Panorama of the Intracellular Molecular Concert Orchestrated by Actinoporins, Pore-Forming Toxins from Sea Anemones

 , , , , , and
, , , , , and 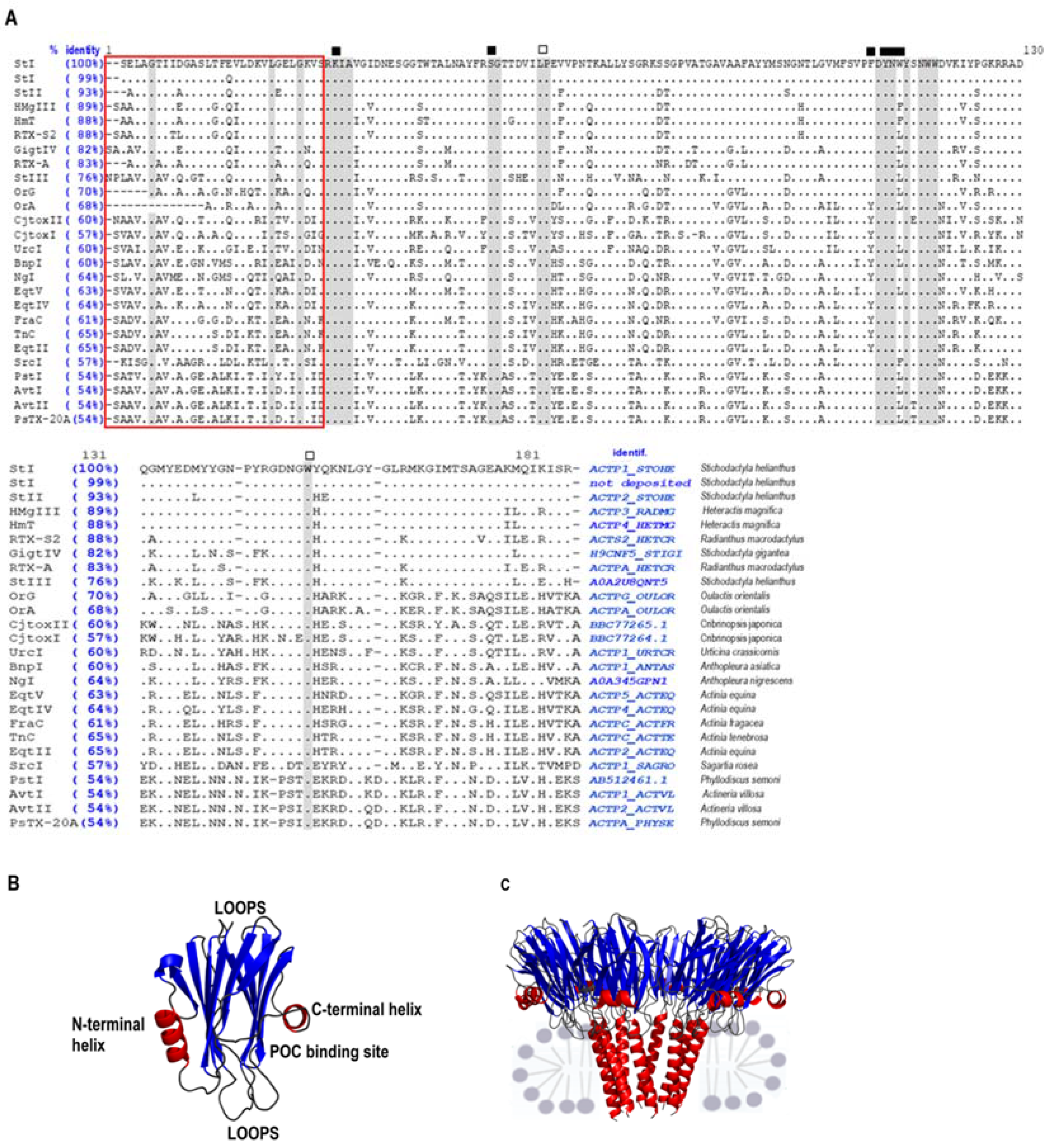{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Actinoporins Are Potent Toxins Produced by Sea Anemones
Mechanism of Pore Formation by APs
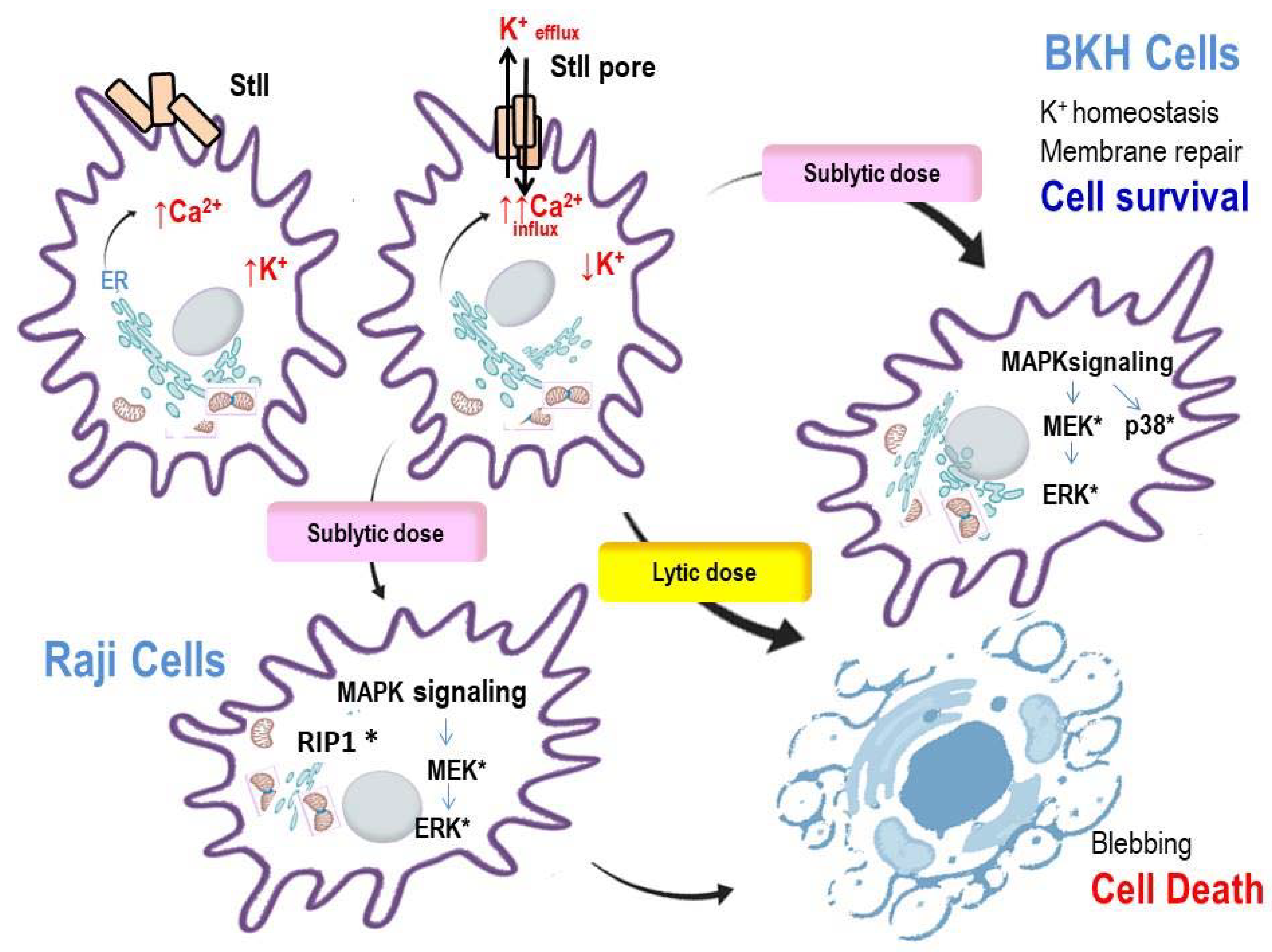
3. Piercing Cells by APs: Cell Death and Survival Mechanisms
3.1. The Colloid-Osmotic Shock Causes Cell Death in Non-Nucleated Erythrocytes
3.2. APs Cytotoxicity on Nucleated Eukaryotic Cells
3.3. Early Signals following Pore-Formation
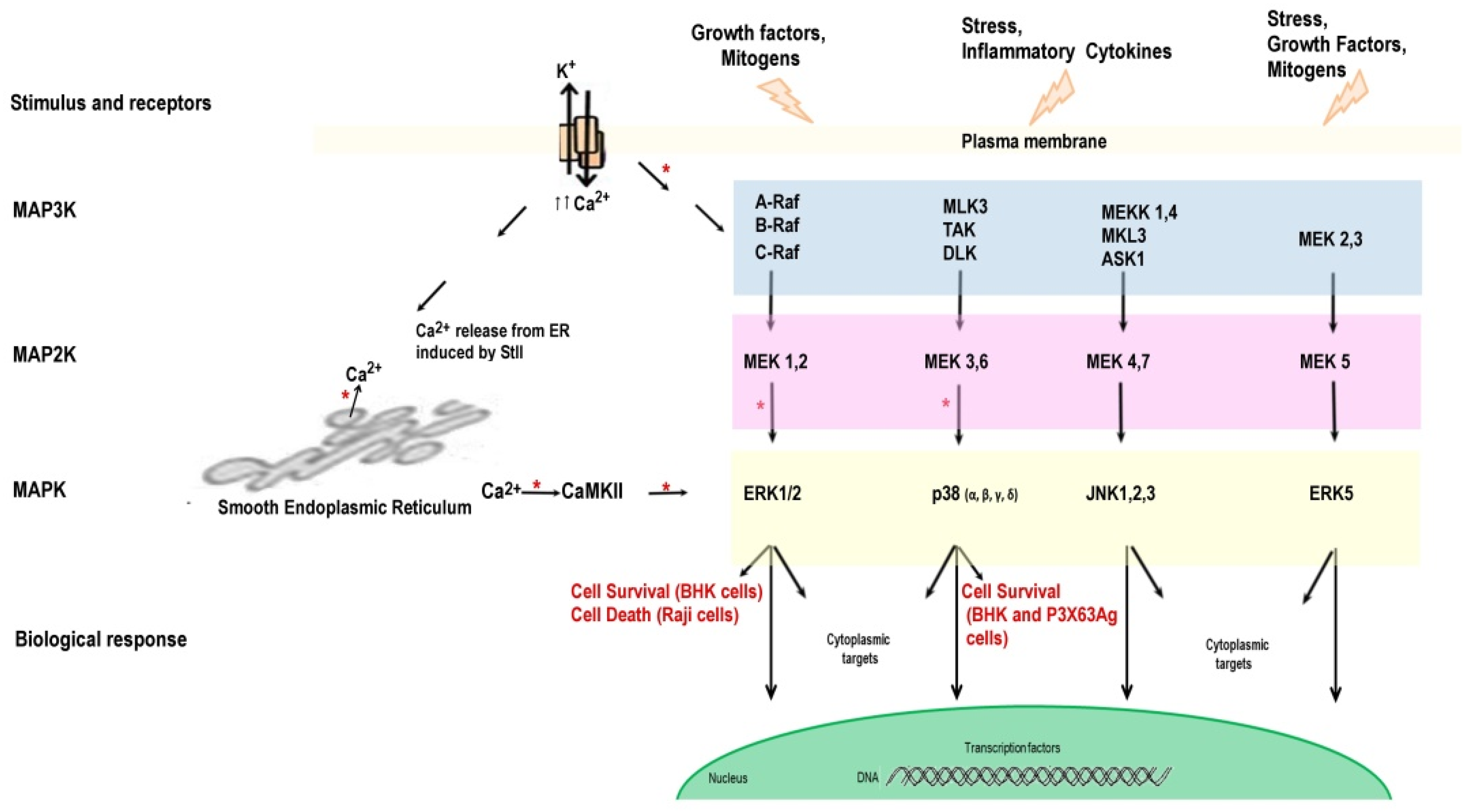
4. Intracellular Signaling Pathways Triggered by APs
4.1. K+ Outflow Promotes the Activation of MAPKs
4.2. Ca2+ Is a Relevant Signaling Mediator of Cell Death
4.3. Signaling via the Pattern Recognition Receptors
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peraro, M.D.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Ros, U.; García-Sáez, A.J. More Than a Pore: The Interplay of Pore-Forming Proteins and Lipid Membranes. J. Membr. Biol. 2015, 248, 545–561. [Google Scholar] [CrossRef]
- Cosentino, K.; Ros, U.; García-Sáez, A.J. Assembling the puzzle: Oligomerization of α-pore forming proteins in membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Draeger, A. Defying death: Cellular survival strategies following plasmalemmal injury by bacterial toxins. Semin. Cell Dev. Biol. 2015, 45, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, S.; Ho, S.; Ros, U.; Lanio, M.E.; Alvarez, C.; van der Goot, F.G. Damage of eukaryotic cells by the pore-forming toxin sticholysin II: Consequences of the potassium efflux. Biochim. Biophys. Acta Biomembr. 2017, 1859, 982–992. [Google Scholar] [CrossRef]
- Bischofberger, M.; Iacovache, I.; Goot, F.G. Pathogenic Pore-Forming Proteins: Function and Host Response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanio, M.E.; Fernández, L.E.; Laborde, R.J.; Cruz, Y.; Luzardo, M.; Mesa, C.; Álvarez, C.M.; Pazos, I.F.; Tejuca, M.; Valle, A.; et al. Vaccine Composition Based on Sticholysin Encapsulated into Liposomes. U.S. Patent US8,697,093,B2, 15 April 2014. [Google Scholar]
- Laborde, R.J.; Sanchez-Ferras, O.; Luzardo, M.C.; Cruz-Leal, Y.; Fernandez, A.; Mesa, C.; Oliver, L.; Canet, L.; Abreu-Butin, L.; Nogueira, C.V.; et al. Novel Adjuvant Based on the Pore-Forming Protein Sticholysin II Encapsulated into Liposomes Effectively Enhances the Antigen-Specific CTL-Mediated Immune Response. J. Immunol. 2017, 198, 2772–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderluh, G.; Macek, P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria). Toxicon 2002, 40, 111–124. [Google Scholar] [CrossRef]
- Kem, W.R. The Biology of Nematocysts; Hessinger, D.A., Lenhoff, H.M., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 1988; ISBN 9780123453204. [Google Scholar]
- Anderluh, G.; Lakey, J.H. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 2008, 33, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Tejuca, M.; Anderluh, G.; Dalla Serra, M. Sea anemone cytolysins as toxic components of immunotoxins. Toxicon 2009, 54, 1206–1214. [Google Scholar] [CrossRef]
- Pentón, D.; Pérez-Barzaga, V.; Díaz, I.; Reytor, M.L.; Campos, J.; Fando, R.; Calvo, L.; Cilli, E.M.; Morera, V.; Castellanos-Serra, L.R.; et al. Validation of a mutant of the pore-forming toxin sticholysin-I for the construction of proteinase-activated immunotoxins. Protein Eng. Des. Sel. 2011, 24, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Mutter, N.; Soskine, M.; Huang, G.; Albuquerque, I.; Bernardes, G.J.; Maglia, G. Modular Pore-Forming Immunotoxins with Caged Cytotoxicity Tailored by Directed Evolution. ACS Chem. Biol. 2018, 13, 3153–3160. [Google Scholar] [CrossRef]
