The Role of Gut-Derived, Protein-Bound Uremic Toxins in the Cardiovascular Complications of Acute Kidney Injury

 , ,
, ,
Abstract
:1. Introduction
2. Cardiovascular Complications in AKI Patients
2.1. Cardiovascular Complications during an AKI Episode
2.2. Cardiovascular Complications after an AKI Episode
3. Gut-Derived, Protein-Bound Uremic Toxins and Cardiovascular Dysfunction in Experimental AKI Models
3.1. Indoxyl Sulfate
3.2. Para-Cresyl Sulfate
3.3. Indole-3-Acetic Acid
4. Gut-Derived Protein-Bound Uremic Toxins and Cardiovascular Dysfunctions in Clinical AKI Studies
4.1. Gut-Derived Protein-Bound Uremic Toxin Accumulation in AKI Patients
4.2. Association between Gut-Derived Protein-Bound Uremic Toxins and Cardiovascular Outcomes of AKI Patients
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khwaja, A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute Kidney Injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Waikar, S.S.; Liu, K.D.; Chertow, G.M. Diagnosis, Epidemiology and Outcomes of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2008, 3, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.J.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of Acute Kidney Injury in Critically Ill Patients: The Multinational AKI-EPI Study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World Incidence of AKI: A Meta-Analysis. CJASN 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global Epidemiology and Outcomes of Acute Kidney Injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute Kidney Injury, Mortality, Length of Stay, and Costs in Hospitalized Patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [Green Version]
- Liaño, F.; Junco, E.; Pascual, J.; Madero, R.; Verde, E. The Spectrum of Acute Renal Failure in the Intensive Care Unit Compared with That Seen in Other Settings. The Madrid Acute Renal Failure Study Group. Kidney Int. Suppl. 1998, 66, S16–S24. [Google Scholar]
- Rimes-Stigare, C.; Frumento, P.; Bottai, M.; Mårtensson, J.; Martling, C.-R.; Walther, S.M.; Karlström, G.; Bell, M. Evolution of Chronic Renal Impairment and Long-Term Mortality after de Novo Acute Kidney Injury in the Critically Ill; a Swedish Multi-Centre Cohort Study. Crit. Care 2015, 19, 221. [Google Scholar] [CrossRef] [Green Version]
- Linder, A.; Fjell, C.; Levin, A.; Walley, K.R.; Russell, J.A.; Boyd, J.H. Small Acute Increases in Serum Creatinine Are Associated with Decreased Long-Term Survival in the Critically Ill. Am. J. Respir. Crit. Care Med. 2014, 189, 1075–1081. [Google Scholar] [CrossRef]
- Sawhney, S.; Marks, A.; Fluck, N.; Levin, A.; Prescott, G.; Black, C. Intermediate and Long-Term Outcomes of Survivors of Acute Kidney Injury Episodes: A Large Population-Based Cohort Study. Am. J. Kidney Dis. 2017, 69, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druml, W.; Mitch, W.E. Metabolic Abnormalities in Acute Renal Failure. Semin. Dial. 2007, 9, 484–490. [Google Scholar] [CrossRef]
- Rydzewska-Rosołowska, A.; Sroka, N.; Kakareko, K.; Rosołowski, M.; Zbroch, E.; Hryszko, T. The Links between Microbiome and Uremic Toxins in Acute Kidney Injury: Beyond Gut Feeling-A Systematic Review. Toxins 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Graboski, A.L.; Redinbo, M.R. Gut-Derived Protein-Bound Uremic Toxins. Toxins 2020, 12, 590. [Google Scholar] [CrossRef]
- Di Lullo, L.; Bellasi, A.; Barbera, V.; Cozzolino, M.; Russo, D.; De Pascalis, A.; Santoboni, F.; Villani, A.; De Rosa, S.; Colafelice, M.; et al. Acute Kidney Injury, Type-3 cardiorenal syndrome, Biomarkers, Renal Replacement Therapy. G. Ital. Nefrol. 2016, 33, gin/33.3.2. [Google Scholar]
