 0.36 | 4740.05 | p: | 0.83 | 0.11 | 0.06 | 4730.48 | 0.43 | 19.12 | 7.05 × 10−5 * |
| ω: | 0.00 | 1.00 | ω: | 0.15 | 1.00 | 3.82 | ||||||
| HA | p: | 0.45 | 0.55 | 2976.76 | p: | 0.52 | 0.00 | 0.48 | 2975.36 | 0.62 | 2.81 | 0.25 |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 1.38 | ||||||
| Kunitz (1) | p: | 0.82 | 0.18 | 1502.79 | p: | 0.82 | 0.11 | 0.08 | 1502.79 | 0.33 | 0.00 | 1.00 |
| ω: | 0.21 | 1.00 | ω: | 0.21 | 1.00 | 1.00 | ||||||
| Kunitz (2) | p: | 0.44 | 0.56 | 944.01 | p: | 0.18 | 0.44 | 0.38 | 916.66 | 3.14 | 54.70 | 1.32 × 10−12 * |
| ω: | 0.05 | 1.00 | ω: | 0.00 | 1.00 | 7.95 | ||||||
| LAAO | p: | 0.44 | 0.56 | 6029.83 | p: | 0.39 | 0.43 | 0.17 | 5970.56 | 1.03 | 118.53 | 0.00* |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 4.00 | ||||||
| NGF (1, 2) | p: | 0.51 | 0.49 | 1366.53 | p: | 0.39 | 0.46 | 0.15 | 1342.54 | 1.30 | 47.98 | 3.81 × 10−11 * |
| ω: | 0.07 | 1.00 | ω: | 0.13 | 1.00 | 5.87 | ||||||
| PLA2 (1) | p: | 0.47 | 0.53 | 1913.94 | p: | 0.41 | 0.21 | 0.38 | 1877.93 | 1.57 | 72.01 | 2.22 × 10−16 * |
| ω: | 0.01 | 1.00 | ω: | 0.00 | 1.00 | 4.16 | ||||||
| PLA2 (2) | p: | 0.45 | 0.55 | 808.89 | p: | 0.68 | 0.00 | 0.32 | 796.33 | 1.95 | 25.12 | 3.51 × 10−6 * |
| ω: | 0.00 | 1.00 | ω: | 0.15 | 1.00 | 8.00 | ||||||
| PLA2 inhibitor (1) | p: | 0.48 | 0.52 | 1900.98 | p: | 0.50 | 0.00 | 0.50 | 1900.96 | 0.50 | 0.04 | 0.98 |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 1.07 | ||||||
| PLB | p: | 0.56 | 0.44 | 3750.75 | p: | 0.53 | 0.45 | 0.02 | 3742.03 | 0.54 | 17.44 | 1.63 × 10−4 * |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 6.82 | ||||||
| QC | p: | 0.78 | 0.22 | 2194.10 | p: | 0.79 | 0.00 | 0.21 | 2194.09 | 0.21 | 0.01 | 1.00 |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 1.05 | ||||||
| SVMP (1) | p: | 0.48 | 0.52 | 8066.12 | p: | 0.35 | 0.46 | 0.19 | 7910.53 | 1.38 | 311.17 | 0.00 * |
| ω: | 0.04 | 1.00 | ω: | 0.00 | 1.00 | 5.06 | ||||||
| VEGF | p: | 0.31 | 0.69 | 1101.80 | p: | 0.87 | 0.00 | 0.13 | 1101.01 | 0.82 | 1.58 | 0.45 |
| ω: | 0.00 | 1.00 | ω: | 0.49 | 1.00 | 3.83 | ||||||
| VF | p: | 0.53 | 0.47 | 13764.79 | p: | 0.51 | 0.46 | 0.04 | 13,726.91 | 0.61 | 75.76 | 0.00 * |
| ω: | 0.00 | 1.00 | ω: | 0.00 | 1.00 | 4.96 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.-Y.; Sun, Y.; Du, Y.; Li, J.-Q.; Lv, J.-G.; Qu, Y.-F.; Lin, L.-H.; Lin, C.-X.; Ji, X.; Gao, J.-F. Venom of the Annulated Sea Snake Hydrophis cyanocinctus: A Biochemically Simple but Genetically Complex Weapon. Toxins 2021, 13, 548. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080548
Zhao H-Y, Sun Y, Du Y, Li J-Q, Lv J-G, Qu Y-F, Lin L-H, Lin C-X, Ji X, Gao J-F. Venom of the Annulated Sea Snake Hydrophis cyanocinctus: A Biochemically Simple but Genetically Complex Weapon. Toxins. 2021; 13(8):548. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080548
Chicago/Turabian StyleZhao, Hong-Yan, Yan Sun, Yu Du, Jia-Qi Li, Jin-Geng Lv, Yan-Fu Qu, Long-Hui Lin, Chi-Xian Lin, Xiang Ji, and Jian-Fang Gao. 2021. "Venom of the Annulated Sea Snake Hydrophis cyanocinctus: A Biochemically Simple but Genetically Complex Weapon" Toxins 13, no. 8: 548. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080548