- Bayley, H. Nanopore sequencing: From imagination to reality. Clin. Chem. 2015, 61, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Siwy, Z. Nanopore analytics: Sensing of single molecules. Chem. Soc. Rev. 2009, 38, 2360–2384. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Voet, A.; Maglia, G. FraC nanopores with adjustable diameter identify the mass of opposite-charge peptides with 44 dalton resolution. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Caaveiro, J.M.M.; Morante, K.; González-Mañas, J.M.; Tsumoto, K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 2015, 6, 6337. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Caaveiro, J.M.M.; Morante, K.; González-Mañas, J.M.; Tsumoto, K. Haemolytic actinoporins interact with carbohydrates using their lipid-binding module. Philos. Trans. R. Soc. L. B 2017, 372, 20160216. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Anderluh, G.; Maček, P.; Turk, D. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure 2001, 9, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Hinds, M.G.; Zhang, W.; Anderluh, G.; Hansen, P.E.; Norton, R.S. Solution structure of the eukaryotic pore-forming cytolysin equinatoxin II: Implications for pore formation. J. Mol. Biol. 2002, 315, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, C.; Mancheño, J.M.; Martínez, D.; Tejuca, M.; Pazos, F.; Lanio, M.E. Sticholysins, two pore-forming toxins produced by the Caribbean Sea anemone Stichodactyla helianthus: Their interaction with membranes. Toxicon 2009, 54, 1135–1147. [Google Scholar] [CrossRef]
- Alvarez, C.; Ros, U.; Valle, A.; Pedrera, L.; Soto, C.; Hervis, Y.P.; Cabezas, S.; Valiente, P.A.; Pazos, F.; Lanio, M.E. Biophysical and biochemical strategies to understand membrane binding and pore formation by sticholysins, pore-forming proteins from a sea anemone. Biophys. Rev. 2017, 9, 529–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Linares, S.; Rivera-de-Torre, E.; Palacios-Ortega, J.; Gavilanes, J.G.; Martínez-del-Pozo, Á. The metamorphic transformation of a water-soluble monomeric protein into an oligomeric transmembrane pore. Adv. Biomembr. Lipid Self-Assembly 2017, 26, 51–97. [Google Scholar] [CrossRef]
- Mancheño, J.M.; Martín-Benito, J.; Martínez-Ripoll, M.; Gavilanes, J.G.; Hermoso, J.A. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 2003, 11, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Uechi, G.; Toma, H.; Arakawa, T.; Sato, Y. Molecular characterization on the genome structure of hemolysin toxin isoforms isolated from sea anemone Actineria villosa and Phyllodiscus semoni. Toxicon 2010, 56, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Monastyrnaya, M.; Leychenko, E.; Isaeva, M.; Likhatskaya, G.; Zelepuga, E.; Kostina, E.; Trifonov, E.; Nurminski, E.; Kozlovskaya, E. Actinoporins from the sea anemones, tropical Radianthus macrodactylus and northern Oulactis orientalis: Comparative analysis of structure-function relationships. Toxicon 2010, 56, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Tkacheva, E.S.; Leychenko, E.V.; Monastyrnaya, M.M.; Issaeva, M.P.; Zelepuga, E.A.; Anastuk, S.D.; Dmitrenok, P.S.; Kozlovskaya, E.P. New Actinoporins from Sea Anemone Heteractis crispa: Cloning and Functional Expression. Biochemistry 2011, 76, 1131–1139. [Google Scholar] [CrossRef]
- Wang, Y.; Yap, L.L.; Chua, K.L.; Khoo, H.E. A multigene family of Heteractis magnificalysins (HMgs). Toxicon 2008, 51, 1374–1382. [Google Scholar] [CrossRef]
- Anderluh, G.; Križaj, I.; Štrukelj, B.; Gubenšek, F.; Maček, P.; Pungerčar, J. Equinatoxins, pore-forming proteins from the sea anemone Actinia equina, belong to a multigene family. Toxicon 1999, 37, 1391–1401. [Google Scholar] [CrossRef]