- Jentzer, J.C.; Bihorac, A.; Brusca, S.B.; Del Rio-Pertuz, G.; Kashani, K.; Kazory, A.; Kellum, J.A.; Mao, M.; Moriyama, B.; Morrow, D.A.; et al. Contemporary Management of Severe Acute Kidney Injury and Refractory Cardiorenal Syndrome: JACC Council Perspectives. J. Am. Coll. Cardiol. 2020, 76, 1084–1101. [Google Scholar] [CrossRef]
- Chuasuwan, A.; Kellum, J.A. Cardio-Renal Syndrome Type 3: Epidemiology, Pathophysiology, and Treatment. Semin. Nephrol. 2012, 32, 31–39. [Google Scholar] [CrossRef]
- Meng, Z.; Zhao, Y.; Zheng, X.; He, Y. The Relationship Between AKI in Patients With STEMI and Short-Term Mortality: A Propensity Score Matching Analysis. Angiology 2021, 72, 733–739. [Google Scholar] [CrossRef]
- Fox, C.S.; Muntner, P.; Chen, A.Y.; Alexander, K.P.; Roe, M.T.; Wiviott, S.D. Short-Term Outcomes of Acute Myocardial Infarction in Patients with Acute Kidney Injury: A Report from the National Cardiovascular Data Registry. Circulation 2012, 125, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Malik, A.H.; Ali, A.; Yang, Y.C.; Aronow, W.S.; Briasoulis, A. Impact of Acute Kidney Injury on In-Hospital Outcomes among Patients Hospitalized with Acute Heart Failure—A Propensity-Score Matched Analysis. Eur. J. Intern. Med. 2020, 79, 76–80. [Google Scholar] [CrossRef]
- Tsai, T.T.; Patel, U.D.; Chang, T.I.; Kennedy, K.F.; Masoudi, F.A.; Matheny, M.E.; Kosiborod, M.; Amin, A.P.; Messenger, J.C.; Rumsfeld, J.S.; et al. Contemporary Incidence, Predictors, and Outcomes of Acute Kidney Injury in Patients Undergoing Percutaneous Coronary Interventions: Insights from the NCDR Cath-PCI Registry. JACC Cardiovasc. Interv. 2014, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorrilla-Vaca, A.; Ziai, W.; Connolly, E.S.; Geocadin, R.; Thompson, R.; Rivera-Lara, L. Acute Kidney Injury Following Acute Ischemic Stroke and Intracerebral Hemorrhage: A Meta-Analysis of Prevalence Rate and Mortality Risk. Cerebrovasc. Dis. 2018, 45, 1–9. [Google Scholar] [CrossRef]
- Legrand, M.; Rossignol, P. Cardiovascular Consequences of Acute Kidney Injury. N. Engl. J. Med. 2020, 382, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J. Distant Effects of Experimental Renal Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2003, 14, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dépret, F.; Prud’homme, M.; Legrand, M. A Role of Remote Organs Effect in Acute Kidney Injury Outcome. Nephron 2017, 137, 273–276. [Google Scholar] [CrossRef]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.-S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II Causes Hypertension and Cardiac Hypertrophy through Its Receptors in the Kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Tsai, I.-J.; Pan, H.-C.; Liao, H.-W.; Neyra, J.A.; Wu, V.-C.; Chueh, J.S. The Impact of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers on Clinical Outcomes of Acute Kidney Disease Patients: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 665250. [Google Scholar] [CrossRef]
- Prud’homme, M.; Coutrot, M.; Michel, T.; Boutin, L.; Genest, M.; Poirier, F.; Launay, J.-M.; Kane, B.; Kinugasa, S.; Prakoura, N.; et al. Acute Kidney Injury Induces Remote Cardiac Damage and Dysfunction Through the Galectin-3 Pathway. JACC Basic Transl. Sci. 2019, 4, 717–732. [Google Scholar] [CrossRef]
- Cianciolo, G.; Donati, G.; La Manna, G.; Ferri, A.; Cuna, V.; Ubaldi, G.; Corsini, S.; Lanci, N.; Colì, L.; Stefoni, S. The Cardiovascular Burden of End-Stage Renal Disease Patients. Minerva. Urol. Nefrol. 2010, 62, 51–66. [Google Scholar]
