Bacterial Toxins and the Nervous System: Neurotoxins and Multipotential Toxins Interacting with Neuronal Cells

Abstract
:1. Introduction
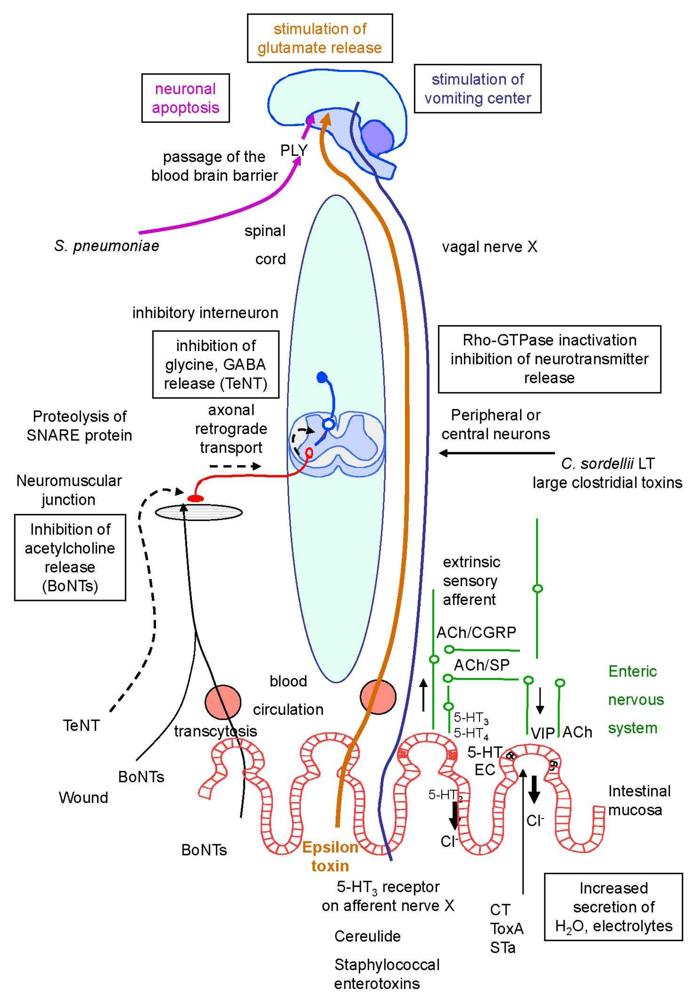
2. The Cellular and Molecular Mechanisms Involved in Neuroexocytosis: An Overview
2.1. An overview of neurotransmission

{kind=link}
{kind=link}
{kind=link}
| Toxin | Bacteria | Structure | Target neuronal cell | Receptor | Activity | Effects | MLD (μg/kg)1 |
|---|---|---|---|---|---|---|---|
| Toxins inhibiting the neuroexocytosis | |||||||
| Botulinum neurotoxins | C. botulinum | single chain protein (150 kDa) | motoneurons | gangliosides (GD1b, GT1b) | proteolysis of SNARE proteins (VAMP, SNAP25, syntaxin) | inhibiton of acetylcholine release (flaccid paralysis) | 0.0003 |
| C. baratii | synaptotagmin, SV2 | ||||||
| C. butyricum | |||||||
| Tetanus neurotoxin | C. tetani | single chain protein (150 kDa) | inhibitory interneurons | gangliosides (GD1b, GT1b) | proteolysis of SNARE protein (VAMP) | inhibition of neurotransmitter release (GABA, glycine) (spastic paralysis) | 0.001 |
| GPI-anchored protein | |||||||
| Lethal toxin | C. sordellii | single chain protein (250 kDa) | potentially all neurons | unknown | inactivation of Rho and Ras-GTPases (glucosylation) | inhibition of neurotransmitter release | 0.1 |
| Toxin B | C. difficile | single chain protein (250 kDa) | potentially all neurons | unknown | inactivation of Rho-GTPases (glucosylation) | inhibition of neurotransmitter release | 32 |
| Pneumolysin | S. pneumoniae | single chain protein (53 kDa) | hippocampal neurons | cholesterol | pore-forming activity | neuronal apoptosis (meningitis) | |
| Enterotoxin | C. perfringens | single chain protein (36 kDa) | enterocyte neurons | pore-forming activity | 80 | ||
| Toxins stimulating neurosecretion | |||||||
| Epsilon toxin | C. perfringens | single chain protein (36 kDa) | hippocampal neurons | unknown | pore-forming activity | stimulation of glutamate release (excitation) | 0.1 |
| Cholera toxin | V. cholerae | AB5 structure | enterochrompaffin cells and enteric neurons | ganglioside GM1 | inactivation of Gsα and activation of adenylate cyclase | 5-HT release (diarrhea) | 250 |
| Heat labile enterotoxin | E. coli | AB5 structure | enterochromaffin cells and enteric neurons | ganglioside GM1 | inactivation of Gsα and activation of adenylate cyclase | 5-HT release (diarrhea) | 250 |
| Toxin A | C. difficile | single chain protein (300 kDa) | enterocytes enteric neurons | membrane glycoprotein | inactivation of Rho-GTPases, other mechanism? | release of inflammatory mediators and neuropeptides (diarrhea) | 0.35 |
| Heat stable enterotoxin | E. coli | short peptide (2–5 kDa) | enterocyte enteric neurons? | guanylate cyclase | GMPc increase,other mechanism? | stimulation of enteric nervous system(diarrhea) | |
| Staphylococcal enterotoxins | S. aureus | single chain protein (25–30 kDa) | enterochromaffin cells, vagal nerve | histocompatibility complex class II molecules | superantigen other mechanism? | 5-HT release stimulation of 5-HT3 receptor (emesis) | 20 (monkey) |
| Cereulide | B. cereus | cyclic dodecadepsipeptide (1.2 kDa) | vagal nerve | 5HT3 receptor | K+ ionophore | stimulation of 5-HT3 receptor (emesis) | |

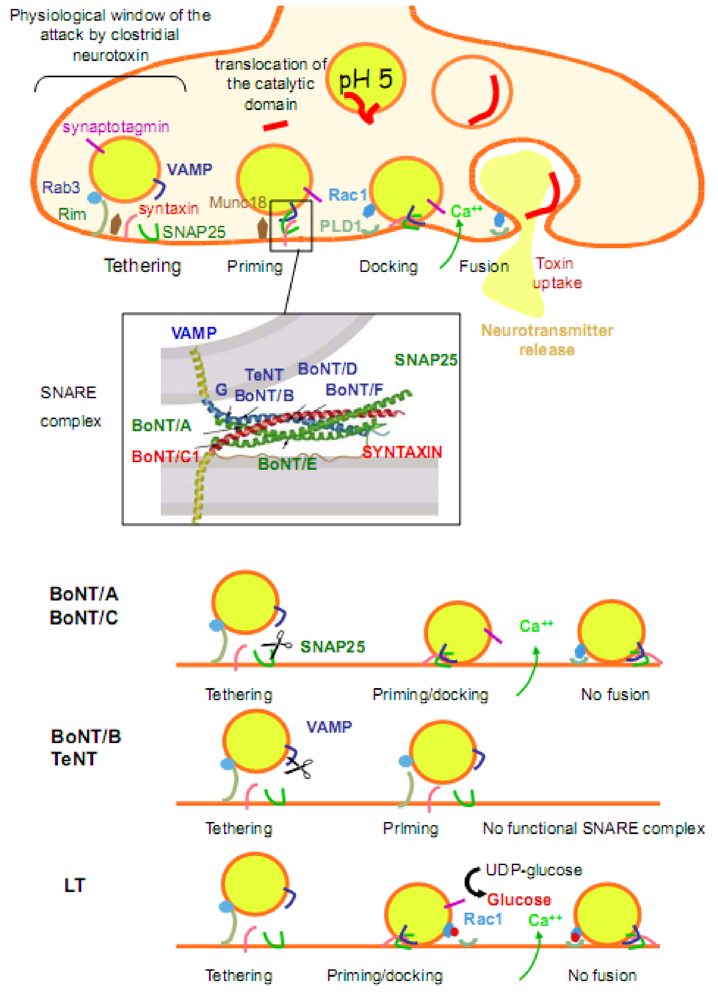
2.2. Mechanisms of exocytosis and SNAREs
2.3. Actin cytoskeleton and small GTPases in exocytotic mechanisms
3. Toxins Inhibiting the Neuroexocytosis
3.1. Toxins which specifically impair the SNARE exocytosis mechanism: Clostridial neurotoxins
3.1.1. Structure
3.1.2. Mode of action
| Neurotransmitter | Model system | References |
|---|---|---|
| Blockage of neurotransmitter and other molecule release by BoNTs in neuronal cells | ||
| Acetylcholine (ACh) | Skeletal muscular junction | [92] |
| Torpedo electric organ | [93] | |
| Aplysia, CNS | [94] | |
| Glutamate | Brain synaptosome | [95] |
| Hind paw/mass spectrometry | [96] | |
| Cultured rat cerebellar neurons/radioassay | [97] | |
| Cultured rat cerebellar neurons/enzymatic assay | [98] | |
| Aspartate | Brain synaptosomes | [99] |
| Gamma aminobutyric acid (GABA) | Brain synaptosomes | [99,100] |
| Glycine | Spinal cord neurons (culture) | [101] |
| Dopamine | Brain synaptosomes | [100,102] |
| Adrenalin | ||
| Noradrenalin | ||
| Serotonin (or 5-HT) | Brain synaptosomes | [103] |
| ATP corelease with ACh | Torpedo synaptosomes | [104] |
| Rat bladder urothelium | [105,106] | |
| Guinea pig stellate neurons | [107] | |
| Nicotinamide adenine dinucleotide (NAD) | Canine mesenteric artery | [108] |
| Human urinary bladder detrusor muscle | [109] | |
| Effects on other neuropeptides | ||
| Substance P (SP) | Inhibition of KCl evoked SP release | |
| Calcitonin gene-related peptide (CGRP) | Inhibition of release in cultured dorsal root ganglia (DRG) neurons | [110,111] |
| Inhibition of release in cultured rat bigeninal nerve cells | [112] | |
| Rat bladder afferent neurons | [113] | |
| Upregulation and increase of CGRP | [114,115,116,117] | |
| Neurotransmitter release resistant to BoNTs | ||
| Vaso intestinal peptide (VIP), CGRP | Periglandular innervation of sweat glands | [118] |
| Neuropeptide Y | Vasoconstrictor neurons afferent to vena cava and uterine artery from guinea pig | [119] |
| SP | Capsain evoked release from cultured DRG | [110] |
| Nitric oxide (NO) Ach | Non vesicular fraction of 5-HT evoked ACh release at bronchiolar smooth muscle (ACh release by epithelial cells ?) | [119,120,121] |
| GABA | Cultured inhibitory hippocampal interneurons (BoNT-resistant SNAP25 related isoform ?) | [122] |
| Blockage of the process release in non-neuronal cells by BoNTs at high concentrations (≥100 nM) | ||
| Catecholamines | Chromaffin cells | [123,124,125] |
| ATP, glutamate | Glial cells: astrocytes or Schwann cells | [126,127,128] |
| Insulin | Pancreatic beta-cells | [129] |
| Store-mediated Ca++ entry | Exocrine pancreas cells | [130] |
| Store-mediated Ca++ entry | Platelets | [131] |
| Blockage of neurotransmitter by TeNT | ||
| Glycine | Cat, rat spinal cord | [132,133] |
| Murine spinal cord cell cultures (complete blockage of evoked and spontaneous release) | [134] | |
| GABA | Rat brain | [135] |
| Pig cerebrocortical synaptosomes | [99] | |
| Rat hippocampal slices | [136,137,138] | |
| Cerebellar cell cultures | [139] | |
| Glutamate | Murine spinal cord cell cultures (partial blockage of evoked release and increase in spontaneous release) | [134] |
| Rat brain | [140] | |
| Cerebellar neuronal cells | [141] | |
| Pig cerebrocortical synaptosomes | [99,142] | |
| Cultured hippocampal neurons (blockage of AMPA receptor insertion via SNARE-dependent exocytosis) | [143,144] | |
| Synaptosomes | [142] | |
| Aspartate | Pig cerebrocortical synaptosomes | [99] |
| Brain synaptosomes | [138] | |
| Brain slices | [145] | |
| Synaptosomes (no inhibition of evoked aspartate release) | [142] | |
| Catecholamines | Cultured brain neurons | [146] |
| Chromaffin cells | [147,148,149,150,151] | |
| Synaptosomes | [152,153] | |
| Rat brain | ||
| Serotonin | Rat brain | [152,153] |
| Synaptosomes | [154] | |
| Synaqptosomes (inhibition of serotonin uptake) | [155,156,157,158] | |
| Acetylcholine | Chromaffin PC12 cells | [159,160] |
| Synaptosomes | [161,162] | |
| Aplysia californica neurons (intraneural injection) | [163] | |
| Met-enkephalin | Pig cerebrocortical synaptosomes | [99] |
| Blockage of exocytosis in non neuronal cells by TeNT | ||
| Glutamate | Astrocytes | [164] |
| Transferrin receptor | CHO cell (cleavage of cellubrevin) | [165] |

3.1.3. Duration of intoxication
3.1.4. Other consequences of SNARE cleavage
3.1.5. Non-proteolytic molecular actions of BoNTs and TeNT
3.2. Toxins which inactivate Rho-GTPases and inhibit neuroexocytosis
3.2.1. Glucosylating clostridial toxins
3.2.2. Neuronal alterations caused by toxin inactivation of Rho GTPases
3.3. Toxins which damage neuronal cells, neuronal apoptosis
4. Toxins Which Stimulate Neurosecretion
4.1. Stimulation of glutamate release: Clostridium perfringens epsilon toxin
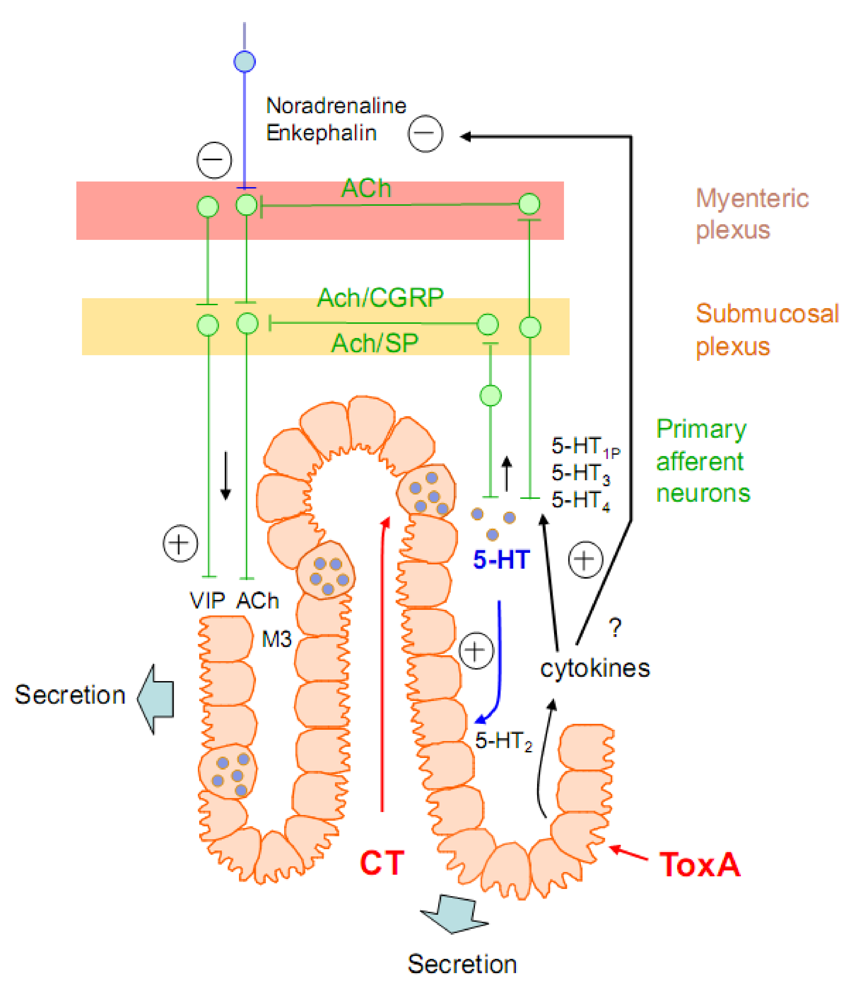
4.2. Stimulation of serotonin release and other neuromediators from the enteric nervous system
4.2.1. Bacterial enterotoxins inducing increased intestinal secretion
4.2.1.1. Cholera toxin
4.2.1.2. Enterotoxins modifying the actin cytoskeleton or inducing an intracellular second messenger

4.2.2. Emetic toxins
4.2.2.1. Bacillus cereus cereulide
4.2.2.2. Staphylococcus enterotoxins
5. Conclusions
References
- Gill, D.M. Bacterial toxins: Lethal amounts. In Toxins and Enzymes; Laskin, A.I., Lechevalier, H.A., Eds.; CRC Press: Cleveland, OH, USA, 1987; Volume 8, pp. 127–135. [Google Scholar]
- Popoff, M.R. Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect. Immun. 1987, 55, 35–43. [Google Scholar]
- Von Eichel-Streiber, C.; Harperath, U.; Bosse, D.; Hadding, U. Purification of two high molecular weigth toxins of Clostridium difficile which are antigenically related. Microb. Pathogen 1987, 2, 307–318. [Google Scholar]
- Meng, X.Q.; Kamiya, S.; Yamakawa, K.; Ogura, H.; Nakamura, S. Purification and characterisation of intracellular toxin A of Clostridium difficile. J. Med. Microbiol. 1993, 38, 69–73. [Google Scholar]
- Burleigh, D.E.; Banks, M.R. Stimulation of intestinal secretion by vasoactive intestinal peptide and cholera toxin. Auton. Neurosci. 2007, 133, 64–75. [Google Scholar]
- Murthy, V.N.; De Camilli, P. Cell biology of the presynaptic terminal. Annu. Rev. Neurosci. 2003, 26, 701–728. [Google Scholar]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; Muller, S.A.; Rammner, B.; Grater, F.; Hub, J.S.; De Groot, B.L.; Mieskes, G.; Moriyama, Y.; Klingauf, J.; Grubmuller, H.; Heuser, J.; Wieland, F.; Jahn, R. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar]
- Kasai, H. Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends Neurosci. 1999, 22, 88–93. [Google Scholar]
- Jahn, R.; Scheller, R.H. SNAREs--engines for membrane fusion. Nat. Rev. Mol. Cell. Biol. 2006, 7, 631–643. [Google Scholar]
- Rizo, J.; Rosenmund, C. Synaptic vesicle fusion. Nat. Struct. Mol. Biol. 2008, 15, 665. [Google Scholar]
- Doussau, F.; Augustine, G.J. The actin cytoskeleton and neurotransmitter release: An overview. Biochimie 2000, 82, 353–363. [Google Scholar]
- Garner, C.C.; Kindler, S.; Gundelfinger, E.D. Molecular determinants of presynaptic active zones. Curr. Opin. Neurobiol. 2000, 10, 321–327. [Google Scholar]
- Petersen, O.H. Localization and regulation of Ca2+ entry and exit pathways in exocrine gland cells. Cell Calcium 2003, 33, 337–344. [Google Scholar]
- Rettig, J.; Neher, E. Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science 2002, 298, 781–785. [Google Scholar]
- Chapman, E.R. Synaptotagmin: A Ca2+ sensor that triggers exocytosis? Nat. Rev. Mol. CellBiol. 2002, 3, 498–508. [Google Scholar] [CrossRef]
- Koh, T.W.; Bellen, H.J. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003, 26, 413–422. [Google Scholar]
- Bhalla, A.; Chicka, M.C.; Tucker, W.C.; Chapman, E.R. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol. 2006, 13, 323–330. [Google Scholar]
- Martens, S.; Kozlov, M.M.; McMahon, H.T. How synaptotagmin promotes membrane fusion. Science 2007, 316, 1205–1208. [Google Scholar]
- Groffen, A.; Friedrich, R.; Brian, E.C.; Ashery, U.; Verhage, M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J. Neurochem. 2006, 97, 818–833. [Google Scholar]
- Augustine, G.J. How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 2001, 11, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Segev, N. Ypt/rab gtpases: Regulators of protein trafficking. Sci. STKE 2001, 2001, RE11. [Google Scholar]
- Eitzen, G. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 2003, 1641, 175–181. [Google Scholar]
- Bader, M.F.; Doussau, F.; Chasserot-Golaz, S.; Vitale, N.; Gasman, S. Coupling actin and membrane dynamics during calcium-regulated exocytosis: A role for Rho and ARF GTPases. Biochim. Biophys. Acta 2004, 1742, 37–49. [Google Scholar]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar]
- Gasman, S.; Chasserot-Golaz, S.; Malacombe, M.; Way, M.; Bader, M.F. Regulated exocytosis in neuroendocrine cells: A role for subplasmalemmal Cdc42/N -WASP-induced actin filaments. Mol. Biol. Cell 2004, 15, 520–531. [Google Scholar]
- Li, Q.; Ho, C.S.; Marinescu, V.; Bhatti, H.; Bokoch, G.M.; Ernst, S.A.; Holz, R.W.; Stuenkel, E.L. Facilitation of Ca(2+)-dependent exocytosis by Rac1-GTPase in bovine chromaffin cells. J. Physiol. 2003, 550, 43–145. [Google Scholar]
- Doussau, F.; Gasman, S.; Humeau, Y.; Vitiello, F.; Popoff, M.R.; Boquet, P.; Bader, M.F.; Poulain, B. A Rho-related GTPase is involved in Ca++-dependent neurotransmitter exocytosis. J. Biol. Chem. 2000, 275, 7764–7770. [Google Scholar]
- Humeau, Y.; Popoff, M.R.; Kojima, H.; Dousseau, F.; Poulain, B. Rac GTPase plays an essential role in exocytosis by controlling the fusion competence in release sites. J. Neurosci. 2002, 22, 7968–7981. [Google Scholar]
- Momboisse, F.; Lonchamp, E.; Calco, V.; Ceridono, M.; Vitale, N.; Bader, M.F.; Gasman, S. betaPIX-activated Rac1 stimulates the activation of phospholipase D, which is associated with exocytosis in neuroendocrine cells. J. Cell. Sci. 2009, 122, 798–806. [Google Scholar]
- Vitale, N.; Chasserot-Golaz, S.; Bailly, Y.; Morinaga, N.; Frohman, M.A.; Bader, M.F. Calcium-regulated exocytosis of dense-core vesicles requires the activation of ADP-ribosylation factor (ARF)6 by ARF nucleotide binding site opener at the plasma membrane. J. Cell. Biol. 2002, 159, 79–89. [Google Scholar]
- Bielinski, D.F.; Pyun, H.Y.; Linko-Stentz, K.; Macara, I.G.; Fine, R.E. Protein Ral and Rab3a are major GTP-binding proteins of axonal rapid transport and synaptic vesicles and do not redistribute following depolarization stimulated synaptosomal exocytosis. Biochim. Biophys. Acta 1993, 1151, 246–256. [Google Scholar]
- Polzin, A.; Shipitsin, M.; Goi, T.; Feig, L.A.; Turner, T.J. Ral-GTPase influences the regulation of the readily releasable pool of synaptic vesicles. Mol. Cell Biol. 2002, 22, 1714–1722. [Google Scholar]
- Luo, J.Q.; Liu, X.; Frankel, P.; Rotunda, T.; Ramos, M.; Flom, J.; Jiang, H.; Feig, L.A.; Morris, A.J.; Kahn, R.A.; Foster, D.A. Functional association between Arf and RalA in active phospholipase D complex. Proc. Natl. Acad. Sci. USA 1998, 95, 3632–3637. [Google Scholar]
- Choi, W.S.; Kim, Y.M.; Combs, C.; Frohman, M.A.; Beaven, M.A. Phospholipases D1 and D2 regulate different phases of exocytosis in mast cells. J. Immunol. 2002, 168, 5682–5689. [Google Scholar]
- Humeau, Y.; Vitale, N.; Chasserot-Golaz, S.; Dupont, J.L.; Du, G.; Frohman, M.A.; Bader, M.F.; Poulain, B. A role for phospholipase D1 in neurotransmitter release. Proc. Natl. Acad. Sci. (USA) 2001, 98, 15300–15305. [Google Scholar]
- Humeau, Y.; Doussau, F.; Popoff, M.R.; Benfenati, F.; Poulain, B. Fast changes in the functional status of release sites during short-term plasticity: Involvement of a frequency-dependent bypass of Rac at Aplysia synapses. J. Physiol. 2007, 583, 983–1004. [Google Scholar]
- Vitale, N.; Caumont, A.S.; Chasserot-Golaz, S.; Du, G.; Wu, S.; Sciorra, V.A.; Morris, A.J.; Frohman, M.A.; Bader, M.F. Phospholipase D1: A key factor for the exocytic machinery in neuroendocrine cells. EMBOJ. 2001, 20, 2424–2434. [Google Scholar]
- Chernomordik, L.V.; Kozlov, M.M. Protein-lipid interplay in fusion and fission of biological membranes. Ann. Rev. Biochem. 2003, 72, 175–207. [Google Scholar]
- Bigalke, H.; Shoer, L.F. Clostridial neurotoxins. In Bacterial Protein Toxins; Aktories, K., Just, I., Eds.; Springer: Berlin, Germany, 2000; pp. 407–443. [Google Scholar]
- Herreros, J.; Lalli, G.; Montecucco, C.; Schiavo, G. Pathophysiological properties of clostridial neurotoxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J.E., Freer, J.H., Eds.; Academic Press: London, UK, 1999; Volume 2, pp. 202–228. [Google Scholar]
- Humeau, Y.; Doussau, F.; Grant, N.J.; Poulain, B. How botulinum and tetanus neurotoxins block neurotransmitter. Biochimie 2000, 82, 427–446. [Google Scholar]
- Meunier, F.A.; Herreros, J.; Schiavo, G.; Poulain, B.; Molgo, J. Molecular mechanism of action of botulinal neurotoxins and the synaptic remodeling they induce in vivo at the skeletal neuromuscular junction. In Handbook of Neurotoxicology; Massaro, J., Ed.; Humana Press: Totowa, NJ, USA, 2002; Volume 1, pp. 305–347. [Google Scholar]
- Meunier, F.A.; Schiavo, G.; Molgo, J. Botulinum neurotoxins: From paralysis to recovery of functional neuromuscular trasnmission. J. Physiol. 2002, 96, 105–113. [Google Scholar]
- Poulain, B.; Popoff, M.R.; Molgo, J. How do the botulinum neurotoxins block neurotransmitter release: From botulism to the molecular mechanism of action. Botulinum. J. 2008, 1, 14–87. [Google Scholar]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar]
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; Jackson, P.J.; Marks, J.D. Genetic diversity among Botulinum Neurotoxin-producing clostridial strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar]
- Smith, T.J.; Hill, K.K.; Foley, B.T.; Detter, J.C.; Munk, A.C.; Bruce, D.C.; Doggett, N.A.; Smith, L.A.; Marks, J.D.; Xie, G.; Brettin, T.S. Analysis of the neurotoxin complex genes in clostridium botulinum A1-A4 and B1 strains: BoNT/A3, /Ba4 and /B1 clusters are located within plasmids. PLoS ONE 2007, 2, e1271. [Google Scholar]
- Smith, T.J.; Lou, J.; Geren, N.; Forsyth, M.; Tsai, R.; La Porte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; Marks, J.D. Sequence variation within botulinum neurotoxin serotypes impacts antibody binding and neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar]
- Arndt, E.R.; Jacobson, M.J.; Abola, E.E.; Forsyth, C.M.; Tepp, W.H.; Marks, J.D.; Johnson, E.A.; Stevens, E.S. A structural perspective of the sequence variability within botulinum neurotoxin subtypes A1–A4. J. Mol. Biol. 2006, 362, 733–742. [Google Scholar]
- Chen, Y.; Korkeala, H.; Aarnikunnas, J.; Lindstrom, M. Sequencing the botulinum neurotoxin gene and related genes in Clostridium botulinum type E strains reveals orfx3 and a novel type E neurotoxin subtype. J. Bacteriol. 2007, 189, 8643–8650. [Google Scholar]
- Carter, A.T.; Paul, C.; Mason, D.R.; Twine, S.M.; Alston, M.J.; Logan, S.M.; Austin, J.W.; Peck, M.W. Independent evolution of neurotoxin and flagellar genetic loci in proteolytic Clostridium botulinum. BMC Genomics 2009, 10, 115. [Google Scholar]
- Popoff, M.R.; Marvaud, J.C. Structural and genomic features of clostridial neurotoxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 2nd; Alouf, J.E., Freer, J.H., Eds.; Academic Press: London, UK, 1999; Volume 2, pp. 174–201. [Google Scholar]
- Hasegawa, K.; Watanabe, T.; Suzuki, T.; Yamano, A.; Oikawa, T.; Sato, Y.; Kouguchi, H.; Yoneyama, T.; Niwa, K.; Ikeda, T.; Ohyama, T. A novel subunit structure of clostridium botulinum serotype D toxin complex with three extended arms. J. Biol. Chem. 2007, 282, 24777–24783. [Google Scholar]
- Lietzow, M.A.; Gielow, E.T.; Le, D.; Zhang, J.; Verhagen, M.F. Subunit stoichiometry of the Clostridium botulinum type A neurotoxin complex determined using denaturing capillary electrophoresis. Protein J. 2008, 27, 420–425. [Google Scholar]
- Call, J.E.; Cooke, P.H.; Miller, A.J. In situ characterization of Clostridium botulinum neurotoxin synthesis and export. J. Appl. Bacteriol. 1995, 79, 257–263. [Google Scholar]
- Emsley, P.; Fotinou, C.; Black, I.; Fairweather, N.F.; Charles, I.G.; Watts, C.; Hewitt, E.; Isaacks, N.W. The structures of the Hc fragment of Tetanus Toxin with carbohydrate subunit complexes provide insight into ganglioside binding. J. Biol. Chem. 2000, 275, 8889–8894. [Google Scholar]
- Lacy, D.B.; Stevens, R.C. Sequence homology and structural analysis of the clostridial neurotoxins. J. Mol. Biol. 1999, 291, 1091–1104. [Google Scholar]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; Das Gupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar]
- Umland, T.C.; Wingert, L.M.; Swaminathan, S.; Furey, W.F.; Schmidt, J.J.; Sax, M. The structure of the receptor binding fragment Hc of tetanus neurotoxin. Nat. Struct. Biol. 1997, 4, 788–792. [Google Scholar]
- Fotinou, C.; Emsley, P.; Black, I.; Ando, H.; Ishida, H.; Kiso, M.; Sinha, K.A.; Fairweather, N.F.; Isaacs, N.W. The crystal structure of Tetanus toxin Hc fragment complexed with a synthetic GT1b analogue suggests cross-linking between ganglioside receptors and the toxin. J. Biol. Chem. 2001, 276, 3274–3281. [Google Scholar]
- Breidenbach, M.A.; Brunger, A.T. 2.3 A crystal structure of tetanus neurotoxin light chain. Biochemistry 2005, 44, 7450–7457. [Google Scholar]
- Fu, Z.; Chen, S.; Baldwin, M.R.; Boldt, G.E.; Crawford, A.; Janda, K.D.; Barbieri, J.T.; Kim, J.J. Light chain of botulinum neurotoxin serotype A: Structural resolution of a catalytic intermediate. Biochemistry 2006, 45, 8903–8911. [Google Scholar]
- Swaminathan, S.; Eswaramoorthy, S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat. Struct. Biol. 2000, 7, 693–699. [Google Scholar]
- Stenmark, P.; Dupuy, J.; Imamura, A.; Kiso, M.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathogen 2008, 4, e1000129. [Google Scholar]
- Kumaran, D.; Eswaramoorthy, S.; Furey, W.; Navaza, J.; Sax, M.; Swaminathan, S. Domain organization in Clostridium botulinum neurotoxin type E is unique: Its implication in faster translocation. J. Mol. Biol. 2009, 386, 233–245. [Google Scholar]
- Maksymowych, A.B.; Simpson, L.L. Binding and transcytosis of botulinum neurotoxin by polarized human carcinoma cells. J. Biol. Chem. 1998, 273, 21950–21957. [Google Scholar]
- Maksymowych, A.B.; Simpson, L.I. Structural features of the botulinum neurotoxin molecule that govern binding and transcytosis across polarized human intestinal epithelial cells. J. Pharmacol. Exp. Ther. 2004, 210, 633–641. [Google Scholar]
- Ahsan, C.R.; Hajnoczky, G.; Maksymowych, A.B.; Simpson, L.L. Visualization of binding and transcytosis of botulinum toxin by human intestinal epithelial cells. J. Pharmacol. Exp. Ther. 2005, 315, 1028–1035. [Google Scholar]
- Couesnon, A.; Pereira, Y.; Popoff, M.R. Receptor-mediated transcytosis of botulinum neurotoxin A through intestinal cell monolayers. Cell Microbiol. 2008, 10, 375–387. [Google Scholar] [PubMed]
- Matsumura, T.; Jin, Y.; Kabumoto, Y.; Takegahara, Y.; Oguma, K.; Lencer, W.I.; Fujinaga, Y. The HA proteins of botulinum toxin disrupt intestinal epithelial intercellular junctions to increase toxin absorption. Cel Microbiol. 2007, 10, 355–364. [Google Scholar]
- Jin, Y.; Takegahara, Y.; Sugawara, Y.; Matsumura, T.; Fujinaga, Y. Disruption of the epithelial barrier by botulinum haemagglutinin (HA) proteins—differences in cell tropism and the mechanism of action between HA proteins of types A or B, and HA proteins of type C. Microbiology 2009, 155, 35–45. [Google Scholar]
- Wellhöner, H.H. Clostridial toxins and the central nervous system: Studies on in situ tissues. In Botulinum Neurotoxin and Tetanus Toxin; Simpson, L.L., Ed.; Academic Press: San Diego, CA, USA, 1989; pp. 231–253. [Google Scholar]
- Manning, K.A.; Erichsen, J.T.; Evinger, C. Retrograde transneuronal transport properties of fragment C of tetanus toxin. Neuroscience 1990, 34, 251–263. [Google Scholar]
- Rossetto, O.; Seveso, M.; Caccin, P.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: Turning bad guys into good by research. Toxicon 2001, 39, 27–41. [Google Scholar]
- Dong, M.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Janz, R.; Chapman, E.R. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol. Biol. Cell. 2008, 19, 522–637. [Google Scholar]
- Dong, M.; Tepp, W.H.; Liu, H.; Johnson, E.A.; Chapman, E.R. Mechanism of botulinum neurotoxin B and G entry into hippocampal neurons. J. Cell. Biol. 2007, 179, 1511–1522. [Google Scholar]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar]
- Mahrhold, S.; Rummel, A.; Bigalke, H.; Davletov, B.; Binz, T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006, 580, 2011–2014. [Google Scholar]
- Nishiki, T.; Kamata, Y.; Nemoto, Y.; Omori, A.; Ito, T.; Takahashi, M.; Kozaki, S. Identification of protein receptor for Clostridium botulinum type B neurotoxin in rat brain synaptosomes. J. Biol. Chem. 1994, 269, 10498–10503. [Google Scholar]
- Rummel, A.; Hafner, K.; Mahrhold, S.; Darashchonak, N.; Holt, M.; Jahn, R.; Beermann, S.; Karnath, T.; Bigalke, H.; Binz, T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J. Neurochem. 2009, 110, 1942–1954. [Google Scholar]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar]
- Herreros, J.; Ng, T.; Schiavo, G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol. Biol. Cell 2001, 12, 2947–2960. [Google Scholar]
- Munro, P.; Kojima, H.; Dupont, J.L.; Bossu, J.L.; Poulain, B.; Boquet, P. High sensitivity of mouse neuronal cells to tetanus toxin requires a GPI-anchored protein. Biochem. Biophys. Res. Comm. 2001, 289, 623–629. [Google Scholar]
- Rummel, A.; Bade, S.; Alves, J.; Bigalke, H.; Binz, T. Two carbohydrate binding sites in the Hcc-domain of tetanus neurotoxin are required for toxicity. J. Mol. Biol. 2003, 326, 835–847. [Google Scholar]
- Rummel, A.; Eichner, T.; Weil, T.; Karnath, T.; Gutcaits, A.; Mahrhold, S.; Sandhoff, K.; Proia, R.L.; Acharya, K.R.; Bigalke, H.; Binz, T. Identification of the protein receptor binding site of botulinum neurotoxins B and G proves the double-receptor concept. Proc. Natl. Acad. Sci. USA 2007, 104, 359–364. [Google Scholar]
- Rummel, A.; Mahrhold, S.; Bigalke, H.; Binz, T. The Hcc-domain of botulinum neurotoxins A and B exhibits a singular gangliosside binding site displaying serotype specific carbohydrate interaction. Mol. Microbiol. 2004, 51, 631–643. [Google Scholar]
- Chen, C.; Fu, Z.; Kim, J.J.; Barbieri, J.T.; Baldwin, M.R. Gangliosides as high affinity receptors for tetanus neurotoxin. J. Biol. Chem. 2009, 284, 26569–26577. [Google Scholar]
- Tsukamoto, K.; Kozai, Y.; Ihara, H.; Kohda, T.; Mukamoto, M.; Tsuji, T.; Kozaki, S. Identification of the receptor-binding sites in the carboxyl-terminal half of the heavy chain of botulinum neurotoxin types C and D. Microb. Pathog. 2008, 44, 484–493. [Google Scholar]
- Muraro, L.; Tosatto, S.; Motterlini, L.; Rossetto, O.; Montecucco, C. The N-terminal half of the receptor domain of botulinum neurotoxin A binds to microdomains of the plasma membrane. Biochem. Biophys. Res. Commun. 2009, 380, 76–80. [Google Scholar]
- Yowler, B.C.; Schengrund, C.L. Botulinum neurotoxin A changes conformation upon binding to ganglioside GT1b. Biochemistry 2004, 43, 9725–9731. [Google Scholar]
- Chen, C.; Baldwin, M.R.; Barbieri, J.T. Molecular basis for tetanus toxin coreceptor interactions. Biochemistry 2008, 47, 7179–7186. [Google Scholar]
- Burgen, A.S.; Dickens, F.; Zatman, L.J. The action of botulinum toxin on the neuro-muscular junction. J. Physiol. 1949, 109, 10–24. [Google Scholar]
- Dunant, Y.; Esquerda, J.E.; Loctin, F.; Marsal, J.; Muller, D. Botulinum toxin inhibits quantal acetylcholine release and energy metabolism in the Torpedo electric organ. J. Physiol. 1987, 385, 677–692. [Google Scholar]
- Poulain, B.; Tauc, L.; Maisey, E.A.; Wadsworth, J.D.; Mohan, P.M.; Dolly, J.O. Neurotransmitter release is blocked intracellularly by botulinum neurotoxin, and this requires uptake of both toxin polypeptides by a process mediated by the larger chain. Proc. Natl. Acad. Sci. USA 1988, 85, 4090–4094. [Google Scholar]
- Sanchez-Prieto, J.; Sihra, T.S.; Evans, D.; Ashton, A.; Dolly, J.O.; Nicholls, D.G. Botulinum toxin A blocks glutamate exocytosis from guinea-pig cerebral cortical synaptosomes. Eur. J. Biochem. 1987, 165, 675–681. [Google Scholar]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar]
- Foran, P.G.; Mohammed, N.; Lisk, G.O.; Nagwaney, S.; Lawrence, G.W.; Johnson, E.; Smith, L.; Aoki, K.R.; Dolly, O.J. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E and F compared with the long lasting type A. J. Biol. Chem. 2003, 278, 1363–1371. [Google Scholar]
- Khairallah, G.; Andreoletti, J.B.; Jover, E.; Simon, E. Measurement of botulinum toxin activity: Towards a new cellular culture assay? Ann. Chir. Plast. Esthet. 2008, 53, 42–49. [Google Scholar]
- McMahon, H.T.; Foran, P.; Dolly, J.O.; Verhage, M.; Wiegant, V.M.; Nicholls, D.G. Tetanus toxin and botulinum toxins type A and B inhibit glutamate, gamma-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. Clues to the locus of action. J. Biol. Chem. 1992, 267, 21338–21343. [Google Scholar] [PubMed]
- Ashton, A.C.; Dolly, J.O. Characterization of the inhibitory action of botulinum neurotoxin type A on the release of several transmitters from rat cerebrocortical synaptosomes. J. Neurochem. 1988, 50, 1808–1816. [Google Scholar]
- Neale, E.A.; Bowers, L.M.; Jia, M.; Bateman, K.E.; Williamson, L.C. Botulinum neurotoxin A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J. Cell. Biol. 1999, 147, 1249–1260. [Google Scholar]
- Maisey, E.A.; Wadsworth, J.D.; Poulain, B.; Shone, C.C.; Melling, J.; Gibbs, P.; Tauc, L.; Dolly, J.O. Involvement of the constituent chains of botulinum neurotoxins A and B in the blockade of neurotransmitter release. Eur. J. Biochem. 1988, 177, 683–691. [Google Scholar]
- Najib, A.; Pelliccioni, P.; Gil, C.; Aguilera, J. Clostridium neurotoxins influence serotonin uptake and release differently in rat brain synaptosomes. J. Neurochem. 1999, 72, 1991–1998. [Google Scholar]
- Marsal, J.; Egea, G.; Solsona, C.; Rabasseda, X.; Blasi, J. Botulinum toxin type A blocks the morphological changes induced by chemical stimulation on the presynaptic membrane of Torpedo synaptosomes. Proc. Natl. Acad. Sci. USA 1989, 86, 372–376. [Google Scholar]
- Khera, M.; Somogyi, G.T.; Kiss, S.; Boone, T.B.; Smith, C.P. Botulinum toxin A inhibits ATP release from bladder urothelium after chronic spinal cord injury. Neurochem. Int. 2004, 45, 987–993. [Google Scholar]
- Smith, C.P.; Vemulakonda, V.M.; Kiss, S.; Boone, T.B.; Somogyi, G.T. Enhanced ATP release from rat bladder urothelium during chronic bladder inflammation: Effect of botulinum toxin A. Neurochem. Int. 2005, 47, 291–297. [Google Scholar]
- Tompkins, J.D.; Parsons, R.L. Exocytotic release of ATP and activation of P2X receptors in dissociated guinea pig stellate neurons. Am. J. Physiol. Cell Physiol. 2006, 291, C1062–C1071. [Google Scholar]
- Smyth, L.M.; Breen, L.T.; Mutafova-Yambolieva, V.N. Nicotinamide adenine dinucleotide is released from sympathetic nerve terminals via a botulinum neurotoxin A-mediated mechanism in canine mesenteric artery. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H1818–H1825. [Google Scholar]
- Breen, L.T.; Smyth, L.M.; Yamboliev, I.A.; Mutafova-Yambolieva, V.N. beta-NAD is a novel nucleotide released on stimulation of nerve terminals in human urinary bladder detrusor muscle. Am. J. Physiol. Renal. Physiol. 2006, 290, F486–F495. [Google Scholar]
- Welch, M.J.; Purkiss, J.R.; Foster, K.A. Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon 2000, 38, 245–258. [Google Scholar]
- Duggan, M.J.; Quinn, C.P.; Chaddock, J.A.; Purkiss, J.R.; Alexander, F.C.; Doward, S.; Fooks, S.J.; Friis, L.M.; Hall, Y.H.; Kirby, E.R.; Leeds, N.; Moulsdale, H.J.; Dickenson, A.; Green, G.M.; Rahman, W.; Suzuki, R.; Shone, C.C.; Foster, K.A. Inhibition of release of neurotransmitters from rat dorsal root ganglia by a novel conjugate of a Clostridium botulinum toxin A endopeptidase fragment and Erythrina cristagalli lectin. J. Biol. Chem. 2002, 277, 34846–34852. [Google Scholar]
- Durham, P.L.; Cady, R.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache: J. Head and Face Pain 2004, 44, 35–42. [Google Scholar] [CrossRef]
- Rapp, D.E.; Turk, K.W.; Bales, G.T.; Cook, S.P. Botulinum toxin type an inhibits calcitonin gene-related peptide release from isolated rat bladder. J. Urol. 2006, 175, 1138–1142. [Google Scholar]
- Hassan, S.M.; Jennekens, F.G.; Wieneke, G.; Veldman, H. Calcitonin gene-related peptide-like immunoreactivity, in botulinum toxin-paralysed rat muscles. Neuromuscul. Disord. 1994, 4, 489–496. [Google Scholar]
- Meunier, F.A.; Colasante, C.; Faille, L.; Gastard, M.; Molgo, J. Upregulation of calcitonin gene-related peptide at mouse motor nerve terminals poisoned with botulinum type-A toxin. Pflugers. Arch. 1996, 431(Suppl. 2), R297–R298. [Google Scholar]
- Sala, C.; Andreose, J.S.; Fumagalli, G.; Lomo, T. Calcitonin gene-related peptide: Possible role in formation and maintenance of neuromuscular junctions. J. Neurosci. 1995, 15, 520–528. [Google Scholar]
- Tarabal, O.; Caldero, J.; Ribera, J.; Sorribas, A.; Lopez, R.; Molgo, J.; Esquerda, J.E. Regulation of motoneuronal calcitonin gene-related peptide (CGRP) during axonal growth and neuromuscular synaptic plasticity induced by botulinum toxin in rats. Eur. J. Neurosci. 1996, 8, 829–836. [Google Scholar]
- Swartling, C.; Naver, H.; Pihl-Lundin, I.; Hagforsen, E.; Vahlquist, A. Sweat gland morphology and periglandular innervation in essential palmar hyperhidrosis before and after treatment with intradermal botulinum toxin. J. Am. Acad. Dermatol. 2004, 51, 739–745. [Google Scholar]
- Morris, J.L.; Jobling, P.; Gibbins, I.L. Botulinum neurotoxin A attenuates release of norepinephrine but not NPY from vasoconstrictor neurons. Am. J. Physiol. Heart. Circ. Physiol. 2002, 283, H2627–H2635. [Google Scholar]
- Jones, O.M.; Brading, A.F.; Mortensen, N.J. Mechanism of action of botulinum toxin on the internal anal sphincter. Br. J. Surg. 2004, 91, 224–228. [Google Scholar]
- Moffatt, J.D.; Cocks, T.M.; Page, C.P. Role of the epithelium and acetylcholine in mediating the contraction to 5-hydroxytryptamine in the mouse isolated trachea. Br. J. Pharmacol. 2004, 141, 1159–1166. [Google Scholar]
- Verderio, C.; Pozzi, D.; Pravettoni, E.; Inverardi, F.; Schenk, U.; Coco, S.; Proux-Gillardeaux, V.; Galli, T.; Rossetto, O.; Frassoni, C.; Matteoli, M. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 2004, 41, 599–610. [Google Scholar]
- Penner, R.; Neher, E.; Dreyer, F. Intracellularly injected tetanus toxin inhibits exocytosis in bovine adrenal chromaffin cells. Nature 1986, 324, 76–78. [Google Scholar]
- Ahnert-Hilger, G.; Bader, M.F.; Bhakdi, S.; Gratzl, M. Introduction of macromolecules into bovine adrenal medullary chromaffin cells and rat pheochromocytoma cells (PC12) by permeabilization with streptolysin O: Inhibitory effect of tetanus toxin on catecholamine secretion. J. Neurochem. 1989, 52, 1751–1758. [Google Scholar]
- Ahnert-Hilger, G.; Weller, U.; Dauzenroth, M.E.; Habermann, E.; Gratzl, M. The tetanus toxin light chain inhibits exocytosis. FEBS Lett. 1989, 242, 245–248. [Google Scholar]
- Abdipranoto, A.; Liu, G.J.; Werry, E.L.; Bennett, M.R. Mechanisms of secretion of ATP from cortical astrocytes triggered by uridine triphosphate. Neuroreport 2003, 14, 2177–2181. [Google Scholar]
- Araque, A.; Li, N.; Doyle, R.T.; Haydon, P.G. SNARE protein-dependent glutamate release from astrocytes. J. Neurosci. 2000, 20, 666–673. [Google Scholar]
- Verderio, C.; Coco, S.; Rossetto, O.; Montecucco, C.; Matteoli, M. Internalization and proteolytic action of botulinum toxins in CNS neurons and astrocytes. J. Neurochem. 1999, 73, 372–379. [Google Scholar]
- Regazzi, R.; Sadoul, K.; Meda, P.; Kelly, R.B.; Halban, P.A.; Wollheim, C.B. Mutational analysis of VAMP domains implicated in Ca2+-induced insulin exocytosis. EMBO J. 1996, 15, 6951–6959. [Google Scholar]
- Rosado, J.A.; Redondo, P.C.; Salido, G.M.; Sage, S.O.; Pariente, J.A. Cleavage of SNAP-25 and VAMP-2 impairs store-operated Ca2+ entry in mouse pancreatic acinar cells. Am. J. Physiol. Cell. Physiol. 2005, 288, C214–C221. [Google Scholar]
- Redondo, P.C.; Harper, A.G.; Salido, G.M.; Pariente, J.A.; Sage, S.O.; Rosado, J.A. A role for SNAP-25 but not VAMPs in store-mediated Ca2+ entry in human platelets. J. Physiol. 2004, 558, 99–109. [Google Scholar]
- Semba, T.; Kano, M. Glycine in the spinal cord of cats with local tetanus rigidity. Science 1969, 164, 571–572. [Google Scholar]
- Fedinec, A.A.; Shank, R.P. Effect of tetanus toxin on the content of glycine, gamma-aminobutyric acid, glutamate, glutamine and aspartate in the rat spinal cord. J. Neurochem. 1971, 18, 2229–2234. [Google Scholar]
- Williamson, L.C.; Fitzgerald, S.C.; Neale, E.A. Differential effects of tetanus toxin on inhibitory and excitatory neurotransmitter release from mammalian spinal cord cells in culture. J. Neurochem. 1992, 59, 2148–2157. [Google Scholar]
- Habermann, E. Inhibition by tetanus and botulinum A toxin of the release of [3H]noradrenaline and [3H]GABA from rat brain homogenate. Experientia 1988, 44, 224–226. [Google Scholar]
- Collingridge, G.L.; Davies, J. Reversible effects of tetanus toxin on striatal-evoked responses and [3H]-gamma-aminobutyric acid release in the rat substantia nigra. Br. J. Pharmacol. 1982, 76, 403–411. [Google Scholar]
- Collingridge, G.L.; Thompson, P.A.; Davies, J.; Mellanby, J. In vitro effect of tetanus toxin on GABA release form rat hippocampal slices. J. Neurochem. 1981, 37, 1039–1041. [Google Scholar]
- Albus, U.; Habermann, E. Tetanus toxin inhibits the evoked outflow of an inhibitory (GABA) and an excitatory (D-aspartate) amino acid from particulate brain cortex. Toxicon 1983, 21, 97–110. [Google Scholar]
- Pearce, B.R.; Gard, A.L.; Dutton, G.R. Tetanus toxin inhibition of K+-stimulated [3H]GABA release from developing cell cultures of the rat cerebellum. J. Neurochem. 1983, 40, 887–890. [Google Scholar]
- Van Vliet, B.J.; Sebben, M.; Dumuis, A.; Gabrion, J.; Bockaert, J.; Pin, J.P. Endogenous amino acid release from cultured cerebellar neuronal cells: effect of tetanus toxin on glutamate release. J. Neurochem. 1989, 52, 1229–1239. [Google Scholar]
- Bagetta, G.; Nistico, G. Glutamate transmission is involved in the mechanisms of neuronal degeneration produced by intrahippocampal tetanus toxin in rats. Toxicol. Lett. 1992, 64-65, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Bradford, S.E.; Nadler, J.V. Aspartate release from rat hippocampal synaptosomes. Neuroscience 2004, 128, 751–765. [Google Scholar]
- Lu, W.; Man, H.; Ju, W.; Trimble, W.S.; MacDonald, J.F.; Wang, Y.T. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 2001, 29, 243–254. [Google Scholar]
- Lindlbauer, R.; Mohrmann, R.; Hatt, H.; Gottmann, K. Regulation of kinetic and pharmacological properties of synaptic NMDA receptors depends on presynaptic exocytosis in rat hippocampal neurones. J. Physiol. 1998, 508, 495–502. [Google Scholar]
- Fleck, M.W.; Barrionuevo, G.; Palmer, A.M. Release of D, L-threo-beta-hydroxyaspartate as a false transmitter from excitatory amino acid-releasing nerve terminals. Neurochem. Int. 2001, 39, 75–81. [Google Scholar]
- Habermann, E.; Muller, H.; Hudel, M. Tetanus toxin and botulinum A and C neurotoxins inhibit noradrenaline release from cultured mouse brain. J. Neurochem. 1988, 51, 522–527. [Google Scholar]
- Figliomeni, B.; Grasso, A. Tetanus toxin affects the K+-stimulated release of catecholamines from nerve growth factor-treated PC12 cells. Biochem. Biophys. Res. Commun. 1985, 128, 249–256. [Google Scholar]
- Bansal, M.K.; Phillips, J.H.; van Heyningen, S. The inhibition by pertussis and tetanus toxins of evoked catecholamine release from intact and permeabilized bovine adrenal chromaffin cells. FEBS Lett. 1990, 276, 165–168. [Google Scholar]
- Stecher, B.; Hens, J.; Weller, U.; Gratzl, M.; Gispen, W.H.; De Graan, P.N. Noradrenaline release from permeabilized synaptosomes is inhibited by the light chain of tetanus toxin. FEBS Lett. 1992, 312, 192–194. [Google Scholar]
- Ashton, A.C.; Dolly, J.O. Microtubules and microfilaments participate in the inhibition of synaptosomal noradrenaline release by tetanus toxin. J. Neurochem. 1997, 68, 649–658. [Google Scholar]
- Tuz, K.; Pasantes-Morales, H. Hyposmolarity evokes norepinephrine efflux from synaptosomes by a depolarization- and Ca2+-dependent exocytotic mechanism. Eur. J. Neurosci. 2005, 22, 1636–1642. [Google Scholar]
- Britton, P.; Whitton, P.S.; Bowery, N.G. Effect of tetanus toxin on basal and evoked release of 5-hydroxytryptamine and dopamine in rat hippocampus in vivo. Brain Res. 1995, 673, 331–334. [Google Scholar]
- Whitton, P.S.; Britton, P.; Bowery, N.G. Tetanus toxin alters 5-hydroxytryptamine, dopamine, and their metabolites in rat hippocampus measured by in vivo microdialysis. Neurosci. Lett. 1992, 144, 95–98. [Google Scholar]
- Gobbi, M.; Facchiano, F.; Frittoli, E.; Luini, A.; Mennini, T. Tetanus toxin inhibits depolarization-induced [3H]serotonin release from rat brain cortex synaptosomes. Neurosci. Lett. 1993, 151, 205–208. [Google Scholar]
- Inserte, J.; Najib, A.; Pelliccioni, P.; Gil, C.; Aguilera, J. Inhibition by tetanus toxin of sodium-dependent, high-affinity [3H]5-hydroxytryptamine uptake in rat synaptosomes. Biochem. Pharmacol. 1999, 57, 111–120. [Google Scholar]
- Gil, C.; Najib, A.; Aguilera, J. Serotonin transport is modulated differently by tetanus toxin and growth factors. Neurochem. Int. 2003, 42, 535–542. [Google Scholar]
- Pelliccioni, P.; Gil, C.; Najib, A.; Sarri, E.; Picatoste, F.; Aguilera, J. Tetanus toxin modulates serotonin transport in rat-brain neuronal cultures. J. Mol. Neurosci. 2001, 17, 303–310. [Google Scholar]
- Najib, A.; Pelliccioni, P.; Gil, C.; Aguilera, J. Serotonin transporter phosphorylation modulated by tetanus toxin. FEBS Lett. 2000, 486, 136–142. [Google Scholar]
- Sandberg, K.; Berry, C.J.; Eugster, E.; Rogers, T.B. A role for cGMP during tetanus toxin blockade of acetylcholine release in the rat pheochromocytoma (PC12) cell line. J. Neurosci. 1989, 9, 3946–3954. [Google Scholar]
- Sandberg, K.; Berry, C.J.; Rogers, T.B. Studies on the intoxication pathway of tetanus toxin in the rat pheochromocytoma (PC12) cell line. Binding, internalization, and inhibition of acetylcholine release. J. Biol. Chem. 1989, 264, 5679–5686. [Google Scholar] [PubMed]
- Egea, G.; Rabasseda, X.; Solsona, C.; Marsal, J.; Bizzini, B. Tetanus toxin blocks potassium-induced transmitter release and rearrangement of intramembrane particles at pure cholinergic synaptosomes. Toxicon 1990, 28, 311–318. [Google Scholar]
- Bigalke, H.; Dimpfel, W.; Habermann, E. Suppression of 3H-acetylcholine release from primary nerve cell cultures by tetanus and botulinum-A toxin. Naunyn. Schmiedebergs. Arch. Pharmacol. 1978, 303, 133–138. [Google Scholar]
- Mochida, S.; Poulain, B.; Weller, U.; Habermann, E.; Tauc, L. Light chain of tetanus toxin intracellularly inhibits acetylcholine release at neuro-neuronal synapses, and its internalization is mediated by heavy chain. FEBS Lett. 1989, 253, 47–51. [Google Scholar]
- Kang, N.; Xu, J.; Xu, Q.; Nedergaard, M.; Kang, J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 2005, 94, 4121–4130. [Google Scholar]
- Galli, T.; Chilcote, T.; Mundigl, O.; Binz, T.; Niemann, H.; De Camilli, P. Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J. Cell. Biol. 1994, 125, 1015–1024. [Google Scholar]
- Lalli, G.; Bohnert, S.; Deinhardt, K.; Verastegui, C.; Schiavo, G. The journey of tetanus and botulinum neurotoxins in neurons. Trends Microbiol. 2003, 11, 431–437. [Google Scholar]
- Lalli, G.; Schiavo, G. Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neutrophin receptor p75NTR. J. Cell Biol. 2002, 156, 233–239. [Google Scholar]
- Bohnert, S.; Deinhardt, K.; Salinas, S.; Schiavo, G. Uptake and transport of clostridium neurotoxins. In The Sourcebook of Comprehensive Bacterial Protein Toxins, 3rd; Alouf, J.E., Popoff, M.R., Eds.; Elsevier Academic Press: Amsterdam, The Netherland, 2006; pp. 390–408. [Google Scholar]
- Bohnert, S.; Schiavo, G. Tetanus toxin is transported in a novel neuronal compartment characterized by a specialized pH regulation. J. Biol. Chem. 2005, 280, 42336–42344. [Google Scholar]
- Deinhardt, K.; Berminghausen, O.; Willison, H.J.; Hopkins, C.R.; Schiavo, G. Tetanus toxin is internalized by a sequential clathrin-dependent mechanism initiated within lipid microdomains and independent of epsin1. J. Cell. Biol. 2006, 174, 459–471. [Google Scholar]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar]
- Li, Y.; Foran, P.; Lawrence, G.; Mohammed, N.; Chan-Kwo-Chion, C.; Lisk, G.; Aoki, R.; Dolly, O. Recombinant forms of tetanus toxin engineered for examining and exploiting neuronal trafficking pathways. J. Biol. Chem. 2001, 276, 31394–31401. [Google Scholar]
- Maskos, U.; Kissa, K.; St Cloment, C.; Brulet, P. Retrograde trans-synaptic transfer of green fluorescent protein allows the genetic mapping of neuronal circuits in transgenic mice. Proc. Natl. Acad.Sci. USA 2002, 99, 10120–10125. [Google Scholar]
- Galloux, M.; Vitrac, H.; Montagner, C.; Raffestin, S.; Popoff, M.R.; Chenal, A.; Forge, V.; Gillet, D. Membrane Interaction of botulinum neurotoxin A translocation (T) domain. The belt region is a regulatory loop for membrane interaction. J. Biol. Chem. 2008, 283, 27668–27676. [Google Scholar] [PubMed]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar]
- Fischer, A.; Montal, M. Crucial role of the disulfide bridge between botulinum neurotoxin light and heavy chains in protease translocation across membranes. J. Biol. Chem. 2007, 282, 29604–29611. [Google Scholar]
- Montal, M. Translocation of botulinum neurotoxin light chain protease by the heavy chain protein-conducting channel. Toxicon 2009, 9, 565–569. [Google Scholar] [CrossRef]
- Ratts, R.; Trujillo, C.; Bharti, A.; vanderSpek, J.; Harrison, R.; Murphy, J.R. A conserved motif in transmembrane helix 1 of diphtheria toxin mediates catalytic domain delivery to the cytosol. Proc. Natl. Acad. Sci. USA 2005, 102, 15635–15640. [Google Scholar]
- Tucker, W.C.; Weber, T.; Chapman, E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 2004, 304, 435–438. [Google Scholar]
- Sakaba, T.; Stein, A.; Jahn, R.; Neher, E. Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science 2005, 309, 491–494. [Google Scholar]
- Lynch, K.L.; Gerona, R.R.; Kielar, D.M.; Martens, S.; McMahon, H.T.; Martin, T.F. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol. Biol. Cell. 2008, 19, 5093–5103. [Google Scholar]
- Gerona, R.R.; Larsen, E.C.; Kowalchyk, J.A.; Martin, T.F. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 2000, 275, 6328–6336. [Google Scholar]
- Apland, J.P.; Adler, M.; Oyler, G.A. Inhibition of neurotransmitter release by peptides that mimic the N-terminal domain of SNAP-25. J. Protein. Chem. 2003, 22, 147–153. [Google Scholar]
- Gutierrez, R.; Garcia, T.; Gonzalez, I.; Sanz, B.; Hernandez, P.E.; Martin, R. A quantitative PCR-ELISA for the rapid enumeration of bacteria in refrigerated raw milk. J. Appl. MIcrobiol. 1997, 83, 518–523. [Google Scholar]
- Keller, J.E.; Neale, E.A. The role of the synaptic protein snap-25 in the potency of botulinum neurotoxin type A. J. Biol. Chem. 2001, 276, 13476–13482. [Google Scholar]
- Chen, Y.A.; Scales, S.J.; Jagath, J.R.; Scheller, R.H. A discontinuous SNAP-25 C-terminal coil supports exocytosis. J. Biol. Chem. 2001, 276, 28503–28508. [Google Scholar]
- Chen, Y.A.; Scales, S.J.; Patel, S.M.; Doung, Y.C.; Scheller, R.H. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 1999, 97, 165–174. [Google Scholar]
- Schuette, C.G.; Hatsuzawa, K.; Margittai, M.; Stein, A.; Riedel, D.; Kuster, P.; Konig, M.; Seidel, C.; Jahn, R. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 2858–2863. [Google Scholar]
- Bajohrs, M.; Rickman, C.; Binz, T.; Davletov, B. A molecular basis underlying differences in the toxicity of botulinum serotypes A and E. EMBO Rep. 2004, 5, 1090–1095. [Google Scholar]
- Salem, N.; Faundez, V.; Horng, J.T.; Kelly, R.B. A v-SNARE participates in synaptic vesicle formation mediated by the AP3 adaptor complex. Nat. Neurosci. 1998, 1, 551–556. [Google Scholar]
- Cornille, F.; Deloye, F.; Fournie-Zaluski, M.C.; Roques, B.P.; Poulain, B. Inhibition of neurotransmitter release by synthetic proline-rich peptides shows that the N-terminal domain of vesicle-associated membrane protein/synaptobrvin is critical for neuro-exocytosis. J. Biol. Chem. 1995, 270, 16826–16832. [Google Scholar]
- Foran, P.; Lawrence, G.W.; Shone, C.C.; Foster, K.A.; Dolly, J.O. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chro-maffin cells: Correlation with its blockade of catecholamine release. Biochemistry 1996, 35, 2630–2636. [Google Scholar]
- Vaidyanathan, V.V.; Yoshino, K.; Jahnz, M.; Dorries, C.; Bade, S.; Nauenburg, S.; Niemann, H.; Binz, T. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: Domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J. Neurochem. 1999, 72, 327–337. [Google Scholar]
- O'Connor, V.; Heuss, C.; De Bello, W.M.; Dresbach, T.; Charlton, M.P.; Hunt, J.H.; Pellegrini, L.L.; Hodel, A.; Burger, M.M.; Betz, H.; Augustine, G.J.; Schafer, T. Disruption of syntaxin-mediated protein interactions blocks neurotransmitter secretion. Proc. Natl. Acad. Sci. USA 1997, 94, 12186–12191. [Google Scholar]
- Capogna, M.; McKinney, R.A.; O'Connor, V.; Gahwiler, B.H.; Thompson, S.M. Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J. Neurosci. 1997, 17, 7190–7202. [Google Scholar]
- Williamson, L.C.; Halpern, J.L.; Montecucco, C.; Brown, J.E.; Neale, E.A. Clostridial neurotoxins and substrate proteolysis in intact neurons: Botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J. Biol. Chem. 1996, 271, 7694–7699. [Google Scholar]
- Poulain, B.; Stiles, B.G.; Popoff, M.R.; Molgo, J. Attack of the nervous system by clostridial toxins: Physical findings, cellular and molecular actions. In The Sourcebook of Bacterial Protein Toxins, 3rd; Alouf, J.E., Popoff, M.R., Eds.; Elsevier Academic Press: Amsterdam, The Netherland, 2006; pp. 348–389. [Google Scholar]
- Keller, J.E.; Neale, E.A.; Oyler, G.; Adler, M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999, 456, 137–142. [Google Scholar]
- O'Sullivan, G.A.; Mohammed, N.; Foran, P.G.; Lawrence, G.W.; Dolly, J.O. Rescue of exocytosis in botulinum toxin A-poisoned chromaffin cells by expression of cleavage-resistant SNAP-25. J. Biol. Chem. 1999, 274, 36897–36904. [Google Scholar]
- Fernandez-Salas, E.; Steward, L.E.; Ho, H.; Garay, P.E.; Sun, S.W.; Gilmore, M.A.; Ordas, J.V.; Wang, J.; Francis, J.; Aoki, K.R. Plasma membrane localization signals in the light chain of botulinum neurotoxin. Proc. Natl. Acad. Sci. USA 2004, 101, 3208–3213. [Google Scholar]
- Hayashi, T.; McMahon, H.; Yamashi, S.; Binz, T.; Hata, Y.; Südhof, T.C.; Niemann, H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994, 13, 5051–5061. [Google Scholar]
- Pellegrini, L.L.; O'Connor, V.; Lottspeich, F.; Betz, H. Clostridial neurotoxins compromise the stability of a low energy SNARE complex mediating NSF activation of synaptic vesicle fusion. EMBO J. 1995, 14, 4705–4713. [Google Scholar]
- Cohen, R.; Atlas, D. R-type voltage-gated Ca(2+) channel interacts with synaptic proteins and recruits synaptotagmin to the plasma membrane of Xenopus oocytes. Neuroscience 2004, 128, 831–841. [Google Scholar]
- Degtiar, V.E.; Scheller, R.H.; Tsien, R.W. Syntaxin modulation of slow inactivation of N-type calcium channels. J. Neurochem. 2000, 20, 4355–4367. [Google Scholar]
- Stanley, E.F. Syntaxin I modulation of presynaptic calcium channel inactivation revealed by botulinum toxin C1. Eur. J. Neurosci. 2003, 17, 1303–1305. [Google Scholar]
- Wiser, O.; Trus, M.; Hernandez, A.; Renstrom, E.; Barg, S.; Rorsman, P.; Atlas, D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc. Natl. Acad. Sci. USA 1999, 96, 248–253. [Google Scholar]
- Bergsman, J.B.; Tsien, R.W. Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J. Neurosci. 2000, 20, 4368–4378. [Google Scholar]
- Aleu, J.; Blasi, J.; Solsona, C.; Marsal, J. Calcium-dependent acetylcholine release from Xenopus oocytes: Simultaneous ionic currents and acetylcholine release recordings. Eur. J. Neurochem. 2002, 8, 1442–1448. [Google Scholar]
- Stanley, E.F.; Mirotznik, R.R. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature 1997, 385, 340–343. [Google Scholar]
- Ji, J.; Tsuk, S.; Salapatek, A.M.; Huang, X.; Chikvashvili, D.; Pasyk, E.A.; Kang, Y.; Sheu, L.; Tsushima, R.; Diamant, N.; Trimble, W.S.; Lotan, I.; Gaisano, H.Y. The 25-kDa synaptosome-associated protein (SNAP-25) binds and inhibits delayed rectifier potassium channels in secretory cells. J. Biol. Chem. 2002, 277, 20195–20204. [Google Scholar]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar]
- Lewis, R.S. The molecular choreography of a store-operated calcium channel. Nature 2007, 446, 284–287. [Google Scholar]
- Yao, Y.; Ferrer-Montiel, A.V.; Montal, M.; Tsien, R.Y. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell 1999, 98, 475–485. [Google Scholar]
- Alderton, J.M.; Ahmed, S.A.; Smith, L.A.; Steinhardt, R.A. Evidence for a vesicle-mediated maintenance of store-operated calcium channels in a human embryonic kidney cell line. Cell Calcium 2000, 28, 161–169. [Google Scholar]
- Rosado, J.A.; Redondo, P.C.; Sage, S.O.; Pariente, J.A.; Salido, G.M. Store-operated Ca2+ entry: Vesicle fusion or reversible trafficking and de novo conformational coupling? J. Cell Physiol. 2005, 205, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Woodard, G.E.; Salido, G.M.; Rosado, J.A. Enhanced exocytotic-like insertion of Orai1 into the plasma membrane upon intracellular Ca2+ store depletion. Am. J. Physiol. Cell Physiol. 2008, 294, C1323–C1331. [Google Scholar]
- Fili, O.; Michaelevski, I.; Bledi, Y.; Chikvashvili, D.; Singer-Lahat, D.; Boshwitz, H.; Linial, M.; Lotan, I. Direct interaction of a brain voltage-gated K+ channel with syntaxin 1A: Functional impact on channel gating. J. Neurosci. 2001, 21, 1964–1974. [Google Scholar]
- Michaelevski, I.; Chikvashvili, D.; Tsuk, S.; Singer-Lahat, D.; Kang, Y.; Linial, M.; Gaisano, H.Y.; Fili, O.; Lotan, I. Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. J. Biol. Chem. 2003, 278, 34320–34330. [Google Scholar] [PubMed]
- Tsuk, S.; Michaelevski, I.; Bentley, G.N.; Joho, R.H.; Chikvashvili, D.; Lotan, I. Kv2.1 channel activation and inactivation is influenced by physical interactions of both syntaxin 1A and the syntaxin 1A/soluble N-ethylmaleimide-sensitive factor-25 (t-SNARE) complex with the C terminus of the channel. Mol. Pharmacol. 2005, 67, 480–488. [Google Scholar] [PubMed]
- Rossetto, O.; Morbiato, L.; Caccin, P.; Rigoni, M.; Montecucco, C. Presynaptic enzymatic neurotoxins. J. Neurochem. 2006, 97, 1534–1545. [Google Scholar]
- Li, Y.; Foran, P.; Fairweather, N.F.; de Paiva, A.; Weller, U.; Dougan, G.; Dolly, J.O. A single mutation in the recombinant light chain of tetanus toxin abolishes its proteolytic activity and removes the toxicity seen after reconstitution with native heavy chain. Biochemistry 1994, 33, 7014–7020. [Google Scholar]
- Yamasaki, S.; Baumeister, A.; Binz, T.; Blasi, J.; Link, E.; Cornille, F.; Roques, B.; Fykse, E.M.; Südhof, T.C.; Jahn, R.; Niemann, H. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J. Biol. Chem. 1994, 269, 12764–12772. [Google Scholar]
- Ashton, A.C.; Li, Y.; Doussau, F.; Weller, U.; Dougan, G.; Poulain, B.; Dolly, O. Tetanus toxin inhibits neuroexocytosis even when its Zn2+-dependent proteasea ctivity is removed. J. Biol. Chem. 1995, 270, 31386–31390. [Google Scholar]
- Niemann, H.; Blasi, J.; Jahn, R. Clostridial neurotoxins: New tools for dissecting exocytosis. Trends Cell Biol. 1994, 4, 179–185. [Google Scholar]
- De Paiva, A.; Ashton, A.C.; Foran, P.; Schiavo, G.; Montecucco, C.; Dolly, J.O. Botulinum A like type B and tetanus toxins fulfils criteria for being a zinc-dependent protease. J. Neurochem. 1993, 61, 2338–2341. [Google Scholar]
- Cenci Di Bello, I.; Poulain, B.; Shone, C.C.; Tauc, L.; Dolly, J.O. Antagonism of the intracellular action of botulinum neurotoxin type A with monoclonal antibodies that map to light-chain epitopes. Eur. J. Biochem. 1994, 219, 161–169. [Google Scholar]
- Facchiano, F.; Benfenati, F.; Valtorta, F.; Luini, A. Covalent modification of synapsin I by a tetanus toxin-activated transglutaminase. J. Biol. Chem. 1993, 268, 4588–4591. [Google Scholar]
- Facchiano, F.; Luini, A. Tetanus toxin potently stimulates tissue transglutaminase. A possible mechanism of neurotoxicity. J. Biol. Chem. 1992, 267, 13267–13271. [Google Scholar] [PubMed]
- Coffield, J.A.; Considine, R.V.; Jeyapaul, J.; Maksymowych, A.B.; Zhang, R.D.; Simpson, L.L. The role of transglutaminase in the mechanism of action of tetanus toxin. J. Biol. Chem. 1994, 269, 24454–24458. [Google Scholar]
- Gobbi, M.; Frittoli, E.; Mennini, T. Role of transglutaminase in [3H]5-HT release from synaptosomes and in the inhibitory effect of tetanus toxin. Neurochem. Int. 1996, 29, 129–134. [Google Scholar]
- Fesus, L.; Piacentini, M. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 27, 534–539. [Google Scholar]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar]
- Maggio, N.; Sellitti, S.; Capano, C.P.; Papa, M. Tissue-transglutaminase in rat and human brain: light and electron immunocytochemical analysis and in situ hybridization study. Brain Res. Bull. 2001, 56, 173–182. [Google Scholar]
- Walther, D.J.; Peter, J.U.; Winter, S.; Holtje, M.; Paulmann, N.; Grohmann, M.; Vowinckel, J.; Alamo-Bethencourt, V.; Wilhelm, C.S.; Ahnert-Hilger, G.; Bader, M. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 2003, 115, 851–862. [Google Scholar]
- Driscoll, H.K.; Adkins, C.D.; Chertow, T.E.; Cordle, M.B.; Matthews, K.A.; Chertow, B.S. Vitamin A stimulation of insulin secretion: effects on transglutaminase mRNA and activity using rat islets and insulin-secreting cells. Pancreas 1997, 15, 69–77. [Google Scholar]
- Pastuszko, A.; Wilson, D.F.; Erecinska, M. A role for transglutaminase in neurotransmitter release by rat brain synaptosomes. J. Neurochem. 1986, 46, 499–508. [Google Scholar]
- Humeau, Y.; Doussau, F.; Vittello, F.; Greengard, P.; Benfenati, F.; Poulain, B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J. Neurosci. 2001, 21, 4195–4206. [Google Scholar]
- Baldelli, P.; Fassio, A.; Valtorta, F.; Benfenati, F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J. Neurosci. 2007, 27, 13520–13531. [Google Scholar]
- Presek, P.; Jessen, S.; Dreyer, F.; Jarvie, P.E.; Findik, D.; Dunkley, P.R. Tetanus toxin inhibits depolarization-stimulated protein phosphorylation in rat cortical synaptosomes: Effect on synapsin I phosphorylation and translocation. J. Neurochem. 1992, 59, 1336–1343. [Google Scholar]
- Dayanithi, G.; Stecher, B.; Höhne-Zell, B.; Yamasaki, S.; Binz, T.; Weller, U.; Niemann, H.; Gratzl, M. Exploring the functional domain and the target of the tetanus toxin light chain in neurophysial terminals. Neuroscience 1994, 58, 423–431. [Google Scholar]
- DasGupta, B.R.; Tepp, W. Protease activity of botulinum neurotoxin type E and its light chain: Cleavage of actin. Biochem. Biophys. Res. Commun. 1993, 190, 470–474. [Google Scholar]
- Marxen, P.; Bigalke, H. Tetanus and botulinum A toxins inhibit stimulated F-actin rearrangement in chromaffin cells. Neuroreport 1991, 2, 33–36. [Google Scholar]
- Eisel, U.; Reynolds, K.; Riddick, M.; Zimmer, A.; Niemann, H.; Zimmer, A. Tetanus toxin light chain expression in Sertoli cells of transgenic mice causes alterations of the actin cytoskeleton and disrupts spermatogenesis. EMBO J. 1993, 12, 3365–3372. [Google Scholar]
- Ishida, H.; Zhang, X.; Erickson, K.; Ray, P. Botulinum toxin type A targets RhoB to inhibit lysophosphatidic acid-stimulated actin reorganization and acetylcholine release in nerve growth factor-treated PC12 cells. J. Pharmacol. Exp. Ther. 2004, 310, 881–889. [Google Scholar]
- Nevins, A.K.; Thurmond, D.C. A Direct interaction between Cdc42 and vesicle-associated membrane protein 2 Regulates SNARE-dependent insulin exocytosis. J. Biol. Chem. 2005, 280, 1944–1952. [Google Scholar]
- Aguilera, J.; Yavin, E. In vivo translocation and down-regulation of protein kinase C following intraventricular administration of tetanus toxin. J. Neurochem. 1990, 54, 339–342. [Google Scholar]
- Gil, C.; Ruiz-Meana, M.; Alava, M.; Yavin, E.; Aguilera, J. Tetanus toxin enhances protein kinase C activity translocation and increases polyphosphoinositide hydrolysis in rat cerebral cortex preparations. J. Neurochem. 1998, 70, 1636–1643. [Google Scholar]
- Gil, C.; Chaib-Oukadour, I.; Pelliccioni, P.; Aguilera, J. Activation of signal transduction pathways involving trkA, PLCgamma-1, PKC isoforms and ERK-1/2 by tetanus toxin. FEBS Lett. 2000, 481, 177–182. [Google Scholar]
- Gil, C.; Chaib-Oukadour, I.; Aguilera, J. C-terminal fragment of tetanus toxin heavy chain activates Akt and MEK/ERK signalling pathways in a Trk receptor-dependent manner in cultured cortical neurons. Biochem. J. 2003, 15, 613–620. [Google Scholar]
- Chaib-Oukadour, I.; Gil, C.; Aguilera, J. The C-terminal domain of the heavy chain of tetanus toxin rescues cerebellar granule neurones from apoptotic death: Involvement of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. J. Neurochem. 2004, 90, 1227–1236. [Google Scholar]
- Chaib-Oukadour, I.; Gil, C.; Rodriguez-Alvarez, J.; Ortega, A.; Aguilera, J. Tetanus toxin H(C) fragment reduces neuronal MPP+ toxicity. Mol. Cell Neurosci. 2009, 41, 297–303. [Google Scholar]
- Mendieta, L.; Venegas, B.; Moreno, N.; Patricio, A.; Martinez, I.; Aguilera, J.; Limon, I.D. The carboxyl-terminal domain of the heavy chain of tetanus toxin prevents dopaminergic degeneration and improves motor behavior in rats with striatal MPP(+)-lesions. Neurosci. Res. 2009, 65, 98–106. [Google Scholar]
- Chaib-Oukadour, I.; Gil, C.; Aguilera, J. The C-terminal domain of the heavy chain of tetanus toxin rescues cerebellar granule neurones from apoptotic death: Involvement of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. J. Neurochem. 2004, 90, 1227–1236. [Google Scholar]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: The ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature (London) 1995, 375, 500–503. [Google Scholar]
- Just, I.; Wilm, M.; Selzer, J.; Rex, G.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 1995, 270, 13932–13936. [Google Scholar]
- Popoff, M.R.; Chaves-Olarte, E.; Lemichez, E.; Von Eichel-Streiber, C.; Thelestam, M.; Chardin, P.; Cussac, D.; Antonny, B.; Chavrier, P.; Flatau, G.; Giry, M.; de Gunzburg, J.; Boquet, P. Ras, Rap, and rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J. Biol. Chem. 1996, 271, 10217–10224. [Google Scholar]
- Hermann, C.; Ahmadian, M.R.; Hofmann, F.; Just, I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J. Biol. Chem. 1998, 273, 16134–16139. [Google Scholar]
- Vetter, I.R.; Hofmann, F.; Wohlgemuth, S.; Hermann, C.; Just, I. Structural consequences of monoglucosylation of Ha-Ras by Clostridium sordellii lethal toxin. J. Mol. Biol. 2000, 301, 1091–1095. [Google Scholar]
- Popoff, M.R.; Bouvet, P. Clostridial toxins. Future Microbiol. 2009, 4, 1021–1064. [Google Scholar]
- Aktories, K.; Just, I. Clostridial Rho-inhibiting protein toxins. Curr. Top Microbiol. Immunol. 2005, 291, 113–145. [Google Scholar]
- Vogelsgesang, M.; Pautsch, A.; Aktories, K. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 374, 347–360. [Google Scholar]
- Djouder, N.; Aneiros, E.; Cavalie, A.; Aktories, K. Effects of large clostridial cytotoxins on activation of RBL 2H3-hm1 mast cells indicate common and different roles of Rac in FcepsilonRI and M1-receptor signaling. J. Pharmacol. Exp. Ther. 2003, 304, 1243–1250. [Google Scholar]
- Gasman, S.; Chasserot-Golaz, S.; Popoff, M.R.; Aunis, D.; Bader, M.F. Involvement of Rho GTPases in calcium-regulated exocytosis from adrenal chromaffin cells. J. Cell Sci. 1999, 112, 4763–4771. [Google Scholar]
- Kowluru, A.; Li, G.; Rabaglia, M.E.; Segu, V.B.; Hofmann, F.; Aktories, K.; Metz, S.A. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cells. Biochem. Pharmacol. 1997, 54, 1097–1108. [Google Scholar]
- Prepens, U.; Just, I.; von Eichel-Streiber, C.; Aktories, K. Inhibition of Fc epsilon-RI-mediated activation of rat basophilic leukemia cells by Clostridium difficile toxin B (monoglucosyltransferase). J. Biol. Chem. 1996, 271, 7324–7329. [Google Scholar]
- Barbier, J.; Popoff, M.R.; Molgo, J. Degeneration and regeneration of murine skeletal neuromuscular junctions after intramuscular injection with a sublethal dose of Clostridium sordellii lethal toxin. Infect. Immun. 2004, 72, 3120–3128. [Google Scholar]
- Geny, B.; Khum, H.; Fitting, C.; Zarantonelli, L.; Mazuet, C.; Cayet, N.; Szatanik, M.; Prevost, M.C.; Cavaillon, J.M.; Huerre, M.; Popoff, M.R. Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am. J. Pathol. 2007, 170, 1003–1017. [Google Scholar]
- Pothoulakis, C.; Castagliuolo, I.; LaMont, J.T. Nerves and intestinal mast cells modulate responses to enterotoxins. News Physiol. Sci. 1998, 13, 58–63. [Google Scholar]
- Djouder, N.; Prepens, U.; Aktories, K.; Cavalie, A. Inhibition of calcium release-activated calcium current by Rac/Cdc42-inactivating clostridial cytotoxins in RBL cells. J. Biol. Chem. 2000, 275, 18732–18738. [Google Scholar]
- Short, B.; Barr, F.A. Membrane traffic: Exocyst III--makes a family. Curr. Biol. 2002, 12, R18–R20. [Google Scholar]
- Ben El Hadj, N.; Popoff, M.R.; Marvaud, J.C.; Payrastre, B.; Boquet, P.; Geny, B. G-protein-stimulated phospholipase D activity is inhibited by lethal toxin from Clostridium sordellii in HL-60 cells. J. Biol. Chem. 1999, 274, 14021–14031. [Google Scholar]
- Hammond, K.; Caputo, G.A.; London, E. Interaction of the membrane-inserted diphtheria toxin T domain with peptides and its possible implications for chaperone-like T domain behavior. Biochemistry 2002, 41, 3243–3253. [Google Scholar]
- Meyer, D.K.; Olenik, C.; Hofmann, F.; Barth, H.; Leemhuis, J.; Brunig, I.; Aktories, K.; Norenberg, W. Regulation of somatodendritic GABAA receptor channels in rat hippocampal neurons: Evidence for a role of the small GTPase Rac1. J. Neurosci. 2000, 20, 6743–6751. [Google Scholar]
- Murray, H.J.; O'Connor, J.J. A role for monomeric G-proteins in synaptic plasticity in the rat dentate gyrus in votro. Brain Res. 2004, 1000, 85–91. [Google Scholar]
- Triller, A.; Choquet, D. Synaptic structure and diffusion dynamics of synaptic receptors. Biol. Cell 2003, 95, 465–476. [Google Scholar]
- Linseman, D.A.; Laessig, T.; Meintzer, M.K.; McClure, M.; Barth, H.; Aktories, K.; Heidenreich, K.A. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J. Biol. Chem. 2001, 276, 39123–39131. [Google Scholar]
- Marriott, H.M.; Mitchell, T.J.; Dockrell, D.H. Pneumolysin: A double-edged sword during the host-pathogen interaction. Curr. Mol. Med. 2008, 8, 497–509. [Google Scholar]
- Rossjohn, J.; Gilbert, R.J.; Crane, D.; Morgan, P.J.; Mitchell, T.J.; Rowe, A.J.; Andrew, P.W.; Paton, J.C.; Tweten, R.K.; Parker, M.W. The molecular mechanism of pneumolysin, a virulence factor from Streptococcus pneumoniae. J. Mol. Biol. 1998, 284, 449–461. [Google Scholar]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar]
- Mitchell, T.J. Pneumolysin: Structure, function, and role in disease. In The Sourcebook of Bacterial Protein Toxins, 3rd; Alouf, J.E., Popoff, M.R., Eds.; Elsevier Academic Press: Amsterdam, The Netherland, 2006; pp. 680–699. [Google Scholar]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J. Biol. Chem. 2007, 282, 15709–15716. [Google Scholar]
- Shepard, L.; Shatursky, O.; Johnson, A.; Tweten, R. The mechanism of pore assembly for a cholesterol-dependent cytolysin: formation of a large prepore complex precedes the insertion of hte transmembrane b-hairpins. Biochemistry 2000, 39, 10284–10293. [Google Scholar]
- Dang, T.X.; Hotze, E.M.; Rouiller, I.; Tweten, R.K.; Wilson-Kubalek, E.M. Prepore to pore transition of a cholesterol-dependent cytolysin visualized by electron microscopy. J. Struct. Biol. 2005, 150, 100–108. [Google Scholar]
- Ramachandran, R.; Tweten, R.K.; Johnson, A.E. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit β-strand alignment. Nat. Struct. Mol. Biol. 2004, 11, 697–705. [Google Scholar]
- Heuck, A.P.; Savva, C.G.; Holzenburg, A.; Johnson, A.E. Conformational changes that effect oligomerization and initiate pore formation are triggered throughout perfringolysin O upon binding to cholesterol. J. Biol. Chem. 2007, 282, 22629–22637. [Google Scholar]
- Rossjohn, J.; Polekhina, G.; Feil, S.C.; Morton, C.J.; Tweten, R.K.; Parker, M.W. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 2007, 367, 1227–1236. [Google Scholar]
- Ramachandran, R.; Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Biol. 2002, 9, 823–827. [Google Scholar]
- Heuck, A.P.; Hotze, E.M.; Tweten, R.K.; Johnson, A.E. Mechanism of membrane insertion of a multimeric beta-barrel protein: Perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell. 2000, 6, 1233–1242. [Google Scholar]
- Heuck, A.P.; Tweten, R.K.; Johnson, A.E. β-barrel pore-forming toxins: Intriguing dimorphic proteins. Biochemistry 2001, 40, 9065–9073. [Google Scholar]
- Shatursky, O.; Heuck, A.; Shepard, L.; Rossjhon, J.; Parker, M.; Johnson, A.; Tweten, R. The mechanism of membrane insertion of a cholesterol-dependent cytolysin: A novel paradigm for pore-forming toxins. Cell 1999, 99, 293–299. [Google Scholar]
- Hotze, E.M.; Heuck, A.P.; Czajkowsky, D.M.; Shao, Z.; Johnson, A.E.; Tweten, R.K. Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem. 2002, 277, 11597–11605. [Google Scholar]
- Rossjohn, J.; Feil, S.C.; McKinstry, W.J.; Tweten, R.K.; Parker, M.W. Structure of a cholesterol-binding thiol-activated cytolysin and a model of its membrane form. Cell 1997, 89, 685–692. [Google Scholar]
- Czajkowsky, D.M.; Hotze, E.M.; Shao, Z.; Tweten, R.K. Vertical collapse of a cytolysin prepore moves its transmembrane beta-hairpins to the membrane. Embo J. 2004, 23, 3206–3215. [Google Scholar]
- Hirst, R.A.; Kadioglu, A.; O'Callaghan, C.; Andrew, P.W. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin. Exp. Immunol. 2004, 138, 195–201. [Google Scholar]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J. Clin. Invest. 2002, 109, 19–27. [Google Scholar]
- Braun, J.S.; Hoffmann, O.; Schickhaus, M.; Freyer, D.; Dagand, E.; Bermpohl, D.; Mitchell, T.J.; Bechmann, I.; Weber, J.R. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect. Immun. 2007, 75, 4245–4254. [Google Scholar]
- Stringaris, A.K.; Geisenhainer, J.; Bergmann, F.; Balshusemann, C.; Lee, U.; Zysk, G.; Mitchell, T.J.; Keller, B.U.; Kuhnt, U.; Gerber, J.; Spreer, A.; Bahr, M.; Michel, U.; Nau, R. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol. Dis. 2002, 11, 355–368. [Google Scholar]
- Goldstein, J.; Morris, W.E.; Loidl, C.F.; Tironi-Farinatti, C.; McClane, B.A.; Uzal, F.A.; Fernandez Miyakawa, M.E. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS One 2009, 4, e7065. [Google Scholar]
- Losada-Eaton, D.M.; Uzal, F.A.; Fernandez Miyakawa, M.E. Clostridium perfringens epsilon toxin is absorbed from different intestinal segments of mice. Toxicon 2008, 51, 1207–1213. [Google Scholar]
- Finnie, J.W. Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: A review. Aust. Vet J. 2003, 81, 219–221. [Google Scholar]
- Payne, D.; Williamson, E.D.; Titball, R.W. The Clostridium perfringens epsilon-toxin. Rev. Med. Microbiol. 1997, 8, S28–S30. [Google Scholar]
- Nagahama, M.; Sakurai, J. Distribution of labeled Clostridium perfringens epsilon toxin in mice. Toxicon 1991, 29, 211–217. [Google Scholar]
- Nagahama, M.; Sakurai, J. High-affinity binding of Clostridium perfringens epsilon-toxin to rat brain. Infect. Immun. 1992, 60, 1237–1240. [Google Scholar]
- Zhu, C.; Ghabriel, M.N.; Blumbergs, P.C.; Reilly, P.L.; Manavis, J.; Youssef, J.; Hatami, S.; Finnie, J.W. Clostridium perfringens prototoxin-induced alteration of endothelial barrier antigen (EBA) immunoreactivity at the blood brain barrier (BBB). Exp. Neurol. 2001, 169, 72–82. [Google Scholar]
- Buxton, D. The use of an imunoperoxidase technique to investigate by light and electron microscopy the sites of binding of Clostridium welchii type D e-toxin in mice. J. Med. Microbiol. 1978, 11, 289–292. [Google Scholar]
- Finnie, J.W.; Blumbergs, P.C.; Manavis, J. Neuronal damage produced in rat brains by Clostridium perfringens type D epsilon-toxin. J. Comp. Path. 1999, 120, 415–420. [Google Scholar]
- Miyamoto, O.; Minami, J.; Toyoshima, T.; Nakamura, T.; Masada, T.; Nagao, S.; Negi, T.; Itano, T.; Okabe, A. Neurotoxicity of Clostridium perfringens epsilon-toxin for the rat hipocampus via glutamanergic system. Infect. Immun. 1998, 66, 2501–2508. [Google Scholar]
- Miyamoto, O.; Sumitami, K.; Nakamura, T.; Yamagani, S.; Miyatal, S.; Itano, T.; Negi, T.; Okabe, A. Clostridium perfringens epsilon toxin causes excessive release of glutamate in the mouse hippocampus. FEMS Microbiol. Lett. 2000, 189, 109–113. [Google Scholar]
- Cole, A.R.; Gibert, M.; Popoff, M.R.; Moss, D.S.; Titball, R.W.; Basak, A. Clostridium perfringens ε-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat. Struct. Mol. Biol. 2004, 11, 797–798. [Google Scholar]
- Knapp, O.; Maier, E.; Benz, R.; Geny, B.; Popoff, M.R. Identification of the channel-forming domain of Clostridium perfringens Epsilon-toxin (ETX). Biochim. Biophys. Acta 2009, 1788, 2584–2593. [Google Scholar]
- Cole, A. Structural studies on epsilon toxin from Clostridium perfringens. In Protein Toxins of the Genus Clostridium and Vaccination; Duchesnes, C., Mainil, J., Popoff, M.R., Titball, R., Eds.; Presses de la Faculté de Médecine Vétérinaire: Liège, Montreal, Canada, 2003; p. 95. [Google Scholar]
- Miyata, S.; Minami, J.; Tamai, E.; Matsushita, O.; Shimamoto, S.; Okabe, A. Clostridium perfringens ε-toxin forms a heptameric pore within the detergent-insoluble microdomains of Madin-Darby Canine Kidney Cells and rat synaptosomes. J. Biol. Chem. 2002, 277, 39463–39468. [Google Scholar]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon-toxin induces a rapid change in cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar]
- Petit, P.; Breard, J.; Montalescol, V.; Ben El Hadj, N.; Levade, T.; Popoff, M.R.; Geny, B. Lethal toxin from Clostridium sordellii induces apoptotic cell death by disruption of mitochondrial homeostasis in HL-60 cells. Cell. Miccrobiol. 2003, 5, 761–771. [Google Scholar]
- Miyata, S.; Matsushita, O.; Minami, J.; Katayama, S.; Shimamoto, S.; Okabe, A. Cleavage of C-terminal peptide is essential for heptamerization of Clostridium perfringens ε-toxin in the synaptosomal membrane. J. Biol. Chem. 2001, 276, 13778–13783. [Google Scholar]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens Epsilon Toxin rapidly decreases membrane barrier permeability of polarized MDCK Cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; Vandewalle, A. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal. Physiol. 2007, 293, F927–F937. [Google Scholar]
- Farthing, M.J. Enterotoxins and the enteric nervous system--a fatal attraction. Int. J. Med. Microbiol. 2000, 290, 491–496. [Google Scholar]
- Farthing, M.J.G.; Casburn-Jones, A.; Banks, M.R. Enterotoxins, enteric nerves, and intestinal secretion. Curr. Gastroenterol. Rep. 2004, 6, 177–180. [Google Scholar]
- Hirst, T.R.; D'Souza, J.M. Vibrio cholerae and Escherichia coli thermolabile enterotoxin. In The Sourcebook of Bacterial Protein Toxins, 3rd; Alouf, J.E., Popoff, M.R., Eds.; Elmsevier Academic Press: Amsterdam, The Netherland, 2006; pp. 270–290. [Google Scholar]
- Holmes, R.K.; Jobling, M.G.; Conell, T.D. Cholera toxin and related enterotoxins of Gram-negative bacteria. In Bacterial Toxins and Virulence Factors in Disease; Moss, J., Iglewski, B., Vaughan, M., Tu, A.T., Eds.; Marcel Dekker: New York, NY, USA, 1995; pp. 225–255. [Google Scholar]
- de Haan, L.; Hirst, T.R. Cholera toxin: A paradigm for multi-functional engagement of cellular mechanisms. Mol. Membr. Biol. 2004, 21, 77–92. [Google Scholar]
- Nichols, B.J. A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 2002, 4, 374–378. [Google Scholar]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.E.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulum. Mol. Biol. Cell 2003, 14, 4783–4793. [Google Scholar]
- Johannes, L.; Tenza, D.; Antony, C.; Goud, B. Retrograde transport of KDEL-bearing B-fragment of Shiga toxin. J. Biol. Chem. 1997, 272, 19554–19561. [Google Scholar]
- Lundgren, O. 5-Hydroxytryptamine, enterotoxins, and intestinal fluid secretion. Gastroenterology 1998, 115, 1009–1012. [Google Scholar]
- Turvill, J.L.; Mourad, F.H.; Farthing, M.J. Crucial role for 5-HT in cholera toxin but not Escherichia coli heat-labile enterotoxin-intestinal secretion in rats. Gastroenterology 1998, 115, 883–890. [Google Scholar]
- Mourad, F.H.; O'Donnell, L.J.; Dias, J.A.; Ogutu, E.; Andre, E.A.; Turvill, J.L.; Farthing, M.J. Role of 5-hydroxytryptamine type 3 receptors in rat intestinal fluid and electrolyte secretion induced by cholera and Escherichia coli enterotoxins. Gut 1995, 37, 340–345. [Google Scholar]
- Bearcroft, C.P.; Perrett, D.; Farthing, M.J. 5-hydroxytryptamine release into human jejunum by cholera toxin. Gut 1996, 39, 528–531. [Google Scholar]
- Nilsson, O.; Cassuto, J.; Larsson, P.A.; Jodal, M.; Lidberg, P.; Ahlman, H.; Dahlstrom, A.; Lundgren, O. 5-Hydroxytryptamine and cholera secretion: A histochemical and physiological study in cats. Gut 1983, 24, 542–548. [Google Scholar]
- Beubler, E.; Horina, G. 5-HT2 and 5-HT3 receptor subtypes mediate cholera toxin-induced intestinal fluid secretion in the rat. Gastroenterology 1990, 99, 83–89. [Google Scholar]
- Buchheit, K.H. Inhibition of cholera toxin-induced intestinal secretion by the 5-HT3 receptor antagonist ICS 205–930. Naunyn Schmiedebergs Arch. Pharmacol. 1989, 339, 704–705. [Google Scholar]
- Beattie, D.T.; Smith, J.A. Serotonin pharmacology in the gastrointestinal tract: A review. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 181–203. [Google Scholar]
- Cooke, H.J. Neurotransmitters in neuronal reflexes regulating intestinal secretion. Ann. NY Acad. Sci. 2000, 915, 77–80. [Google Scholar]
- Lundgren, O. Enteric nerves and diarrhoea. Pharmacol. Toxicol. 2002, 90, 109–120. [Google Scholar]
- Turvill, J.L.; Connor, P.; Farthing, M.J. Neurokinin 1 and 2 receptors mediate cholera toxin secretion in rat jejunum. Gastroenterology 2000, 119, 1037–1044. [Google Scholar]
- Castagliuolo, I.; Lamont, J.T.; Letourneau, R.; Kelly, C.; O'Keane, J.C.; Jaffer, A.; Theoharides, T.C.; Pothoulakis, C. Neuronal involvement in the intestinal effects of Clostridium difficile Toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterology 1994, 107, 657–665. [Google Scholar]
- Pothoulakis, C.; Lamont, J.T. Microbes and microbial toxins: paradigms for microbial-mucosa interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G178–G183. [Google Scholar] [PubMed]
- Castagliuolo, I.; Riegler, M.; Pasha, A.; Nikulasson, S.; Lu, B.; Gerard, C.; Gerard, N.P.; Pothoulakis, C. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile-induced enteritis. J. Clin. Invest. 1998, 101, 1547–1550. [Google Scholar]
- Castagliuolo, I.; Wang, C.C.; Valenick, L.; Pasha, A.; Nikulasson, S.; Carraway, R.E.; Pothoulakis, C. Neurotensin is a proinflammatory peptide in colonic inflammation. J. Clin. Invest. 1999, 103, 843–849. [Google Scholar]
- Xia, Y.; Hu, H.Z.; Liu, S.; Pothoulakis, C.; Wood, J.D. Clostridium difficile toxin A excites enteric neurones and suppresses sympathetic neurotransmision in the guinea pig. Gut 2000, 46, 481–486. [Google Scholar]
- McClane, B.A. Clostridium perfringens enterotoxin. In The Cmprehensive Sourcebook of Bacterial Protein Toxins, 3rd; Alouf, J.E., Popoff, M.R., Eds.; Elsevier Academic Press: Amsterdam, The Netherland, 2006; pp. 763–778. [Google Scholar]
- Senda, T.; Sugimoto, N.; Horiguchi, