Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea

1
Synthetic Bioengineering Research Group, Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 6, 1-1-1 Higashi, Tsukuba 305-8566, Japan
2
United Graduate School of Drug Discovery and Medical Information Sciences, Gifu University, 1-1 Yanagido, Gifu 501-1193, Japan
Toxins 2021, 13(9), 621; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13090621
Submission received: 12 July 2021
/
Revised: 29 August 2021
/
Accepted: 1 September 2021
/
Published: 3 September 2021
(This article belongs to the Special Issue Toxinologic and Pharmacological Investigation of Venomous Arthropods)
Abstract
:Inhibitor cystine knot (ICK) peptides are knotted peptides with three intramolecular disulfide bonds that affect several types of ion channels. Some are proteolytically stable and are promising scaffolds for drug development. GTx1-15 is an ICK peptide that inhibits the voltage-dependent calcium channel Cav3.1 and the voltage-dependent sodium channels Nav1.3 and Nav1.7. As a model molecule to develop an ICK peptide drug, we investigated several important pharmaceutical characteristics of GTx1-15. The stability of GTx1-15 in rat and human blood plasma was examined, and no GTx1-15 degradation was observed in either rat or human blood plasma for 24 h in vitro. GTx1-15 in blood circulation was detected for several hours after intravenous and intramuscular administration, indicating high stability in plasma. The thermal stability of GTx1-15 as examined by high thermal incubation and protein thermal shift assays indicated that GTx1-15 possesses high heat stability. The cytotoxicity and immunogenicity of GTx1-15 were examined using the human monocytic leukemia cell line THP-1. GTx1-15 showed no cytotoxicity or immunogenicity even at high concentrations. These results indicate that GTx1-15 itself is suitable for peptide drug development and as a peptide library scaffold.
Key Contribution: The inhibitor cystine knot peptide GTx1-15 is thermally stable, stable in plasma in vitro and in vivo and shows no detectable cytotoxicity or antigenicity. It is thus a suitable lead molecule for the development of ion channel-blocking drugs.
1. Introduction
Ion channels play essential roles in a wide range of biological processes (including neural function, muscle contraction and circulation in many physiological systems [1]) and have become attractive pharmaceutical targets related to various conditions such as hypertension, diabetes, pain and cancer [2,3,4,5].
Bioactive peptides from various venomous animals serve as a basis for peptide drug development; examples include ziconotide from cone snails for pain control [6] and exendin-4 from the Gila monster lizard for diabetes treatment [7]. Spider venoms contain various types of bioactive peptides that have attracted attention as peptide drug candidates [8,9,10,11,12,13,14].
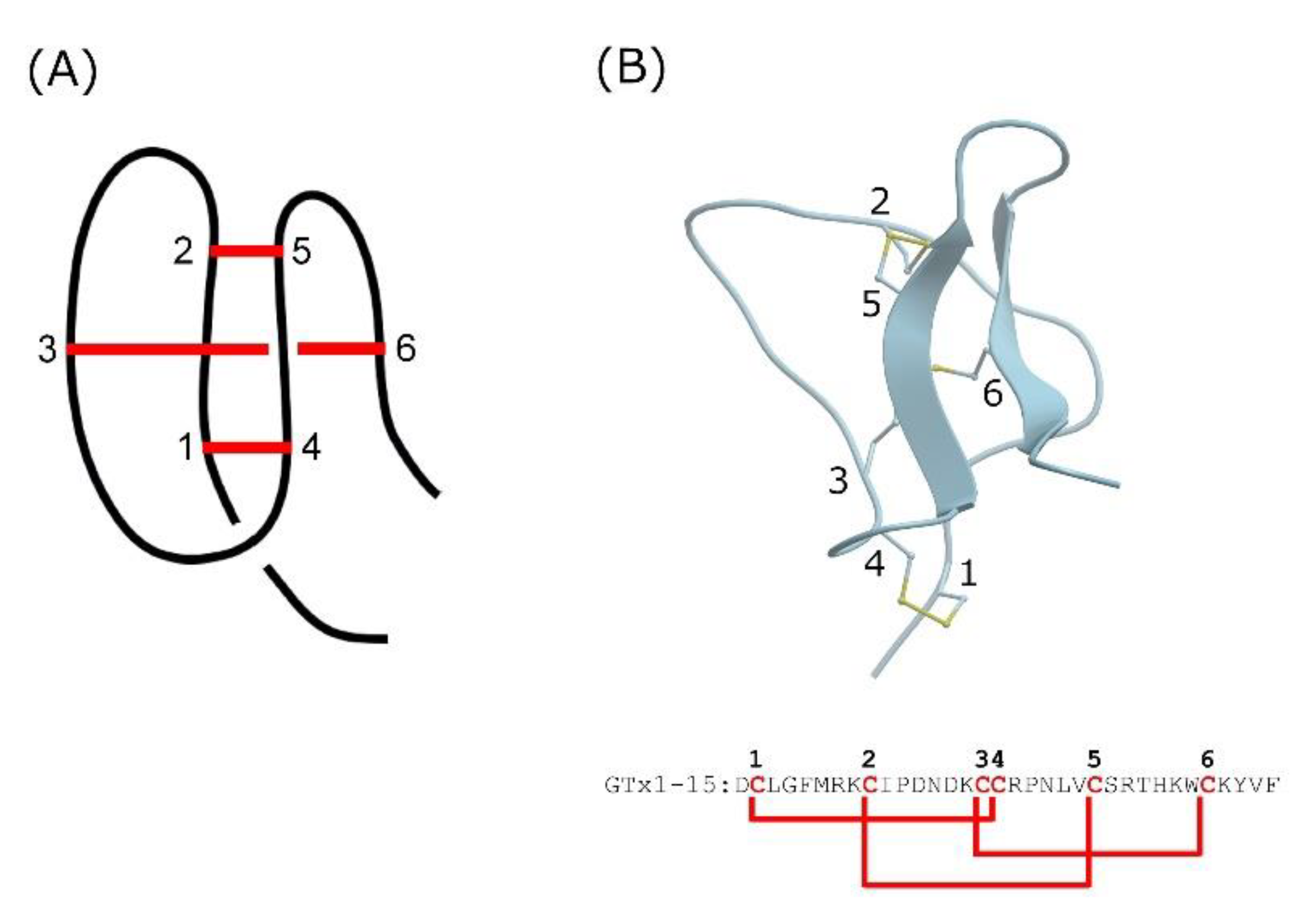
Short peptides cannot form structural motifs and are likely to be readily degraded by proteases. Knottins, also known as cystine-knot mini-proteins, are characteristic structural motifs due to multiple covalent bonds formed between side chain thiols of cysteine residues that are spatially distant from each other [15]. Knottins have a compact spherical structure with a very small hydrophobic core due to these disulfide bonds (Figure 1A). The knotted rigid structure is a common feature of knottins and is thought to endow them with unusual proteolytic, thermal and chemical stability [16,17].
Inhibitor cystine knot (ICK) peptides are a class of knottins, which are known to affect several types of ion channel, including voltage-dependent potassium, sodium and calcium channels, as well as mechanosensitive channels [12,18,19]. GTx1-15 is an ICK peptide identified from venom of the tarantula Grammostola rosea [20] and thought to have a compact structure due to intramolecular disulfide bonds (Figure 1B). GTx1-15 inhibits both the low-voltage activated calcium channel Cav3.1 [20] and sodium channels Nav1.3 and Nav1.7 [21].
We previously reported the proteolytic resistance of several ICK peptides such as ProTxI, ProTxII, GsMTx4 and GTx1-15 [22]. GTx1-15 is not degraded by pepsin, trypsin, chymotrypsin and elastase. Based on the proteolytic enzyme resistance of GTx1-15, it is conceivable that GTx1-15 could be used as a template for the development of drugs targeting the gastrointestinal tract. For example, linaclotide is an ICK peptide that targets the adenylate cyclase 2C receptor in the gastrointestinal tract for the treatment of irritable bowel syndrome with constipation [23].
Here, to clarify the eligibility of GTx1-15 as a template for drug development, we report the stability of GTx1-15 in rat and human plasma and its thermal stability. In addition to its stability in vitro, GTx1-15 concentrations in circulating blood after intravenous (i.v.), intramuscular (i.m.) and oral administration were also monitored in vivo. Furthermore, the cytotoxicity and antigenicity of GTx1-15 were determined.