- Claro, L.; Moreno-Amaral, A.; Gadotti, A.; Dolenga, C.; Nakao, L.; Azevedo, M.; de Noronha, L.; Olandoski, M.; de Moraes, T.; Stinghen, A.; et al. The Impact of Uremic Toxicity Induced Inflammatory Response on the Cardiovascular Burden in Chronic Kidney Disease. Toxins 2018, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.K.; Gammelager, H.; Jacobsen, C.-J.; Hjortdal, V.E.; Layton, J.B.; Rasmussen, B.S.; Andreasen, J.J.; Johnsen, S.P.; Christiansen, C.F. Acute Kidney Injury and Long-Term Risk of Cardiovascular Events After Cardiac Surgery: A Population-Based Cohort Study. J. Cardiothorac. Vasc. Anesth. 2015, 29, 617–625. [Google Scholar] [CrossRef]
- Parikh, C.R.; Puthumana, J.; Shlipak, M.G.; Koyner, J.L.; Thiessen-Philbrook, H.; McArthur, E.; Kerr, K.; Kavsak, P.; Whitlock, R.P.; Garg, A.X.; et al. Relationship of Kidney Injury Biomarkers with Long-Term Cardiovascular Outcomes after Cardiac Surgery. JASN 2017, 28, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Hsu, C.; Yang, J.; Tan, T.C.; Zheng, S.; Ordonez, J.D.; Liu, K.D. Acute Kidney Injury and Risk of Heart Failure and Atherosclerotic Events. CJASN 2018, 13, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, N.; Matheny, M.E.; Greevy, R.A.; Eden, S.K.; Perkins, A.M.; Parr, S.K.; Fly, J.; Abdel-Kader, K.; Himmelfarb, J.; Hung, A.M.; et al. Acute Kidney Injury and Risk of Incident Heart Failure Among US Veterans. Am. J. Kidney Dis. 2018, 71, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Tecson, K.M.; Hashemi, H.; Afzal, A.; Gong, T.A.; Kale, P.; McCullough, P.A. Community-Acquired Acute Kidney Injury as a Risk Factor of de Novo Heart Failure Hospitalization. Cardiorenal Med. 2019, 9, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.-C.; Wu, C.-H.; Huang, T.-M.; Wang, C.-Y.; Lai, C.-F.; Shiao, C.-C.; Chang, C.-H.; Lin, S.-L.; Chen, Y.-Y.; Chen, Y.-M.; et al. Long-Term Risk of Coronary Events after AKI. J. Am. Soc. Nephrol. 2014, 25, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, S.; Kang, M.W.; Yoo, H.-W.; Han, K.; Kim, Y.; Lee, J.P.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; et al. Postdischarge Long-Term Cardiovascular Outcomes of Intensive Care Unit Survivors Who Developed Dialysis-Requiring Acute Kidney Injury after Cardiac Surgery. J. Crit. Care 2019, 50, 92–98. [Google Scholar] [CrossRef]
- Odutayo, A.; Wong, C.X.; Farkouh, M.; Altman, D.G.; Hopewell, S.; Emdin, C.A.; Hunn, B.H. AKI and Long-Term Risk for Cardiovascular Events and Mortality. JASN 2017, 28, 377–387. [Google Scholar] [CrossRef]
- Silver, S.A.; Harel, Z.; McArthur, E.; Nash, D.M.; Acedillo, R.; Kitchlu, A.; Garg, A.X.; Chertow, G.M.; Bell, C.M.; Wald, R. Causes of Death after a Hospitalization with AKI. JASN 2018, 29, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Hsu, R.K.; Yang, J.; Ordonez, J.D.; Zheng, S.; Go, A.S. Elevated BP after AKI. JASN 2016, 27, 914–923. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Parikh, C.R.; Himmelfarb, J.; Chinchilli, V.M.; Liu, K.D.; Coca, S.G.; Garg, A.X.; Hsu, C.; Siew, E.D.; Wurfel, M.M.; et al. A Prospective Cohort Study of Acute Kidney Injury and Kidney Outcomes, Cardiovascular Events, and Death. Kidney Int. 2021, 99, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Omotoso, B.A.; Abdel-Rahman, E.M.; Xin, W.; Ma, J.Z.; Scully, K.W.; Arogundade, F.A.; Balogun, R.A. Acute Kidney Injury (AKI) Outcome, a Predictor of Long-Term Major Adverse Cardiovascular Events (MACE). Clin. Nephrol. 2016, 85, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T. Uraemic Syndrome of Chronic Kidney Disease: Altered Remote Sensing and Signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Chaves, L.D.; McSkimming, D.I.; Bryniarski, M.A.; Honan, A.M.; Abyad, S.; Thomas, S.A.; Wells, S.; Buck, M.; Sun, Y.; Genco, R.J.; et al. Chronic Kidney Disease, Uremic Milieu, and Its Effects on Gut Bacterial Microbiota Dysbiosis. Am. J. Physiol. Ren. Physiol. 2018, 315, F487–F502. [Google Scholar] [CrossRef]