- Valle, A.; Alvarado-Mesén, J.; Lanio, M.E.; Álvarez, C.; Barbosa, J.A.R.G.; Pazos, I.F. The multigene families of actinoporins (part I): Isoforms and genetic structure. Toxicon 2015, 103, 176–187. [Google Scholar] [CrossRef]
- Rivera-de-torre, E.; Palacios-ortega, J.; Slotte, J.P.; Gavilanes, J.G.; Martínez-del-Pozo, Á.; García-Linares, S. Functional and Structural Variation among Sticholysins, Pore-Forming Proteins from the Sea Anemone Stichodactyla helianthus. Int. J. Mol. Sci. 2020, 21, 8915. [Google Scholar] [CrossRef] [PubMed]
- Rivera-de-torre, E.; Martínez-del-pozo, Á.; Garb, J.E. Stichodactyla helianthus’ de novo transcriptome assembly: Discovery of a new actinoporin isoform. Toxicon 2018, 150, 105–114. [Google Scholar] [CrossRef]
- Huerta, V.; Morera, V.; Guanche, Y.; Chinea, G.; González, L.J.; Betancourt, L.; Martínez, D.; Alvarez, C.; Lanio, M.E.; Besada, V. Primary structure of two cytolysin isoforms from Stichodactyla helianthus differing in their hemolytic activity. Toxicon 2001, 39, 1253–1256. [Google Scholar] [CrossRef]
- Lanio, M.E.; Morera, V.; Alvarez, C.; Tejuca, M.; Gómez, T.; Pazos, F.; Besada, V.; Martínez, D.; Huerta, V.; Padrón, G.; et al. Purification and characterization of two hemolysins from Stichodactyla helianthus. Toxicon 2000, 39, 187–194. [Google Scholar] [CrossRef]
- Tejuca, M.; Dalla Serra, M.; Ferreras, M.; Lanio, M.E.; Menestrina, G. Mechanism of membrane permeabilization by sticholysin I, a cytolysin isolated from the venom of the sea anemone Stichodactyla helianthus. Biochemistry 1996, 35, 14947–14957. [Google Scholar] [CrossRef] [PubMed]
- Tejuca, M.; Dalla Serra, M.; Potrich, C.; Alvarez, C.; Menestrina, G. Sizing the radius of the pore formed in erythrocytes and lipid vesicles by the toxin sticholysin I from the sea anemone Stichodactyla helianthus. J. Membr. Biol. 2001, 183, 125–135. [Google Scholar] [CrossRef]
- Belmonte, G.; Pederzolli, C.; Macek, P.; Menestrina, G. Pore Formation by the Sea Anemone Cytolysin Equinatoxin II in Red Blood Cells and Model Lipid Membranes. J. Membr. Biol. 1993, 131, 11–22. [Google Scholar] [CrossRef]
- Anderluh, G.; Barlic, A.; Potrich, C.; Macek, P.; Menestrina, G. Lysine 77 is a Key Residue in Aggregation of Equinatoxin II, a Pore-forming Toxin from Sea Anemone Actinia equina. J. Membr. Biol. 2000, 173, 47–55. [Google Scholar] [CrossRef]
- Mechaly, A.E.; Bellomio, A.; Gil-Cartón, D.; Morante, K.; Valle, M.; González-Mañas, J.M.; Guerin, D.M.A. Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure 2011, 19, 181–191. [Google Scholar] [CrossRef] [Green Version]
- García-Linares, S.; Castrillo, I.; Bruix, M.; Menéndez, M.; Alegre-Cebollada, J.; Martínez-del-Pozo, Á.; Gavilanes, J.G. Three-dimensional structure of the actinoporin sticholysin I. influence of long-distance effects on protein function. Arch. Biochem. Biophys. 2013, 532, 39–45. [Google Scholar] [CrossRef]
- Morante, K.; Bellomio, A.; Viguera, A.R.; Gonzalez-Manas, J.M.; Tsumoto, K.; Caaveiro, J.M.M. The Isolation of New Pore-Forming Toxins from the Sea Anemone Actinia fragacea Provides Insights into the Mechanisms of Actinoporin Evolution. Toxins 2019, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Hong, Q.; Gutiérrez, I.; Barlic, A.; Malovrh, P.; Kristan, K.; Podlesek, Z.; Macek, P.; Turk, D.; González-Mañas, J.M.; Lakey, J.H.; et al. Two-step Membrane Binding by Equinatoxin II, a Pore-forming Toxin from the Sea Anemone, Involves an Exposed Aromatic Cluster and a Flexible Helix. J. Biol. Chem. 2002, 277, 41916–41924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakrac, B.; Gutiérrez-Aguire, I.; Podlesek, Z.; Sonnen, A.F.P.; Gilbert, R.J.C.; Macek, P.; Lakey, J.H.; Anderluh, G. Molecular Determinants of Sphingomyelin Specificity of a Eukaryotic Pore-forming Toxin. J. Biol. Chem. 2008, 283, 18665–18677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, C.; Pazos, F.; Soto, C.; Laborde, R.; Lanio, M.E. Pore-forming toxins from sea anemones: From protein membrane interaction to its implications for developing biomedical applications. In Advances in Biomembranes and Lipid Self-Assembly; Iglic, A., Rappolt, M., Garcia-Saez, A.J., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 31, pp. 129–183. ISBN 978-0-12-820967-7. [Google Scholar]