2. Results
2.1. GTx1-15 Is Not Degraded in Plasma
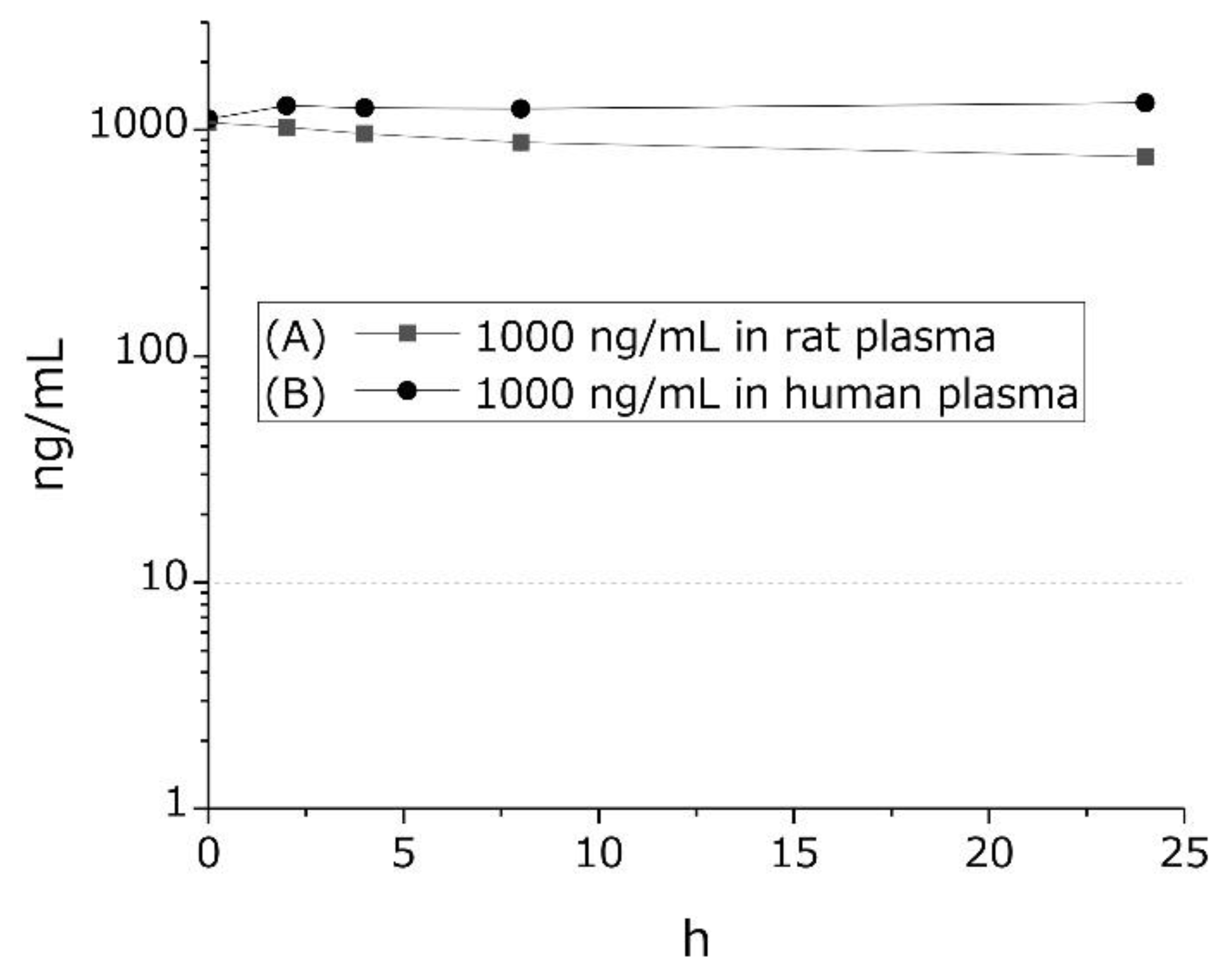
We examined GTx1-15 degradation in rat and human blood plasma. In vitro, 1 μg/mL GTx1-15 was mixed with rat or human blood plasma and incubated at 37 °C for 24 h. In rat plasma, GTx1-15 was only slightly degraded after a 24 h incubation (Figure 2A), while in human plasma, GTx1-15 was not degraded at all after 24 h (Figure 2B).
2.2. GTx1-15 Is Stable in Circulation
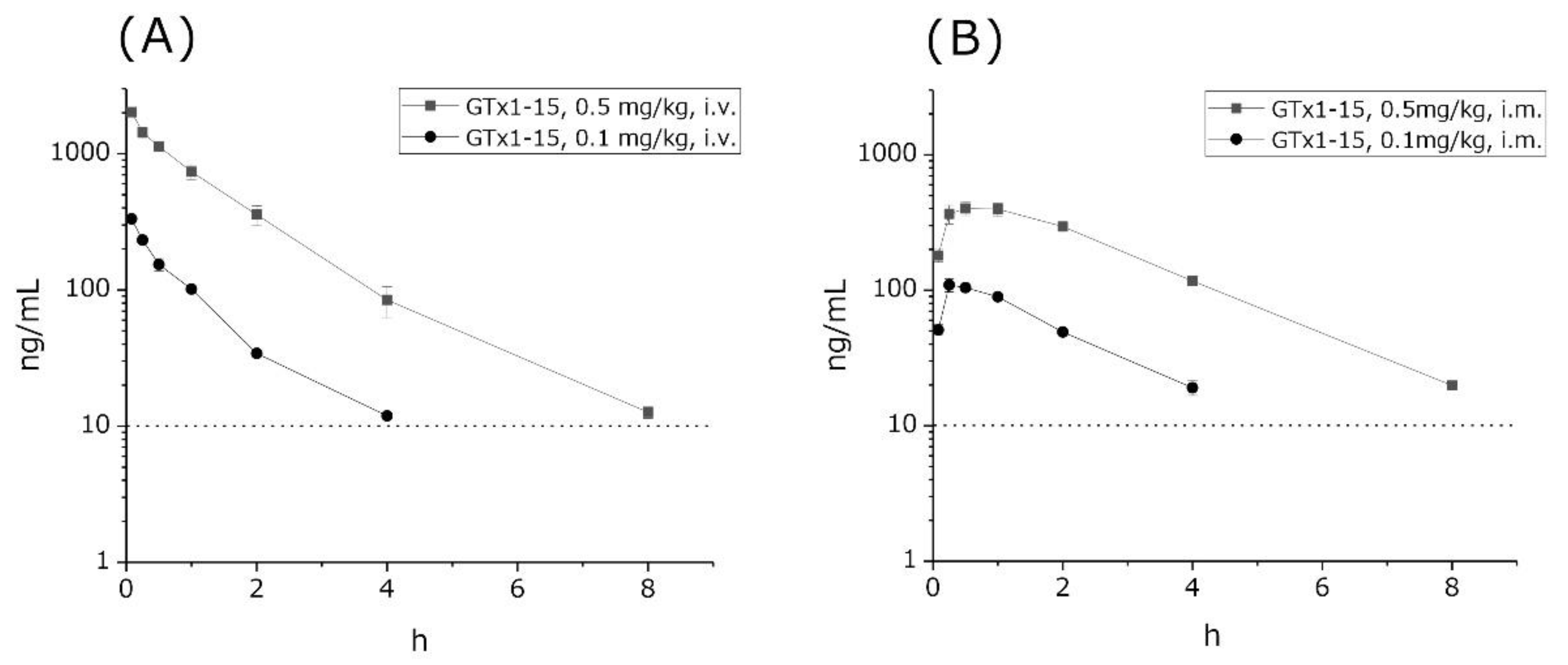
The concentrations of GTx1-15 in circulation were investigated using rats. 0.1 or 0.5 mg/kg GTx1-15 was administered to rats in the femoral vein, via i.m. injection, or orally. Then, we observed GTx1-15 concentrations in blood over 24 h. GTx1-15 concentrations in circulation blood gradually decreased and kept dropping to the detection limit (<10.0 ng/mL) dose-dependently within 4 and 8 h after both i.v. (Figure 3A) and i.m. administration (Figure 3B). After peroral administration with 1 or 5 mg/kg of GTx1-15, it was not detected in blood circulation at any time (data not shown). The half-lives of intravenous GTx1-15 in the blood were 30 and 40 min for the 0.1 mg/kg and 0.5 mg/kg doses, respectively. Intramuscular doses of 0.1 mg/kg and 0.5 mg/kg of GTx1-15 showed the highest blood concentrations at 15 and 30 min, respectively, with half-lives of 2 and 3 h.