- Nakade, Y.; Iwata, Y.; Furuichi, K.; Mita, M.; Hamase, K.; Konno, R.; Miyake, T.; Sakai, N.; Kitajima, S.; Toyama, T.; et al. Gut Microbiota–Derived D-Serine Protects against Acute Kidney Injury. JCI Insight 2018, 3, e97957. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kim, C.J.; Go, Y.S.; Lee, H.Y.; Kim, M.-G.; Oh, S.W.; Cho, W.Y.; Im, S.-H.; Jo, S.K. Intestinal Microbiota Control Acute Kidney Injury Severity by Immune Modulation. Kidney Int. 2020, 98, 932–946. [Google Scholar] [CrossRef]
- de Andrade, L.S.; Ramos, C.I.; Cuppari, L. The Cross-Talk between the Kidney and the Gut: Implications for Chronic Kidney Disease. Nutrire 2017, 42, 27. [Google Scholar] [CrossRef]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. JASN 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjölund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-Term Antibiotic Treatment Has Differing Long-Term Impacts on the Human Throat and Gut Microbiome. PLoS ONE 2010, 5, e9836. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of Urea in Intestinal Barrier Dysfunction and Disruption of Epithelial Tight Junction in Chronic Kidney Disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Iwata, Y.; Nakade, Y.; Wada, T. Significance of the Gut Microbiota in Acute Kidney Injury. Toxins 2021, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ichijo, M.; Kawabe, T.; Kikuchi, K.; Akiyama, Y.; Toyohara, T.; Suzuki, T.; Suzuki, C.; Asao, A.; Ishii, N.; et al. Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage. Toxins 2020, 12, 547. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, N.V.; Popkov, V.A.; Klimenko, N.S.; Tyakht, A.V.; Baydakova, G.V.; Frolova, O.Y.; Zorova, L.D.; Pevzner, I.B.; Zorov, D.B.; Plotnikov, E.Y. Microbiome-Metabolome Signature of Acute Kidney Injury. Metabolites 2020, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Moturi, K.R.; Wang, L.; Zhang, K.; Yu, C. Gut Derived-Endotoxin Contributes to Inflammation in Severe Ischemic Acute Kidney Injury. BMC Nephrol. 2019, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.K. Kidney–Gut Crosstalk in AKI. Kidney360 2021, 2, 886–889. [Google Scholar] [CrossRef]
- Six, I.; Gross, P.; Rémond, M.C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious Vascular Effects of Indoxyl Sulfate and Reversal by Oral Adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The Uremic Solute Indoxyl Sulfate Induces Oxidative Stress in Endothelial Cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The Uremic Solutes P-Cresol and Indoxyl Sulfate Inhibit Endothelial Proliferation and Wound Repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Tumur, Z.; Niwa, T. Indoxyl Sulfate Inhibits Nitric Oxide Production and Cell Viability by Inducing Oxidative Stress in Vascular Endothelial Cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-KappaB Activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Maciel, R.; Cunha, R.; Busato, V.; Franco, C.; Gregório, P.; Dolenga, C.; Nakao, L.; Massy, Z.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-Bound Uremic Toxins Stimulate Crosstalk between Leukocytes and Vessel Wall. JASN 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic Uremic Solutes Increase Tissue Factor Production in Endothelial Cells by the Aryl Hydrocarbon Receptor Pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiński, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl Sulfate—The Uremic Toxin Linking Hemostatic System Disturbances with the Prevalence of Cardiovascular Disease in Patients with Chronic Kidney Disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Nishijima, F.; Goto, S.; Sugano, M.; Yamato, H.; Kitazawa, R.; Kitazawa, S.; Fukagawa, M. Oral Charcoal Adsorbent (AST-120) Prevents Progression of Cardiac Damage in Chronic Kidney Disease through Suppression of Oxidative Stress. Nephrol. Dial. Transplant. 