- Malovrh, P.; Viero, G.; Dalla Serra, M.; Podlesek, Z.; Lakey, J.H.; Macek, P.; Menestrina, G.; Anderluh, G. A novel mechanism of pore formation: Membrane penetration by the N-terminal amphipathic region of equinatoxin. J. Biol. Chem. 2003, 278, 22678–22685. [Google Scholar] [CrossRef] [Green Version]
- Rojko, N.; Dalla, M.; Maček, P.; Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta Biomembr. 2016, 1858, 446–456. [Google Scholar] [CrossRef]
- Menestrina, G.; Cabiaux, V.; Tejuca, M. Secondary structure of sea anemone cytolysins in soluble and membrane bound form by infrared spectroscopy. Biochem. Biophys. Res. Commun. 1999, 254, 174–180. [Google Scholar] [CrossRef]
- Alvarez, C.; Casallanovo, F.; Shida, C.; Nogueira, L.; Martinez, D.; Tejuca, M.; Pazos, I.F.; Lanio, M.E.; Menestrina, G.; Lissi, E.; et al. Binding of sea anemone pore-forming toxins sticholysins I and II to interfaces—Modulation of conformation and activity, and lipid-protein interaction. Chem. Phys. Lipids 2003, 122, 97–105. [Google Scholar] [CrossRef]
- Alegre-Cebollada, J.; Oñaderra, M.; Gavilanes, J.G.; del Pozo, M. Sea anemone actinoporins: The transition from a folded soluble state to a functionally active membrane-bound oligomeric pore. Curr. Protein Pept. Sci. 2007, 8, 558–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojko, N.; Kristan, K.Č.; Viero, G.; Žerovnik, E.; Maček, P.; Dalla Serra, M.; Anderluh, G. Membrane damage by an α-helical pore-forming protein, equinatoxin II, proceeds through a succession of ordered steps. J. Biol. Chem. 2013, 288, 23704–23715. [Google Scholar] [CrossRef] [Green Version]
- Antonini, V.; Pérez-Barzaga, V.; Bampi, S.; Pentón, D.; Martínez, D.; Dalla Serra, M.; Tejuca, M. Functional Characterization of Sticholysin I and W111C Mutant Reveals the Sequence of the Actinoporin’s Pore Assembly. PLoS ONE 2014, 9, e110824. [Google Scholar] [CrossRef] [Green Version]
- Morante, K.; Bellomio, A.; Gil-carto, D.; Redondo-morata, L.; Scheuring, S.; Valle, M.; Gonza, J.M.; Tsumoto, K. Identification of a Membrane-bound Prepore Species Clarifies the Lytic Mechanism of Actinoporins. J. Biol. Chem. 2016, 291, 19210–19219. [Google Scholar] [CrossRef] [Green Version]
- Palacios-ortega, J.; Rivera-de-Torre, E.; Garc, S.; Gavilanes, J.G.; Mart, A.; Slotte, J.P. Oligomerization of Sticholysins from Förster Resonance Energy Transfer. Biochemistry 2021, 60, 314–323. [Google Scholar] [CrossRef]
- Hervis, Y.P.; Valle, A.; Dunkel, S.; Klare, J.P.; Canet, L.; Lanio, M.E.; Alvarez, C.; Pazos, I.F.; Steinhoff, H.-J. Architecture of the pore forming toxin sticholysin I in membranes. J. Struct. Biol. 2019, 208, 30–42. [Google Scholar] [CrossRef]
- Subburaj, Y.; Ros, U.; Hermann, E.; Tong, R.; García-Sáez, A.J. Toxicity of an α-pore-forming toxin depends on the assembly mechanism on the target membrane as revealed by single molecule imaging. J. Biol. Chem. 2015, 290, 4856–4865. [Google Scholar] [CrossRef] [Green Version]
- Valle, A.; López-Castilla, A.; Pedrera, L.; Martínez, D.; Tejuca, M.; Campos, J.; Fando, R.; Lissi, E.; Alvarez, C.; Lanio, M.E.; et al. Cys mutants in functional regions of Sticholysin I clarify the participation of these residues in pore formation. Toxicon 2011, 58, 8–17. [Google Scholar] [CrossRef]
- Mesa-Galloso, H.; Delgado-Magnero, K.H.; Cabezas, S.; López-Castilla, A.; Hernández-González, J.E.; Pedrera, L.; Alvarez, C.; Peter Tieleman, D.; García-Sáez, A.J.; Lanio, M.E.; et al. Disrupting a key hydrophobic pair in the oligomerization interface of the actinoporins impairs their pore-forming activity. Protein Sci. 2017, 26, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Tejuca, M.; Alvarez, C.; Lanio, M.E.; Pazos, I.F. Effect of different factors on the hemolytic activity of a cytolysin from Stichodactyla helianthus. Biologia 1994, 8, 1–5. [Google Scholar]
- Soto, C.; Bergado, G.; Blanco, R.; Griñán, T.; Rodríguez, H.; Ros, U.; Pazos, F.; Lanio, M.E.; Hernández, A.M.; Alvarez, C. Sticholysin II-mediated cytotoxicity involves the activation of regulated intracellular responses that anticipates cell death. Biochimie 2018, 148, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.D.; Mateo de Acosta, C.; Lage, A. A new immunotoxin built by linking a hemolytic toxin to a monoclonal antibody specific for immature T lymphocytes. Int. J. Cancer 1988, 42, 568–571. [Google Scholar] [CrossRef]
- Avila, A.D.; Mateo de Acosta, C.; Lage, A. A carcinoembryonic antigen-directed immunotoxin built by linking a monoclonal antibody to a hemolytic toxin. Int. J. Cancer 1989, 43, 926–929. [Google Scholar] [CrossRef]
- Tejuca, M.; Díaz, I.; Figueredo, R.; Roque, L.; Pazos, F.; Martínez, D.; Iznaga-Escobar, N.; Pérez, R.; Alvarez, C.; Lanio, M.E. Construction of an immunotoxin with the pore forming protein StI and ior C5, a monoclonal antibody against a colon cancer cell line. Int. Immunopharmacol. 2004, 4, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Menestrina, G. Pore-Forming Proteins from Sea Anemones and the Construction of Immunotoxins for Selective Killing of Harmful Cells in Bio-Organic Compounds: Chemistry and Biomedical Applications; Fingerman, M., Ed.; Science Publishers, Inc: Enfield, NH, USA, 2001; pp. 131–148. [Google Scholar]
- Fedorov, S.; Dyshlovoy, S.; Monastyrnaya, M.; Shubina, L.; Leychenko, E.; Kozlovskaya, E.; Jin, J.; Kwak, J.; Bode, A.M.; Dong, Z.; et al. The anticancer effects of actinoporin RTX-A from the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 2010, 55, 811–8177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvetkina, A.; Malyarenko, O.; Pavlenko, A.; Dyshlovoy, S.; von Amsberg, G.; Ermakova, S.; Leychenko, E. Sea Anemone Heteractis crispa Actinoporin Demonstrates In Vitro Anticancer Activities and Prevents HT-29 Colorectal Cancer Cell Migration. Molecules 2020, 25, 5979. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Mesén, J.; Solano-Campos, F.; Canet, L.; Pedrera, L.; Hervis, Y.P.; Soto, C.; Borbón, H.; Lanio, M.E.; Lomonte, B.; Valle, A.; et al. Cloning, purification and characterization of nigrelysin, a novel actinoporin from the sea anemone Anthopleura nigrescens. Biochimie 2019, 156, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Campos, A.M.; Pazos, F.; Alvarez, C.; Lanio, M.E.; Casallanovo, F.; Schreier, S.; Salinas, R.K.; Vergara, C.; Lissi, E. Properties of St I and St II, two isotoxins isolated from Stichodactyla helianthus: A comparison. Toxicon 2001, 39, 1547–1560. [Google Scholar] [CrossRef]
- Maček, P.; Belmonte, G.; Pederzolli, C.; Menestrina, G. Mechanism of action of equinatoxin II, a cytolysin from the sea anemone Actinia equina L. belonging to the family of actinoporins. Toxicology 1994, 87, 205–227. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Molnar, A.A.; Kozlovskaya, E.P.; Monastyrnaya, M.M. The action of toxin from Radianthus macrodactylus on biological and model membrane permeability. Biol. Membr. 1987, 4, 243–248. [Google Scholar]
- Zorec, R.; Tester, M.; Macek, P.; Mason, W. Cytotoxicity of Equinatoxin II from the Sea Anemone Actinia equina Involves Ion Channel Formation and an Increase in Intracellular Calcium Activity. J. Membr. Biol. 1990, 118, 243–249. [Google Scholar] [CrossRef]
- Celedon, G.; Venegas, F.; Campos, A.M.; Lanio, M.E.; Martinez, D.; Soto, C.; Alvarez, C.; Lissi, E. Role of endogenous channels in red blood cells response to their exposure to the pore forming toxin Sticholysin II. Toxicon 2005, 46, 297–307. [Google Scholar] [CrossRef]
- Celedón, G.; González, G.; Lissi, E.; Cerda, T.; Martínez, D.; Soto, C.; Pupo, M.; Pazos, F.; Lanio, M.E.; Alvarez, C. Effect of calcium on the hemolytic activity of Stichodactyla helianthus toxin sticholysin II on human erythrocytes. Toxicon 2009, 54, 845–850. [Google Scholar] [CrossRef]
- García-Sáez, A.J.; Buschhorn, S.B.; Keller, H.; Anderluh, G.; Simons, K.; Schwille, P. Oligomerization and Pore Formation by Equinatoxin II Inhibit Endocytosis and Lead to Plasma Membrane Reorganization. J. Biol. Chem. 2011, 286, 37768–37777. [Google Scholar] [CrossRef] [Green Version]
- Celedón, G.; González, G.; Barrientos, D.; Pino, J.; Venegas, F.; Lissi, E.A.; Soto, C.; Martinez, D.; Alvarez, C.; Lanio, M.E. Stycholysin II, a cytolysin from the sea anemone Stichodactyla helianthus promotes higher hemolysis in aged red blood cells. Toxicon 2008, 51, 1383–1390. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bischofberger, M.; Frêche, B.; Ho, S.; Parton, R.G.; Goot, F.G. Pore-forming toxins induce multiple cellular responses promoting survival. Cell. Microbiol. 2011, 13, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of Pore-Forming Toxins in Bacterial Infectious Diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Abe, Y.; Maeda, T.; Shimada, H. Parasporin-2 requires GPI-anchored proteins for the efficient cytocidal action to human hepatoma cells. Toxicology 2009, 264, 80–88. [Google Scholar] [CrossRef]
- Barros, L.F.; Kanaseki, T.; Sabirov, R.; Morishima, S.; Castro, J.; Bittner, C.X.; Maeno, E.; Ando-Akatsuka, Y.; Okada, Y. Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ. 2003, 10, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bergman, A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 10, e1005337. [Google Scholar] [CrossRef]
- Gonzalez-Juarbe, N.; Bradley, K.M.; Riegler, A.N.; Reyes, L.F.; Brissac, T.; Park, S.S.; Restrepo, M.I.; Orihuela, C.J. Bacterial Pore-Forming Toxins Promote the Activation of Caspases in Parallel to Necroptosis to Enhance Alarmin Release and Inflammation during Pneumonia. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, N.C.; Doronin, K.; Baldwin, L.K.; Papayannopoulou, T.; Shayakhmetov, D.M. The Transcription Factor IRF3 Triggers “Defensive Suicide” Necrosis in Response to Viral and Bacterial Pathogens. Cell Rep. 2013, 3, 1840–1846. [Google Scholar] [CrossRef] [Green Version]
- McNeela, E.A.; Burke, Á.; Neill, D.R.; Baxter, C.; Fernandes, V.E.; Ferreira, D.; Smeaton, S.; El-Rachkidy, R.; McLoughlin, R.M.; Mori, A.; et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Soletti, R.C.; Alves, T.; Vernal, J.; Terenzi, H.; Anderluh, G.; Borges, H.L.; Gabilan, N.H.; Moura-Neto, V. Inhibition of MAPK/ERK, PKC and CaMKII Signaling Blocks Cytolysin-induced Human Glioma Cell Death. Anticancer Res. 2010, 30, 1209–1215. [Google Scholar] [PubMed]
- Knapp, O.; Maier, E.; Mkaddem, S.B.; Benz, R.; Bens, M.; Chenal, A.; Geny, B.; Vandewalle, A.; Popoff, M.R. Clostridium septicum α-toxin forms pores and induces rapid cell necrosis. Toxicon 2010, 55, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotze, E.M.; Tweten, R.K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. ActaBiomembr. 2012, 1818, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Skals, M.; Leipziger, J.; Praetorius, H.A. Haemolysis induced by α-toxin from Staphylococcus aureus requires P2X receptor activation. Pflugers Arch. 2011, 462, 669–679. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.; de Alemida, P.; Corrote, M. Damage Control: Cellular Mechanisms of Plasma Membrane Repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, A.J.; Perez, F. Plasma membrane repair: The adaptable cell life-insurance. Curr. Opin. Cell Biol. 2017, 47, 99–107. [Google Scholar] [CrossRef]
- Keefe, D.; Shi, L.; Feske, S.; Massol, R.; Navarro, F.; Kirchhausen, T.; Lieberman, J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walev, I.; Martin, E.; Jonas, D.; Mohamadzadeh, M.; Muller-Klieser, W.; Kunz, L.; Bhakdi, S. Staphylococcal α-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun. 1993, 61, 4972–4979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitzer, A.; Wassenaar, T.M.; Walev, I.; Bhakdi, S. Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect. Immun. 1997, 65, 1293–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, F.C.; Kao, C.-Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 2011, 9, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, K.J.; Horbaschek, M.; Lucas, K.; Zimmermann, U.; Sukhorukov, V.L. Electrotransfection of anchorage-dependent mammalian cells. Exp. Cell Res. 2003, 288, 344–353. [Google Scholar] [CrossRef]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus α-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (w)hole story? Cell. Mol.Life. Sci 2008, 65, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Wald, T.; Petry-Podgorska, I.; Fiser, R.; Matousek, T.; Dedina, J.; Osicka, R.; Sebo, P.; Masin, J. Quantification of potassium levels in cells treated with Bordetella adenylate cyclase toxin. Anal. Biochem. 2014, 450, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.L.; Kulkarni, R.; Randis, T.M.; Soman, S.; Kikuchi, A.; Yin, Y.; Ratner, A.J. Phosphatase-dependent regulation of epithelial mitogen-activated protein kinase responses to toxin-induced membrane pores. PLoS ONE 2009, 4, e8076. [Google Scholar] [CrossRef]
- Porta, H.; Cancino-Rodezno, A.; Soberon, M.; Bravo, A. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides 2011, 32, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.; Elion, E.A. MAP kinase pathways. J. Cell Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, M.; Nkhoma, P.; Mulder, N.; Martin, D.P. Integrated molecular characterisation of the MAPK pathways in human cancers reveals pharmacologically vulnerable mutations and gene dependencies. Commun. Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 2009, 385, 503–506. [Google Scholar] [CrossRef]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; Goot, F.G. Caspase-1 Activation of Lipid Metabolic Pathways in Response to Bacterial Pore- Forming Toxins Promotes Cell Survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Bischof, L.J.; Kao, C.; Los, F.C.O.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; Van Der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef]
- Chakravorty, A.; Awad, M.M.; Cheung, J.K.; Hiscox, T.J.; Lyras, D.; Rood, J.I. The pore-forming α-toxin from Clostridium septicum activates the MAPK pathway in a Ras-c-Raf-dependent and independent manner. Toxins 2015, 7, 516–534. [Google Scholar] [CrossRef]
- Soto, C.; De León, L.; Alvarado, J.; Álvarez, F.; Yglesias, A.; Rodríguez, H.; Blanco, R.; Lanio, M.E.; Pazos, I.; Hernández, A.M.; et al. Cell death mechanisms induced by pore forming toxins with special focus on actinoporins. Rev. Cub. C. Biol. 2020, 8, 1–22. [Google Scholar]
- Bischofberger, M.; Gonzalez, M.R.; Goot, F.G. Membrane injury by pore-forming proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The targeting of plasmalemmal ceramide to mitochondria during apoptosis. PLoS ONE 2011, 6, e23706. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Meunier, F.A.; Frangež, R.; Benoit, E.; Ouanounou, G.; Rouzaire-Dubois, B.; Šuput, D.; Molgó, J. Ca2+ and Na+ contribute to the swelling of differentiated neuroblastoma cells induced by equinatoxin-II. Toxicon 2000, 38, 1547–1560. [Google Scholar] [CrossRef]
- Migues, P.V.; Leal, R.B.; Mantovani, M.; Nicolau, M.; Gabilan, N.H. Synaptosomal glutamate release induced by the fraction Bc2 from the venom of the sea anemone Bunodosoma caissarum. Neuroreport 1999, 10, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2alpha Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef]
- Chao, T.S.; Byron, K.L.; Lee, K.M.; Villereal, M.; Rosner, M.R. Activation of MAP kinases by calcium-dependent and calcium-independent pathways. Stimulation by thapsigargin and epidermal growth factor. J. Biol. Chem. 1992, 267, 19876–19883. [Google Scholar] [CrossRef]
- Chuderland, D.; Marmor, G.; Shainskaya, A.; Seger, R. Calcium-Mediated Interactions Regulate the Subcellular Localization of Extracellular Signal-Regulated Kinases (ERKs). Cell. Physiol. Biochem. 2020, 54, 474–492. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 1–7. [Google Scholar] [CrossRef]
- Cruz-Leal, Y.; Grubaugh, D.; Nogueira, C.V.; Lopetegui-González, I.; del Valle, A.; Escalona, F.; Laborde, R.J.; Alvarez, C.; Fernández, L.; Starnbach, M.N.; et al. The Vacuolar Pathway in Macrophages Plays a Major Role in Antigen Cross-Presentation Induced by the Pore-Forming Protein Sticholysin II Encapsulated Into Liposomes. Front. Immunol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laborde, R.J.; Ishimura, M.E.; Abreu-Butin, L.; Nogueira, C.V.; Grubaugh, D.; Cruz-Leal, Y.; Luzardo, M.C.; Fernández, A.; Mesa, C.; Pazos, F.; et al. Sticholysins, pore-forming proteins from a marine anemone can induce maturation of dendritic cells through a TLR4 dependent-pathway. Mol. Immunol. 2021, 131, 144–154. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.A.; Tampe, R.; et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blander, J.M.; Medzhitov, R. Regulation of phagosome maturation by signals from toll-like receptors. Science 2004, 304, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M. Do mature red blood cells die by apoptosis? Trends Genet. 2002, 18, 9525. [Google Scholar] [CrossRef]
- Ávila, A.D.; Calderón, C.F.; Pérez, R.M.; Pons, C.; Pereda, C.M.; Ortiz, A. Construction of an immunotoxin by linking a monoclonal antibody against the human epidermal growth factor receptor and a hemolytic toxin. Biol. Res. 2007, 40, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Potrich, C.; Viero, G.; Tejuca, M.; Anderluh, G.; Macek, P.; Menestrina, G. Construction of new immunotoxins by linking equinatoxin II to monoclonal antibodies via the biotin–avidin interaction. Cytotoxic effects on human tumor cells. Acta Biol. Slov. 2000, 43, 47–51. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, C.; Soto, C.; Cabezas, S.; Alvarado-Mesén, J.; Laborde, R.; Pazos, F.; Ros, U.; Hernández, A.M.; Lanio, M.E. Panorama of the Intracellular Molecular Concert Orchestrated by Actinoporins, Pore-Forming Toxins from Sea Anemones. Toxins 2021, 13, 567. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080567
Alvarez C, Soto C, Cabezas S, Alvarado-Mesén J, Laborde R, Pazos F, Ros U, Hernández AM, Lanio ME. Panorama of the Intracellular Molecular Concert Orchestrated by Actinoporins, Pore-Forming Toxins from Sea Anemones. Toxins. 2021; 13(8):567. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080567
Chicago/Turabian StyleAlvarez, Carlos, Carmen Soto, Sheila Cabezas, Javier Alvarado-Mesén, Rady Laborde, Fabiola Pazos, Uris Ros, Ana María Hernández, and María Eliana Lanio. 2021. "Panorama of the Intracellular Molecular Concert Orchestrated by Actinoporins, Pore-Forming Toxins from Sea Anemones" Toxins 13, no. 8: 567. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080567