2.3. GTx1-15 Is Stable in High-Temperature Environments
Figure 4 indicates that GTx1-15 showed no degradation between 20 °C and 75 °C and was degraded by about 30% with 95 °C incubation for 24 h. Protein thermal shift assays indicated that the Tm B (Boltzmann Tm) values of GTx1-15 and other ICK peptides (ProTxI, ProTxII and GsMTx4) were about 95 °C (Table 1), which means that these ICK peptides show no three-dimensional structural changes in high-temperature environments.
2.4. GTx1-15 Shows No Cytotoxicity or Antigenicity
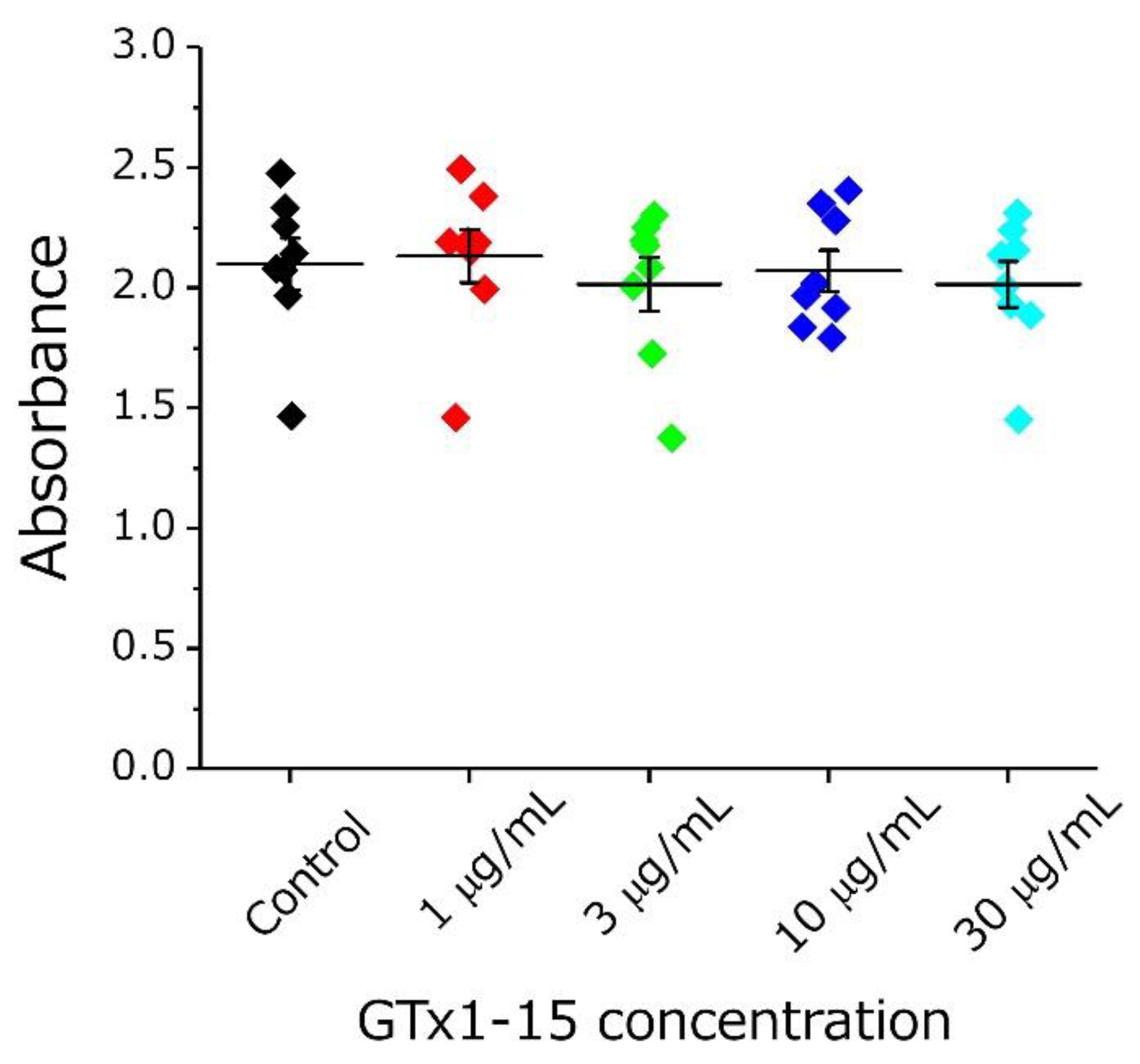
First, the cytotoxicity of GTx1-15 was evaluated using THP-1 cells (a human monocytic leukemia cell line). At all concentrations examined (1, 3, 10 and 30 μg/mL), GTx1-15 had no cytotoxicity (Figure 5).
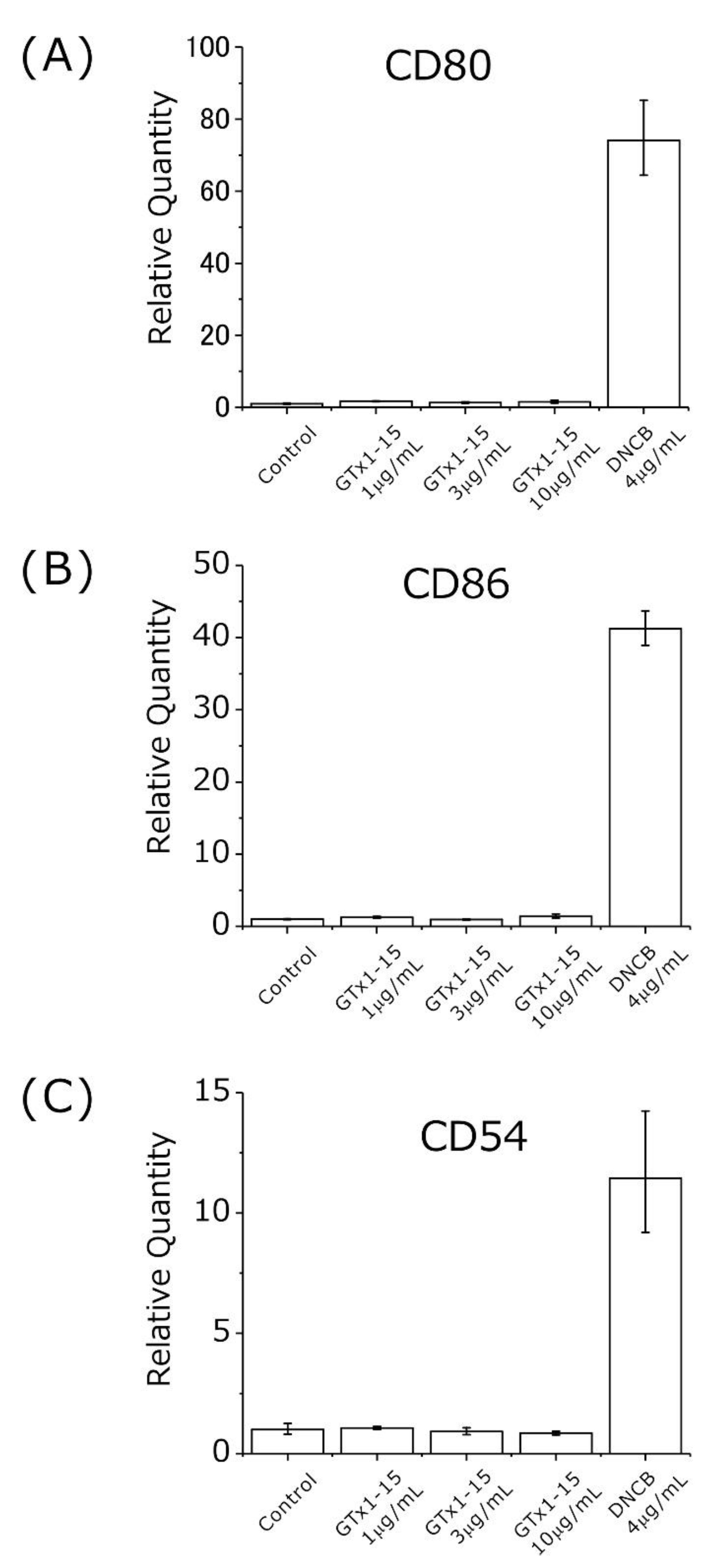
Second, the antigenicity of GTx1-15 was evaluated using THP-1 cells at concentrations of 1, 3 and 10 μg/mL. The concentration of 30 μg/mL used in cytotoxicity assays is too high compared with its effective concentration (30 μg/mL: 7.36 μM; Cav3.1 30% inhibition, 40 ng/mL: 9.8 nM (×751); Nav1.3 50% inhibition, 1.2 μg/mL: 300 nM (×25); Nav1.7 50% inhibition, 102 ng/mL: 25 nM (×294)), so the test concentration of 30 μg/mL in the antigenicity assay was omitted. 2,4-Dinitrochlorobenzene (DNCB) is known as a strong allergen and is used in skin sensitization tests. DNCB induces the expression of CD80/CD86 and CD54 when exposed to THP-1 cells [24,25]. Using DNCB as a positive control, approximately 20–70 times higher expressions of CD80, CD86 and CD54 genes were detected after 24 h of exposure (Figure 6). With 1, 3 and 10 μg/mL of GTx1-15, no expression changes of CD80, CD86 and CD54 genes were detected after 24 h of exposure (Figure 6).
3. Discussion
3.1. The GTx1-15 Molecule Shows High Stability
GTx1-15 concentrations in circulation gradually decreased and kept dropping within 4 and 8 h dose-dependently, meaning that GTx1-15 is stable in blood circulation, which is supported by a previous report showing that GTx1-15 has proteolytic resistance, i.e., GTx1-15 is not degraded by trypsin, chymotrypsin, pepsin or elastase [22].
GTx1-15 inhibits 50% of Nav1.3 current at 300 nM and 50% of Nav1.7 current at 25 nM [21]. GTx1-15 also inhibits 30% of Cav3.1 current at 9.8 nM in Xenopus oocyte two-electrode voltage clamp experiments [20]. Approximately, 9.8 nM of GTx1-15 is equivalent to 40 ng/mL. Four or eight hours after i.v. or i.m. injection, the concentration of GTx1-15 was about 10–20 ng/mL, i.e., 4–8 nM. GTx1-15 might be effective against the target molecules Nav1.7 and Cav3.1 for several hours after i.v. and i.m. injection. Since ICK peptides are thought to act by inserting their hydrophobic side into the cell membrane and binding to the ion channel voltage sensor domain on their hydrophilic side [18,26], it is thought that they remain in the cell membranes of the target tissue even after 8 h.
We previously reported that ProTxII (an ICK peptide) remains in circulation for over 4 h [22]. Similar to those results, GTx1-15 retains an effective concentration in circulation for over several hours. Both ProTxII and GTx1-15 are ICK peptides, indicating that ICK peptides might be stable in plasma and are a promising scaffold for peptide drug development.
After peroral administration of GTx1-15, it was not detected in blood circulation at any time. The limit of quantification of GTx1-15 was 10 ng/mL in this experiment. If GTx1-15 absorption from the gastrointestinal tract is less than this limit, then it would not have been measurable by LC-MS/MS. Some ICK peptides have been shown to permeate through rat intestinal mucosa better than other model drugs [27]. It might be that GTx1-15 is absorbed from the gastrointestinal tract at very low concentrations that are under the limit of quantification. To detect very low concentrations of GTx1-15 in circulation after peroral administration, more sensitive peptide detection methods, such as the use of radio-iodinated GTx1-5, are required. Using iodinated GTx1-15, it would be possible to show the absorption of GTx1-15 from the intestinal tract and its permeability to the brain. Ziconotide (Prialt), which is an ICK peptide from cone snails and is already on the market, is known to transfer to the brain [28]. On the other hand, it is possible that GTX1-15 is not absorbed from the intestinal tract at all. In that case, GTx1-15 and its derivatives could be developed as drugs that target molecules present in the intestinal tract such as linaclotide [23] or used as lead compounds for drugs with improved absorption from the intestinal tract. Modification of GTx1-15 with reference to chlorotoxin, which is used for targeting glioblastoma [29], and crotamine, a cell membrane-permeable peptide [30], may improve intestinal absorption.
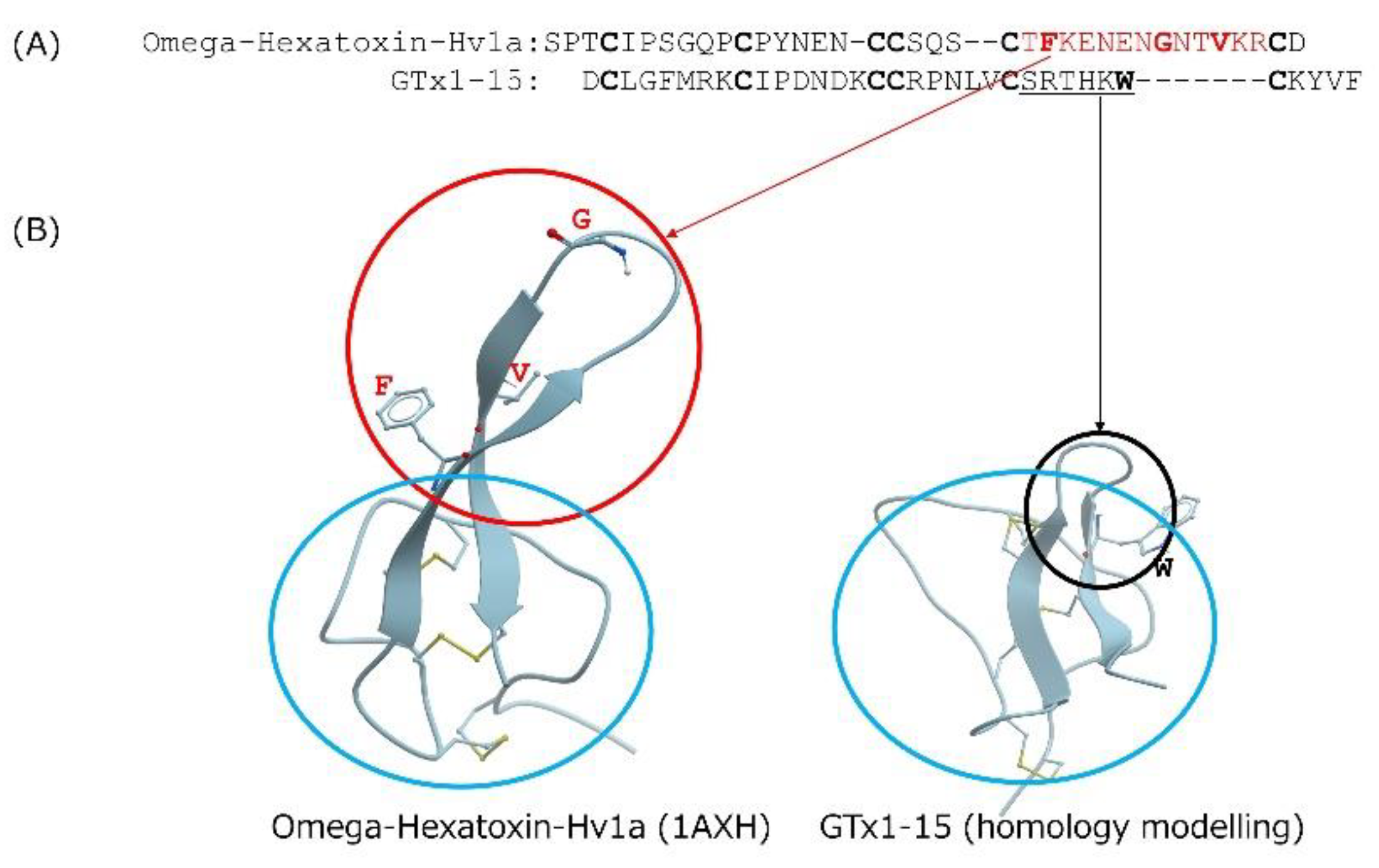
Cyclotides are cystine-knotted peptides in which the N- and C-termini are cyclized. Kalata B1 is a well-known cyclotide that consists of 29 amino acids and is not degraded after heating to temperatures approaching boiling [15]. The insecticidal spider toxin ω-hexatoxin-Hv1a is an ICK peptide whose thermal stability has been investigated [15]. After incubating it for 24 h at 50 °C, 75 °C or 95 °C, ω-hexatoxin-Hv1a is degraded about 20%, 30% and 90%, respectively. In contrast, GTx1-15 degraded only 30% after a 24-h incubation at 95 °C. GTx1-15 thus shows higher thermal stability than ω-hexatoxin-Hv1a. In comparing the amino acid sequences of GTx1-15 and ω-hexatoxin-Hv1a (Figure 7A), the c-terminal region of ω-hexatoxin-Hv1a has a long loop region compared to GTx1-15 (Figure 7B). The long loop of ω-hexatoxin-Hv1a protrudes from the main body consisting of three disulfide bonds, which is different from the compact structure of GTx1-15, and this protrusion is thought to cause instability at high temperatures by forming aggregates. This might be why ω-hexatoxin-Hv1a is more unstable than GTx1-15 at high temperatures.
3.2. GTx1-15 Is Safe with No Cytotoxicity or Antigenicity
It widely accepted that little or no cytotoxicity or antigenicity of peptide drugs is necessary for their safe use. First, we evaluated the cytotoxicity of GTx1-15 using THP-1 cells (a human monocytic leukemia cell line) and a WST-1 cell proliferation assay kit. We quantified the cell proliferation activity and viability to evaluate the cytotoxicity. As a result, GTx1-15 showed no cytotoxicity at any concentration tested.
Second, to clarify whether GTx1-15 is immunogenic, its antigenicity was evaluated using THP-1 cells at concentrations of 1, 3 and 10 μg/mL by detecting CD80, CD86 and CD54 expression using RT-PCR quantification. CD80 and CD86 are expressed in dendritic cells, monocytes and antigen-presenting cells such as macrophages. These molecules bind with CD28 or CD152 expressed in T cells in the case of antigen presentation and participate in the activation or inhibition of T cells as a costimulatory pathway [31]. In contrast, although CD54 binds with CD11/CD18 of T cells and contributes to the costimulatory pathway, CD54 is expresses in various cells such as vascular endothelial cells as well as antigen-presenting cells. These molecules work as adhesion factors and are important in lymphocyte migration [32]. Among these molecules, CD86 and CD54 are used as molecular markers for antigenicity tests [25,25,33]. A GTx1-15 concentration of 30 μg/mL is equivalent to 7.5 μM, and this concentration is about 750 times higher than that (9.8 nM) at which GTx1-15 inhibits 30% of calcium currents expressed in Xenopus oocytes. So, the concentration of 30 μg/mL was omitted from antigenicity assay. In the antigenicity assays, a 24-h exposure to DNCB, as a positive control, induced approximately 20–70 times higher expression of CD80, CD86 and CD54 genes. With 1, 3 and 10 μg/mL of GTx1-15, no expression changes in CD80, CD86 and CD54 genes were detected after 24 h of exposure. These results indicate that antigenicity was not detected after 24 h of exposure with concentrations of GTx1-15 that are 25, 75 and 250 times higher than the 30% inhibition concentration of about 9.8 nM.
Taken together, GTx1-15 will likely have little or no antigenicity if injected systemically in humans, especially for a short-term period, implying that it may be safe to develop GTx1-15 as a pharmaceutical product. To clarify this point, larger-scale experiments using a wide range of healthy cell lines from various body tissues would be necessary.
3.3. GTx1-15 Is a Suitable Scaffold for Peptide Library Construction
Recently, we developed an Escherichia coli periplasmic peptide display technique, PERISS (intra periplasm secretion and selection). In the PERISS method, a target protein (we focused on ion channels) and a peptide library (we focused on an ICK peptide library) are coexpressed in the E. coli inner-membrane and periplasmic space, respectively [34].
It is assumed that membrane proteins that are expressed in the inner membrane have correct three-dimensional structures and correct activity. To verify this assumption, we have developed an E. coli giant spheroplast electrophysiological technique [35]. Using this technique, we can measure induced potassium channel currents. Peptide library screening against correctly expressed ion channels should be able to identify new bioactive peptides.
The E. coli periplasmic space is suitable for the expression of disulfide-rich peptides [34,36,37,38,39], and the ICK peptide scaffold is suitable for directed molecular evolution to generate new peptide drugs [40,41,42,43]. The current study revealed that GTx1-15 possesses high stability (thermal stability and stability in circulation) and is safe at the cellular level (no cytotoxicity or antigenicity). Therefore, peptides screened from a GTx1-15-based peptide library might have the same characteristics of high stability and safety in vivo. We have prepared a peptide library using GTx1-15 as a scaffold to screen with the PERISS method, and after screening with PERISS against an ion channel, several bioactive peptides were successfully obtained [44].
4. Conclusions
ICK peptides are viable drug leads based on their high target specificity, high affinity, and thermal, chemical and proteolytic stability. Our results show that little or no degradation of GTx1-15 occurred in rat or human plasma for 24 h in vitro. GTx1-15 concentrations in circulation gradually decreased and kept dropping within 4 and 8 h, implying that the main clearance pathway might be the renal or other pathways (but not degradation) and pharmacologically effective peptide concentrations remain for several hours compared with other nonmodified peptides. GTx1-15 has no cytotoxicity and no antigenicity as revealed by THP-1 cell exposure tests. GTx1-15 has potential as a lead for a novel class of peptide drugs that are highly stable with low cytotoxicity and antigenicity. Finally, GTx1-15 is suitable as a template in a peptide library for periplasmic peptide display.
5. Materials and Methods
5.1. Peptides and Chemicals
GTx1-15 was obtained from Alomone Labs (Jerusalem, Israel). Human plasma was purchased from KOJIN BIO (Saitama, Japan). The protein thermal shift starter kit was from Thermo Fisher Scientific (Tokyo, Japan). RPMI-1640 with L-glutamine and phenol red, sodium hydrogen carbonate, and Tris-HCl were purchased from Wako Chemicals (Osaka, Japan). A human monocytic leukemia cell line, THP-1 (No. JCRB0112), was from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). Penicillin-streptomycin solution was obtained from Life Technologies Japan Ltd. (Tokyo, Japan). HyClone fetal bovine serum was purchased from GE Healthcare Japan (Tokyo, Japan). Premix WST-1 reagent was purchased from Takara Bio (Shiga, Japan).
5.2. Stability Assays in Animal Plasma In Vitro
To observe peptide stability in vitro, blood was extracted from three 8-week-old male SD rats after anesthesia with isoflurane inhalation, and the plasma was separated by centrifugation at 1850× g for 10 min at 4 °C. Plasma was stored on ice and used within the day of the experiment. Human plasma was stored at −80 °C until use after purchase. GTx1-15 was added to the plasma to make a 1 µg/mL mixture and incubated at 37 °C for 24 h, withdrawing 250 µL aliquots at 0, 2, 4, 8 and 24 h. Samples were kept at −25 °C away from light until analysis. This experiment, including LC-MS/MS sample analysis, was conducted by Nemoto Science Co., Ltd. (Ibaraki, Japan).
5.3. Concentration in Circulation after Intravenous, Intramuscular, and Peroral Administration
To observe GTx1-15 concentrations in blood circulation in vivo, three nonfasted male rats were administered with 0.1 or 0.5 mg/mL/kg of GTx1-15 i.v. or i.m. under isoflurane anesthesia, or peroral administration with 1 or 5 mg/mL/kg of GTx1-15 without any anesthesia. Blood samples of 450 μL were taken from the tail veins at 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24 h after administration using Pasteur pipettes coated with heparin sodium. Plasma was obtained via centrifugation at 10,000× g for 3 min at 4 °C and stored at −25 °C protected from light until analysis. Animal experiments, including LC-MS/MS analysis, were conducted by Nemoto Science Co., Ltd. in accordance with the guidelines of the animal experiment ethics committee (authorization numbers: 2016-0012, 2016-0073, 2017-0121) and under approval of the animal experiment ethics committee of National Institute of Advanced Science and Technology (authorization numbers: A2015-119, A2016-199, A2017-199).