2009, 24, 2089–2095. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does Indoxyl Sulfate, a Uraemic Toxin, Have Direct Effects on Cardiac Fibroblasts and Myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Kumfu, S.; Wang, B.H.; Manabe, M.; Nishijima, F.; Kelly, D.J.; Krum, H.; Kompa, A.R. The Uremic Toxin Adsorbent AST-120 Abrogates Cardiorenal Injury Following Myocardial Infarction. PLoS ONE 2013, 8, e83687. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Chen, J.; Wang, M.; Yan, F. Klotho Alleviates Indoxyl Sulfate-Induced Heart Failure and Kidney Damage by Promoting M2 Macrophage Polarization. Aging 2020, 12, 9139–9150. [Google Scholar] [CrossRef]
- Yisireyili, M.; Shimizu, H.; Saito, S.; Enomoto, A.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Promotes Cardiac Fibrosis with Enhanced Oxidative Stress in Hypertensive Rats. Life Sci. 2013, 92, 1180–1185. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Krum, H. Cardiorenal Syndrome: Role of Protein-Bound Uremic Toxins. J. Ren. Nutr. 2015, 25, 149–154. [Google Scholar] [CrossRef]
- Wu, V.-C.; Young, G.-H.; Huang, P.-H.; Lo, S.-C.; Wang, K.-C.; Sun, C.-Y.; Liang, C.-J.; Huang, T.-M.; Chen, J.-H.; Chang, F.-C.; et al. In Acute Kidney Injury, Indoxyl Sulfate Impairs Human Endothelial Progenitor Cells: Modulation by Statin. Angiogenesis 2013, 16, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Katome, T.; Taguchi, K.; Kobayashi, T. Direct Impairment of the Endothelial Function by Acute Indoxyl Sulfate through Declined Nitric Oxide and Not Endothelium-Derived Hyperpolarizing Factor or Vasodilator Prostaglandins in the Rat Superior Mesenteric Artery. Biol. Pharm. Bull. 2019, 42, 1236–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Taguchi, K.; Kobayashi, T. Acute Exposure to Indoxyl Sulfate Impairs Endothelium-Dependent Vasorelaxation in Rat Aorta. Int. J. Mol. Sci. 2019, 20, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savira, F.; Cao, L.; Wang, I.; Yang, W.; Huang, K.; Hua, Y.; Jucker, B.M.; Willette, R.N.; Huang, L.; Krum, H.; et al. Apoptosis Signal-Regulating Kinase 1 Inhibition Attenuates Cardiac Hypertrophy and Cardiorenal Fibrosis Induced by Uremic Toxins: Implications for Cardiorenal Syndrome. PLoS ONE 2017, 12, e0187459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savira, F.; Kompa, A.R.; Magaye, R.; Xiong, X.; Huang, L.; Jucker, B.M.; Willette, R.N.; Kelly, D.J.; Wang, B.H. Apoptosis Signal-Regulating Kinase 1 Inhibition Reverses Deleterious Indoxyl Sulfate-Mediated Endothelial Effects. Life Sci. 2021, 272, 119267. [Google Scholar] [CrossRef]
- Shen, W.-C.; Liang, C.-J.; Huang, T.-M.; Liu, C.-W.; Wang, S.-H.; Young, G.-H.; Tsai, J.-S.; Tseng, Y.-C.; Peng, Y.-S.; Wu, V.-C.; et al. Indoxyl Sulfate Enhances IL-1β-Induced E-Selectin Expression in Endothelial Cells in Acute Kidney Injury by the ROS/MAPKs/NFκB/AP-1 Pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef]
- Shen, W.-C.; Chou, Y.-H.; Shi, L.-S.; Chen, Z.-W.; Tu, H.-J.; Lin, X.-Y.; Wang, G.-J. AST-120 Improves Cardiac Dysfunction in Acute Kidney Injury Mice via Suppression of Apoptosis and Proinflammatory NF-ΚB/ICAM-1 Signaling. J. Inflamm. Res. 2021, 14, 505–518. [Google Scholar] [CrossRef]
- Tan, X.; Cao, X.; Zhang, P.; Xiang, F.; Teng, J.; Zou, J.; Ding, X. Endoplasmic Reticulum Stress Associated Apoptosis as a Novel Mechanism in Indoxyl Sulfate-induced Cardiomyocyte Toxicity. Mol. Med. Rep. 2018, 18, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.-C.; Chou, Y.-H.; Huang, H.-P.; Sheen, J.-F.; Hung, S.-C.; Chen, H.-F. Induced Pluripotent Stem Cell-Derived Endothelial Progenitor Cells Attenuate Ischemic Acute Kidney Injury and Cardiac Dysfunction. Stem. Cell Res. Ther. 2018, 9, 344. [Google Scholar] [CrossRef]
- Chen, J.-H.; Chao, C.-T.; Huang, J.-W.; Hung, K.-Y.; Liu, S.-H.; Tarng, D.-C.; Chiang, C.-K. Early Elimination of Uremic Toxin Ameliorates AKI-to-CKD Transition. Clin. Sci. 2021, 135, 2643–2658. [Google Scholar] [CrossRef]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic Reticulum Stress Is Activated in Post-Ischemic Kidneys to Promote Chronic Kidney Disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic Sulfotransferase as a Nephropreventing Target by Suppression of the Uremic Toxin Indoxyl Sulfate Accumulation in Ischemic Acute Kidney Injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Uremic Toxins Induce Kidney Fibrosis by Activating Intrarenal Renin–Angiotensin–Aldosterone System Associated Epithelial-to-Mesenchymal Transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl Sulfate Upregulates Renal Expression of MCP-1 via Production of ROS and Activation of NF-ΚB, P53, ERK, and JNK in Proximal Tubular Cells. Life Sci. 2012, 90, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.J.; Sidor, N.A.; Tonial, N.C.; Che, A.; Urquhart, B.L. Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets. Toxins 2021, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The Uremic Retention Solute P-Cresyl Sulfate and Markers of Endothelial Damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Huang, S.-Y.; Wu, C.-C.; Hsu, C.-F. P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin. Toxins 2020, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Chen, Y.; Zhu, Z.; Su, X.; Ni, J.; Du, R.; Zhang, R.; Jin, W. p-Cresyl Sulfate Promotes the Formation of Atherosclerotic Lesions and Induces Plaque Instability by Targeting Vascular Smooth Muscle Cells. Front. Med. 2016, 10, 320–329. [Google Scholar] [CrossRef]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. p-Cresyl Sulfate Aggravates Cardiac Dysfunction Associated With Chronic Kidney Disease by Enhancing Apoptosis of Cardiomyocytes. JAHA 2015, 4, e001852. [Google Scholar] [CrossRef] [Green Version]
- Cerini, C.; Dou, L.; Anfosso, F.; Sabatier, F.; Moal, V.; Glorieux, G.; De Smet, R.; Vanholder, R.; Dignat-George, F.; Sampol, J.; et al. P-Cresol, a Uremic Retention Solute, Alters the Endothelial Barrier Function in Vitro. Thromb. Haemost. 2004, 92, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-Cresylsulphate, the Main in Vivo Metabolite of p-Cresol, Activates Leucocyte Free Radical Production. Nephrol. Dial. Transplant. 2006, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.-A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel Method for Simultaneous Determination of P-Cresylsulphate and p-Cresylglucuronide: Clinical Data and Pathophysiological Implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-Cresyl Sulfate Acutely Impairs Vascular Reactivity and Induces Vascular Remodeling: PARA-CRESYL SULFATE AND VASCULAR DYSFUNCTION. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-H.; Wang, C.-P.; Yu, T.-H.; Tai, P.-Y.; Liang, S.-S.; Hung, W.-C.; Wu, C.-C.; Huang, S.-H.; Lee, Y.-J.; Chen, S.-C. Protein-Bounded Uremic Toxin p-Cresylsulfate Induces Vascular Permeability Alternations. Histochem. Cell Biol. 2018, 149, 607–617. [Google Scholar] [CrossRef]
- Huang, T.-H.; Yip, H.-K.; Sun, C.-K.; Chen, Y.-L.; Yang, C.-C.; Lee, F.-Y. P-Cresyl Sulfate Causes Mitochondrial Hyperfusion in H9C2 Cardiomyoblasts. J. Cell Mol. Med. 2020, 24, 8379–8390. [Google Scholar] [CrossRef]
- Peng, Y.-S.; Ding, H.-C.; Lin, Y.-T.; Syu, J.-P.; Chen, Y.; Wang, S.-M. Uremic Toxin P-Cresol Induces Disassembly of Gap Junctions of Cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. P-Cresyl Sulfate Causes Renal Tubular Cell Damage by Inducing Oxidative Stress by Activation of NADPH Oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Suppression of Klotho Expression by Protein-Bound Uremic Toxins Is Associated with Increased DNA Methyltransferase Expression and DNA Hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.A.; van den Broek, P.H.H.; Wetzels, J.F.M.; van den Brand, J.A.J.G.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.G.; et al. Proximal Tubular Efflux Transporters Involved in Renal Excretion of P-Cresyl Sulfate and p-Cresyl Glucuronide: Implications for Chronic Kidney Disease Pathophysiology. Toxicol. Vitr. 2015, 29, 1868–1877. [Google Scholar] [CrossRef]
- Schroeder, J.C.; DiNatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The Uremic Toxin 3-Indoxyl Sulfate Is a Potent Endogenous Agonist for the Human Aryl Hydrocarbon Receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The Cardiovascular Effect of the Uremic Solute Indole-3 Acetic Acid. JASN 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Dou, L.; Sabatier, F.; Calaf, R.; Cerini, C.; Robert, S.; Camoin-Jau, L.; Charpiot, P.; Argiles, A.; Dignat-George, F.; et al. Levels of Circulating Endothelial Progenitor Cells Are Related to Uremic Toxins and Vascular Injury in Hemodialysis Patients. J. Thromb. Haemost. 2009, 7, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Bennis, Y.; Titeca-Beauport, D.; Caillard, P.; Cluet, Y.; Kamel, S.; Choukroun, G.; Maizel, J.; Liabeuf, S.; Bodeau, S. Two Rapid, Accurate Liquid Chromatography Tandem Mass Spectrometry Methods for the Quantification of Seven Uremic Toxins: An Application for Describing Their Accumulation Kinetic Profile in a Context of Acute Kidney Injury. J. Chromatogr. B 2020, 1152, 122234. [Google Scholar] [CrossRef] [PubMed]
- Veldeman, L.; Vanmassenhove, J.; Van Biesen, W.; Massy, Z.A.; Liabeuf, S.; Glorieux, G.; Vanholder, R. Evolution of Protein-Bound Uremic Toxins Indoxyl Sulphate and p-Cresyl Sulphate in Acute Kidney Injury. Int. Urol. Nephrol. 2019, 51, 293–302. [Google Scholar] [CrossRef]
- Yousef Selim, N.; Farag Mannaa, H.; Atef Sharaki, O.; Zaytoun, T.; Elkholy, N.; Arafat, W. Highlighting Levels of Indoxyl Sulphate among Critically Ill Patients with Acute Nephrotoxicity; Correlations Between Indoxyl Sulphate Levels and Patients’ Characteristics. Rep. Biochem. Mol. Biol. 2021, 10, 266–279. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and Pathologic Concentrations of Uremic Toxins. JASN 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hao, G.; Pan, Y.; Ma, S.; Yang, T.; Shi, P.; Zhu, Q.; Xie, Y.; Ma, S.; Zhang, Q.; et al. Serum Indoxyl Sulfate Is Associated with Mortality in Hospital-Acquired Acute Kidney Injury: A Prospective Cohort Study. BMC Nephrol. 2019, 20, 57. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic Kidney Disease Alters Intestinal Microbial Flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the Accumulation of Uremic Toxins in the Blood and Their Precursors in the Feces after Oral Administration of Lebenin®, a Lactic Acid Bacteria Preparation, to Uremic Patients Undergoing Hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered Microbiome in Chronic Kidney Disease: Systemic Effects of Gut-Derived Uremic Toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Xie, Y.; Sheng, J.; Song, J. Intestinal Microbiota Dysbiosis in Acute Kidney Injury: Novel Insights into Mechanisms and Promising Therapeutic Strategies. Ren. Fail. 2022, 44, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Noel, S.; Pluznick, J.L.; Hamad, A.R.A.; Rabb, H. Gut Microbiota-Kidney Cross-Talk in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, S.G.; Zhang, W.R.; Moledina, D.G.; Coca, S.G.; Jia, Y.; Thiessen-Philbrook, H.; McArthur, E.; Inoue, K.; Koyner, J.L.; Shlipak, M.G.; et al. The Association of Angiogenesis Markers With Acute Kidney Injury and Mortality after Cardiac Surgery. Am. J. Kidney Dis. 2019, 74, 36–46. [Google Scholar] [CrossRef]
- Ravid, J.D.; Kamel, M.H.; Chitalia, V.C. Uraemic Solutes as Therapeutic Targets in CKD-Associated Cardiovascular Disease. Nat. Rev. Nephrol. 2021, 17, 402–416. [Google Scholar] [CrossRef]


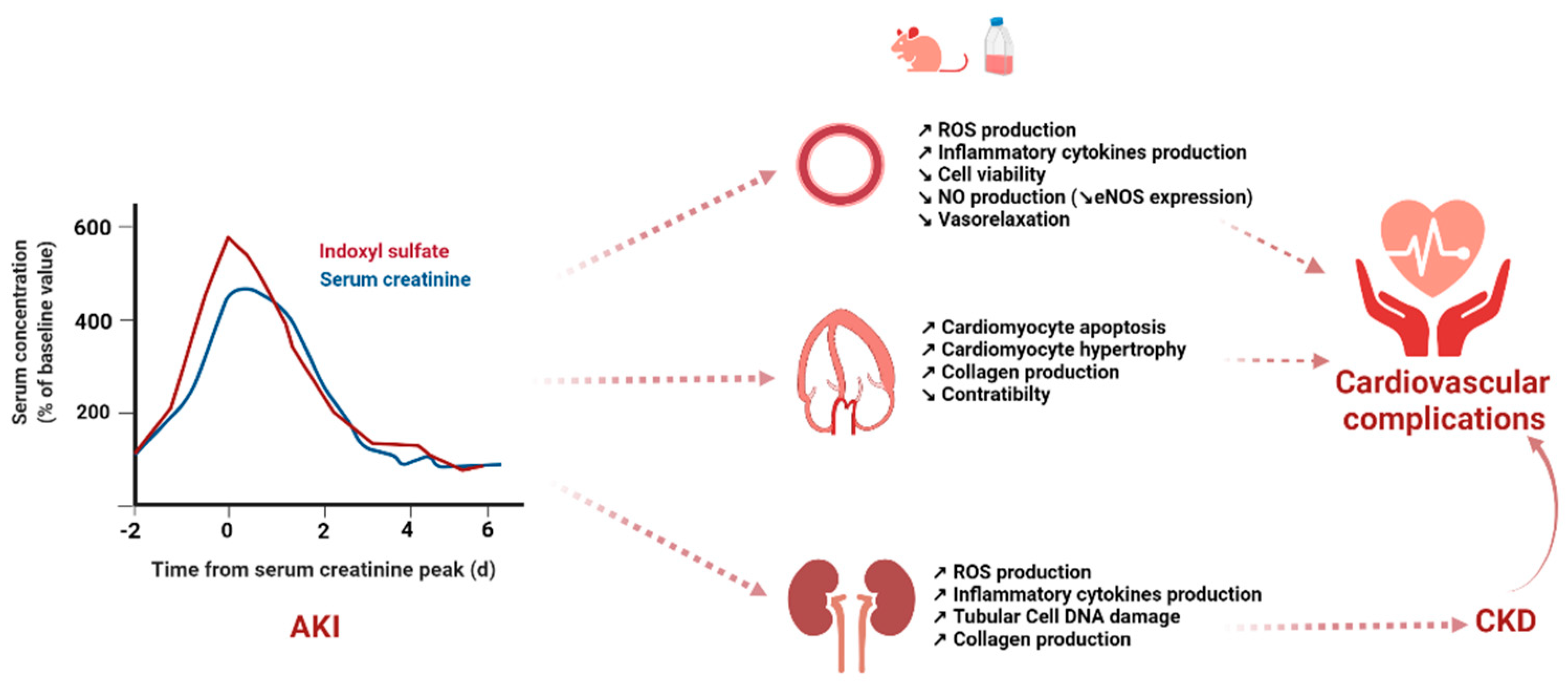{kind=link}
| Authors (Year) Country | Setting | No. of Patients | Staging of AKI | Measurement | Uremic Toxin Level (µg/mL) | Main Results |
|---|---|---|---|---|---|---|
| Wu et al. (2013) [71] Taiwan | Post-cardiac surgery AKI | 41 | AKIN Stage 1: 17 (41.4) Stage 2: 12 (29.3) Stage 3: 12 (29.3) | tIS at AKI diagnosis | tIS mean ± SD 28.78 ± 20.04 | Negative correlation between tIS level above 51.16 µg/mL and the human endothelial progenitor cell count. |
| Veldeman et al. (2019) [104] Belgium | Sepsis in ICU Septic shock (63%) | 194 | RIFLE No AKI: 64 (33.0) Risk: 40 (20.6) Injury: 57 (29.4) Failure: 33 (17.0) | tIS and tPCS at admission (D0) and Dend (D4 or day before drop-out) | tIS at D0 median [IQR] No AKI: 0.258 [0.097–0.610] AKI: 0.64 [0.252–1.802] -Risk: 0.377 [0.231–0.908] -Injury: 0.50 [0.169–1.716] -Failure: 1.785 [0.762–3.400] | -Correlation between severity of AKI and tIS level. -Decrease in tIS in all AKI groups between D0 and Dend. -No change in tIS level observed between D0 and Dend in cases of worsening of kidney function, but it decreased in cases of recovery. |
| Wang et al. (2019) [107] China | Hospital-acquired AKI | 262 | KDIGO Stage 1: 119 (45.5) Stage 2: 63 (24.0) Stage 3: 80 (30.5) | tIS at baseline, AKI diagnosis (n = 262) and 7 days after (n = 89) | tIS at AKI diagnosis mean ± SD 2.7 ± 0.8 | tIS > 2.74 µg/mL was associated with an increased Day 90 mortality rate with an aHR [95%CI] of 2.92 [1.76–4.86] p < 0.001. |
| Andre et al. (2020) [103] France | Post-cardiac surgery AKI | 8 | Time course of tIS, tPCS, and tIAA serum concentration according to that of creatinine | Peak concentration (min–max) tIS: 1.52 (0.35–2.62) tPCS: 9.33 (4.30–16.00) tIAA: 0.97 (0.60–1.80) | Serum creatinine-like accumulation and elimination profiles | |
| Selim et al. (2021) Egypt [105] | Toxic AKI in ICU | 74 | RIFLE Risk: 15 (20.3) Injury: 28 (37.8) Failure: 31 (41.9) | tIS and fIS within 48 h after toxic AKI, then at weeks 1 and 2 (or as ended earlier) | Basal tIS median [range] * tIS 3.33 [0.00–37.31] | Correlation between AKI severity and IS level. Association between basal IS level and AKI recovery at discharge No association between IS level and in-hospital mortality |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caillard, P.; Bennis, Y.; Six, I.; Bodeau, S.; Kamel, S.; Choukroun, G.; Maizel, J.; Titeca-Beauport, D. The Role of Gut-Derived, Protein-Bound Uremic Toxins in the Cardiovascular Complications of Acute Kidney Injury. Toxins 2022, 14, 336. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins14050336
Caillard P, Bennis Y, Six I, Bodeau S, Kamel S, Choukroun G, Maizel J, Titeca-Beauport D. The Role of Gut-Derived, Protein-Bound Uremic Toxins in the Cardiovascular Complications of Acute Kidney Injury. Toxins. 2022; 14(5):336. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins14050336
Chicago/Turabian StyleCaillard, Pauline, Youssef Bennis, Isabelle Six, Sandra Bodeau, Saïd Kamel, Gabriel Choukroun, Julien Maizel, and Dimitri Titeca-Beauport. 2022. "The Role of Gut-Derived, Protein-Bound Uremic Toxins in the Cardiovascular Complications of Acute Kidney Injury" Toxins 14, no. 5: 336. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins14050336