5.4. LC-MS/MS
For LC-MS/MS sample preparation, 100 µL plasma was mixed with 20 µL 50% methanol and 200 µL 4% phosphoric acid. Whole sample mixtures were added to an Oasis HLB 1 cc/10 mg extraction cartridge (Waters, MA, USA) equilibrated with 1 mL methanol and 1 mL distilled water. The column was washed with 1 mL 5% methanol and eluted with 1 mL methanol. The eluate was dried under nitrogen flow and dissolved in 100 µL solvent A/B (30%:70%, v/v) for LC-MS/MS. 10 µL samples were analyzed by a Waters LC-MS/MS unit. ACQUITY UPLC BEH HILIC, 1.7 µm, 2.1 mm I.D. × 100 mm (Milford, MA, USA), was used at a flow rate of 0.3 mL/min by linear gradient elution (solvent A: solvent B = 30%:70%, v/v; solvent A: 0.1% TFA, solvent B: acetonitrile) using electro-spray ionization for Xevo TQ MS (Waters).
5.5. Thermal Stability
Thermal stability was examined by incubating GTx1-15 (24.7 µM, 10 μg/100 μL) for 24 h at various temperatures (20, 37, 50, 75 and 95 °C). After 24 h, samples were immediately stored at −20 °C prior to HPLC fractionation. Each 100 μL sample was diluted with 400 μL 0.05% TFA solution and subsequently separated using a Superiorex ODS column (4.6 × 250 mm, Shiseido) eluted with a linear gradient of 0–30% acetonitrile containing 0.05% TFA at a flow rate of 1 mL/min using AKTA pure25 (GE Healthcare). The peak area measured at wavelength 214 nm, which represents the native GTx1-15 quantity, was calculated using the evaluation function of Unicorn software version 7.1.
5.6. Protein Thermal Shift Assay
The protein thermal shift assay was conducted with the StepOne Real-Time PCR System (Thermo Fisher Scientific) using the Protein Thermal Shift Assay Kit according to the manufacturer’s instructions with Protein Thermal Shift Software version 1. The protein melt reaction mix was added to the wells of a 48-well PCR plate. The plate was heated from 25 to 99 °C with a heating rate of 1 °C/min. The software allows the user to calculate a melting temperature (Tm) from their melt curve data using the Boltzmann equation (Tm B).
5.7. Cytotoxicity Assay
THP-1 cells were cultured in RPMI-1640 containing 10% fetal bovine serum and supplemented with L-glutamine and penicillin/streptomycin. Cells were grown in a humidified 5% CO2 incubator at 37 °C. For cytotoxicity assays, 3 × 104 THP-1 cells were inoculated into single wells of a 96-well plate with 80 μL RPMI-1640 medium. Immediately after inoculation, 20 μL of GTx1-15 solution were added to the wells. Final concentrations of GTx1-15 were 1, 3, 10 and 30 μg/mL. As a control, RPMI-1640 medium was added to the wells instead of GTx1-15 solution. After a 24-h incubation, cytotoxicity was tested according to manufacturer’s instructions. Briefly, 10 μL WST-1 solution was added into each well and incubated for 2 h at 37 °C. After incubation, the absorbance was measured using a microplate reader (Eppendorf, Hamburg, Germany) at 450 nm.
5.8. Antigenicity Assay
THP-1 cells were cultured as described above. For antigenicity assays, 2.4 × 106 THP-1 cells were inoculated into single wells of a 12-well plate with 1.2 mL RPMI-1640 medium. Immediately after inoculation, GTx1-15 solution or DNCB solution were added to each well. Final concentrations of GTx1-15 were 1, 3 and 10 μg/mL, and the DNCB concentration was 4 μg/mL as a positive control. As a negative control, RPMI-1640 was added. After a 24-h incubation, THP-1 cells were collected via centrifugation at 1000× g for 5 min, and then total RNA was extracted from cells using an RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Total RNA was treated with DNase (QIAGEN) for 10 min to digest contaminating genomic DNA and purified by an RNeasy MinElute Cleanup Kit (QIAGEN). Complementary DNA was synthesized with PrimeScript Reverse Transcriptase (Takara Bio) with an oligo (dT)12-18 primer (Invitrogen, Waltham, USA), 10 mM each dNTP mixture (Promega, Madison, USA), and RNase Inhibitor (Takara Bio). Real-time PCR mixtures were prepared with SYBR Premix Ex Taq (Takara Bio) according to the manufacturer’s instructions. The reaction and monitoring were performed with a StepOne Real-Time System (Thermo Fisher Scientific) for 40 cycles of two-step shuttle PCR (95 °C for 5 s, 60 °C for 30 s). Primers for real-time PCR were designed using Roche ProbeFinder version 2.45 (http://qpcr.probefinder.com/roche3.html. This site is stopped at the end of 2020). Primers used for the detection of human CD80, CD86, CD54 and GAPDH are shown in Table 2.
Funding
This work was partly supported by the Japan Science and Technology Agency, Program for Creating Start-ups from Advanced Research and Technology, number ST272005WV.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of National Institute of Advanced Science and Technology (AIST) (protocol code A2015-119, A2016-199, A2017-199 and date of approval, 25 June 2015, 29 June 2016, 23 June 2017, respectively).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request to the corresponding author.
Acknowledgments
We would like to acknowledge technical assistance from Kazuhiko Kurosawa and Masanobu Hayashi.
Conflicts of Interest
The authors of this paper declare that there are no conflict of interest.
References
- Annette, C.D.; Paul, A.I.; Terrence, F.B.; Urs, A.M. Introduction to the Theme “Ion Channels and Neuropharmacology: From the Past to the Future”. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 1–6. [Google Scholar] [CrossRef]
- Lazniewska, J.; Weiss, N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim. Biophys. Acta 2017, 1859, 662–668. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Targeting of Cav3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflügers Arch. 2014, 466, 701–706. [Google Scholar] [CrossRef]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Saha, S. T-Type voltage gated calcium channels: A target in breast cancer? Breast Cancer Res. Treat. 2019, 173, 11–21. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raufman, J.P. Bioactive peptides from lizard venoms. Regul. Pept. 1996, 61, 1–18. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Hasaballah, N.; Vetter, I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology 2017, 127, 4–19. [Google Scholar] [CrossRef]
- Doupnik, C.A. Venom-derived peptides inhibiting Kir channels: Past, present, and future. Neuropharmacology 2017, 127, 161–172. [Google Scholar] [CrossRef]
- Ma, R.; Mahadevappa, R.; Kwok, H.F. Venom-based peptide therapy: Insights into anti-cancer mechanism. Oncotarget 2017, 8, 100908–100930. [Google Scholar] [CrossRef] [Green Version]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef]
- Pennington, M.W.; Czerwinski, A.; Norton, R.S. Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef]
- Saez, N.J.; Herzig, V. Versatile spider venom peptides and their medical and agricultural applications. Toxicon 2019, 158, 109–126. [Google Scholar] [CrossRef]
- Herzig, V.; Cristofori-Armstrong, B.; Israel, M.R.; Nixon, S.A.; Vetter, I.; King, G.F. Animal toxins—Nature’s evolutionary-refined toolkit for basic research and drug discovery. Biochem. Pharmacol. 2020, 181, 114096. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Craik, D.J. Thermal, Chemical, and Enzymatic Stability of the Cyclotide Kalata B1: The Importance of the Cyclic Cystine Knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Herzig, V.; King, G. The Cystine Knot Is Responsible for the Exceptional Stability of the Insecticidal Spider Toxin ω-Hexatoxin-Hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postic, G.; Gracy, J.; Périn, C.; Chiche, L.; Gelly, J.-C. KNOTTIN: The database of inhibitor cystine knot scaffold after 10 years, toward a systematic structure modeling. Nucleic Acids Res. 2018, 46, D454–D458. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, N.; Wu, B.; Ligutti, J.; Cheneval, O.; Agwa, A.J.; Benfield, A.H.; Biswas, K.; Craik, D.J.; Miranda, L.P.; Henriques, S.T.; et al. Peptide-Membrane Interactions Affect the Inhibitory Potency and Selectivity of Spider Toxins ProTx-II and GpTx-1. ACS Chem. Biol. 2019, 14, 118–130. [Google Scholar] [CrossRef]
- Mobli, M.; Undheim, E.A.B.; Rash, L.D. Modulation of Ion Channels by Cysteine-Rich Peptides: From Sequence to Structure. Adv. Pharmacol. 2017, 79, 199–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.; Kimura, T.; Kubo, T. Characterization of voltage-dependent calcium channel blocking peptides from the venom of the tarantula Grammostola rosea. Toxicon 2011, 58, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Meir, A.; Cheriki, R.S.; Kolb, E.; Langut, Y.; Bajayo, N. Novel Peptides Isolated from Spider Venom, and Uses Thereof PCT Patent Application Publication WO/2011/033358, 24 March 2011.
- Kikuchi, K.; Sugiura, M.; Kimura, T. High Proteolytic Resistance of Spider-Derived Inhibitor Cystine Knots. Int. J. Pept. 2015, 2015, 537508. [Google Scholar] [CrossRef] [Green Version]
- Bassotti, G.; Usai-Satta, P.; Bellini, M. Linaclotide for the treatment of chronic constipation. Expert Opin. Pharmacother. 2018, 19, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Ashikaga, T.; Miyazawa, M.; Kosaka, N.; Ito, Y.; Yoneyama, K.; Sono, S.; Itagaki, H.; Toyoda, H.; Suzuki, H. The relationship between CD86/CD54 expression and THP-1 cell viability in an in vitro skin sensitization test—Human cell line activation test (h-CLAT). Cell Biol. Toxicol. 2009, 25, 109–126. [Google Scholar] [CrossRef]
- Bocchietto, E.; Paolucci, C.; Breda, D.; Sabbioni, E.; Burastero, S.E. Human monocytoid THP-1 cell line versus monocyte-derived human immature dendritic cells as in vitro models for predicting the sensitising potential of chemicals. Int. J. Immunopathol. Pharmacol. 2007, 20, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Agwa, A.J.; Tran, P.; Mueller, A.; Tran, H.N.T.; Deuis, J.R.; Israel, M.R.; McMahon, K.L.; Craik, D.J.; Vetter, I.; Schroeder, C.I. Manipulation of a spider peptide toxin alters its affinity for lipid bilayers and potency and selectivity for voltage-gated sodium channel subtype 1.7. J. Biol. Chem. 2020, 295, 5067–5080. [Google Scholar] [CrossRef]
- Werle, M.; Schmitz, T.; Huang, H.L.; Wentzel, A.; Kolmar, H.; Bernkop-Schnurch, A. The potential of cystine-knot microproteins as novel pharmacophoric scaffolds in oral peptide drug delivery. J. Drug Target. 2006, 14, 137–146. [Google Scholar] [CrossRef]
- Newcomb, R.; Abbruscato, T.J.; Singh, T.; Nadasdi, L.; Davis, T.P.; Miljanich, G. Bioavailability of Ziconotide in brain: Influx from blood, stability, and diffusion. Peptides 2000, 21, 491–501. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Falcao, C.B.; Radis-Baptista, G. Crotamine and crotalicidin, membrane active peptides from Crotalus durissus terrificus rattlesnake venom, and their structurally-minimized fragments for applications in medicine and biotechnology. Peptides 2020, 126, 170234. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M.; Manzotti, C.N.; Zheng, Y. What’s the difference between CD80 and CD86? Trends Immunol. 2003, 24, 314–319. [Google Scholar] [CrossRef]
- Stucki, A.; Rivier, A.S.; Gikic, M.; Monai, N.; Schapira, M.; Spertini, O. Endothelial cell activation by myeloblasts: Molecular mechanisms of leukostasis and leukemic cell dissemination. Blood 2001, 97, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Galbiati, V.; Gatti, N.; Marinovich, M.; Galli, C.L.; Corsini, E. Optimization of the THP-1 activation assay to detect pharmaceuticals with potential to cause immune mediated drug reactions. Toxicol. In Vitro 2015, 29, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T. Screening Techniques Using the Periplasmic Expression of Peptide Libraries and Target Molecules. J. Bioanal. Biomed. 2017, 9. [Google Scholar] [CrossRef]
- Kikuchi, K.; Sugiura, M.; Nishizawa-Harada, C.; Kimura, T. The application of the Escherichia coli giant spheroplast for drug screening with automated planar patch clamp system. Biotechnol. Rep. 2015, 7, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Turchetto, J.; Sequeira, A.F.; Ramond, L.; Peysson, F.; Brás, J.L.A.; Saez, N.J.; Duhoo, Y.; Blémont, M.; Guerreiro, C.I.P.D.; Quinton, L.; et al. High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb. Cell Factories 2017, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, A.F.; Turchetto, J.; Saez, N.J.; Peysson, F.; Ramond, L.; Duhoo, Y.; Blémont, M.; Fernandes, V.O.; Gama, L.T.; Ferreira, L.M.A.; et al. Gene design, fusion technology and TEV cleavage conditions influence the purification of oxidized disulphide-rich venom peptides in Escherichia coli. Microb. Cell Factories 2017, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Saez, N.J.; Cristofori-Armstrong, B.; Anangi, R.; King, G.F. A Strategy for Production of Correctly Folded Disulfide-Rich Peptides in the Periplasm of E. coli. Methods Mol. Biol. 2017, 1586, 155–180. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.; Rash, L.D.; Mobli, M.; King, G.F. Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Heitz, A.; Avrutina, O.; Le-Nguyen, D.; Diederichsen, U.; Hernandez, J.F.; Gracy, J.; Kolmar, H.; Chiche, L. Knottin cyclization: Impact on structure and dynamics. BMC Struct. Biol. 2008, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Kolmar, H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Reinwarth, M.; Nasu, D.; Kolmar, H.; Avrutina, O. Chemical synthesis, backbone cyclization and oxidative folding of cystine-knot peptides: Promising scaffolds for applications in drug design. Molecules 2012, 17, 12533–12552. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T. Screening and Fast Identification of ICK Peptides Which Affect Kv2.1 Activity Using Periplasmic Peptide Display Technique PERISS; National Institute of Advanced Industrial Science and Technology (AIST): Tsukuba, Japan, 2021; manuscript in preparation. [Google Scholar]
Figure 1.
Representative structure of knottin and GTx1-15. (A) Cartoon represents peptide backbone and disulfide-bonded cystine-knot core. Red bars indicate disulfide bond connectivity. (B) Three-dimensional structure model and amino acid sequence of GTx1-15. 3D structure model of GTx1-15 was constructed via homology modeling with ICM-PRO (Molsoft, La Jolla, CA, USA) based on NMR structures of HnTx-IV (PDB: 1niy). Red letters indicate cysteine residues and red bars indicate disulfide bond connectivity.
Figure 1.
Representative structure of knottin and GTx1-15. (A) Cartoon represents peptide backbone and disulfide-bonded cystine-knot core. Red bars indicate disulfide bond connectivity. (B) Three-dimensional structure model and amino acid sequence of GTx1-15. 3D structure model of GTx1-15 was constructed via homology modeling with ICM-PRO (Molsoft, La Jolla, CA, USA) based on NMR structures of HnTx-IV (PDB: 1niy). Red letters indicate cysteine residues and red bars indicate disulfide bond connectivity.

Figure 2.
Stability of GTx1-15 in rat or human plasma. GTx1-15 was incubated in rat plasma (A) and in human plasma (B). No degradation was observed in either rat or human plasma in vitro for 24 h. Results are means ± SEM, n = 3. Note that error bars are too small to be visible.
Figure 2.
Stability of GTx1-15 in rat or human plasma. GTx1-15 was incubated in rat plasma (A) and in human plasma (B). No degradation was observed in either rat or human plasma in vitro for 24 h. Results are means ± SEM, n = 3. Note that error bars are too small to be visible.

Figure 3.
Plasma concentrations of GTx1-15 after i.v. or i.m. administration in rats. (A) GTx1-15 concentrations in rat blood after i.v. administration of 0.1 mg/kg or 0.5 mg/kg. GTx1-15 dropped below the detection limit in 4 or 8 h. (B) GTx1-15 concentration in rat blood after i.m. administration of 0.1 mg/kg or 0.5 mg/kg. The peak concentration of GTx1-15 in rat blood circulation was detected at 15 min after 0.1 mg/kg administration and at 30 min and 1 h after 0.5 mg/kg administration. Results are means ± SEM, n = 3. The dotted line indicates the detection limit of GTx1-15 (10 ng/mL) in blood circulation. No animals administered with GTx1-15 via i.v. or i.m. showed any abnormal behavior throughout the experiments.
Figure 3.
Plasma concentrations of GTx1-15 after i.v. or i.m. administration in rats. (A) GTx1-15 concentrations in rat blood after i.v. administration of 0.1 mg/kg or 0.5 mg/kg. GTx1-15 dropped below the detection limit in 4 or 8 h. (B) GTx1-15 concentration in rat blood after i.m. administration of 0.1 mg/kg or 0.5 mg/kg. The peak concentration of GTx1-15 in rat blood circulation was detected at 15 min after 0.1 mg/kg administration and at 30 min and 1 h after 0.5 mg/kg administration. Results are means ± SEM, n = 3. The dotted line indicates the detection limit of GTx1-15 (10 ng/mL) in blood circulation. No animals administered with GTx1-15 via i.v. or i.m. showed any abnormal behavior throughout the experiments.

Figure 4.
Thermal stability of GTx1-15. The calculated chromatogram peak area after incubation at the indicated temperatures for 24 h are shown. GTx1-15 did not degrade at 20 °C, 37 °C or 50 °C. At 75 °C, GTx1-15 degraded about 5%, but not significantly. About a 30% degradation of GTx1-15 was observed at 95 °C. Experiments were repeated in duplicate, and results are indicated as means ± SEM, n = 3. Statistical significance was determined by Dunnett’s multiple test, and p values < 0.05 were considered significant. ** indicates a significant difference p < 0.01.
Figure 4.
Thermal stability of GTx1-15. The calculated chromatogram peak area after incubation at the indicated temperatures for 24 h are shown. GTx1-15 did not degrade at 20 °C, 37 °C or 50 °C. At 75 °C, GTx1-15 degraded about 5%, but not significantly. About a 30% degradation of GTx1-15 was observed at 95 °C. Experiments were repeated in duplicate, and results are indicated as means ± SEM, n = 3. Statistical significance was determined by Dunnett’s multiple test, and p values < 0.05 were considered significant. ** indicates a significant difference p < 0.01.

Figure 5.
Cytotoxicity of GTx1-15. The effect of GTx1-15 on THP-1 cells after a 24-h exposure is shown. After 2 h of WST-1 incubation, the absorbance at 450 nm was measured by a plate reader. No effect of GTx1-15 was observed. Data are means ± SEM, n = 8.
Figure 5.
Cytotoxicity of GTx1-15. The effect of GTx1-15 on THP-1 cells after a 24-h exposure is shown. After 2 h of WST-1 incubation, the absorbance at 450 nm was measured by a plate reader. No effect of GTx1-15 was observed. Data are means ± SEM, n = 8.

Figure 6.
Antigenicity of GTx1-15. The effect of GTx1-15 on THP-1 cells after a 24-h exposure is shown. The expressions of CD80, CD86 and CD54 were quantified by RT-PCR using primers listed in Table 2. No effect of GTx1-15 was observed on CD80 expression (A), CD86 expression (B), or CD54 expression (C). Data are means ± SEM, n = 3. Experiments were repeated in triplicate.
Figure 6.
Antigenicity of GTx1-15. The effect of GTx1-15 on THP-1 cells after a 24-h exposure is shown. The expressions of CD80, CD86 and CD54 were quantified by RT-PCR using primers listed in Table 2. No effect of GTx1-15 was observed on CD80 expression (A), CD86 expression (B), or CD54 expression (C). Data are means ± SEM, n = 3. Experiments were repeated in triplicate.

Figure 7.
Comparison of GTx1-15 and ω-hexatoxin-Hv1a. (A) Amino acid comparison of GTx1-15 and ω-hexatoxin-Hv1a. Cysteine residues are indicated in bold letters. Note that ω-hexatoxin-Hv1a contains a long loop in the C-terminal region of the molecule (amino acid residues shown in red). Hydrophobic amino acid residues are shown in red bold letters. (B) 3D structure comparison of GTx1-15 and ω-hexatoxin-Hv1a. 3D structure models of GTx1-15 was constructed by homology modeling with ICM-PRO (Molsoft, La Jolla, CA) based on NMR structures of HnTx-IV (PDB: 1niy). 3D structure of ω-hexatoxin-Hv1a is based on PDB No. 1AXH. The long loop part of ω-hexatoxin-Hv1a (the area circled in red) protrudes from the main body consisting of three disulfide bonds (the area circled in blue). However, the very small loop part of GTx1-15 (the area circled in black) is different from the ω-hexatoxin-Hv1a long loop. The hydrophobic amino acid residues shown in red bold in (A) are indicated by a single letter at the corresponding position on the ribbon. All hydrophobic amino acid residues are located outside of the blue circle. However, tryptophan, a hydrophobic amino acid residue shown as a bold letter in the small loop of GTx1-15, is located inside the blue loop.
Figure 7.
Comparison of GTx1-15 and ω-hexatoxin-Hv1a. (A) Amino acid comparison of GTx1-15 and ω-hexatoxin-Hv1a. Cysteine residues are indicated in bold letters. Note that ω-hexatoxin-Hv1a contains a long loop in the C-terminal region of the molecule (amino acid residues shown in red). Hydrophobic amino acid residues are shown in red bold letters. (B) 3D structure comparison of GTx1-15 and ω-hexatoxin-Hv1a. 3D structure models of GTx1-15 was constructed by homology modeling with ICM-PRO (Molsoft, La Jolla, CA) based on NMR structures of HnTx-IV (PDB: 1niy). 3D structure of ω-hexatoxin-Hv1a is based on PDB No. 1AXH. The long loop part of ω-hexatoxin-Hv1a (the area circled in red) protrudes from the main body consisting of three disulfide bonds (the area circled in blue). However, the very small loop part of GTx1-15 (the area circled in black) is different from the ω-hexatoxin-Hv1a long loop. The hydrophobic amino acid residues shown in red bold in (A) are indicated by a single letter at the corresponding position on the ribbon. All hydrophobic amino acid residues are located outside of the blue circle. However, tryptophan, a hydrophobic amino acid residue shown as a bold letter in the small loop of GTx1-15, is located inside the blue loop.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Protein thermal shift assay.
| ICK Peptide | Tm B (°C) |
|---|---|
| GTx1-15 | 96.30 ± 0.03 |
| ProTxI | 93.23 ± 0.12 |
| ProTxII | 93.95 ± 0.12 |
| GsMTx4 | 95.26 ± 0.10 |
Tm B: Boltzmann melting temperature.
Table 2.
Primers used to detect human CD80, CD86, CD54, and GAPDH in real-time PCR.
| Gene | Sequence | Amplicon Size (bp) | |
|---|---|---|---|
| CD80 | Forward Reverse | CCTACTGCTTTGCCCCAAGA AAGGGCAAGGTGGGGTAATC | 188 |
| CD86 | Forward Reverse | ACGCGGCTTTTATCTTCACC TCTTCCCTCTCCATTGTGTTGG | 200 |
| CD54 | Forward Reverse | TTGAGGGCACCTACCTCTGT GATCTTCCGCTGGCGGTTAT | 176 |
| GAPDH | Forward Reverse | CCATGGAGAAGGCTGGGG CAAAGTTGTCATGGATGACC | 195 |
CD80: Cluster of differentiation 80, CD86: Cluster of differentiation 86, CD54: Cluster of Differentiation 54, GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase, bp: base pair.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kimura, T. Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea. Toxins 2021, 13, 621. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13090621
AMA Style
Kimura T. Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea. Toxins. 2021; 13(9):621. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13090621
Chicago/Turabian StyleKimura, Tadashi. 2021. "Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea" Toxins 13, no. 9: 621. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13090621
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.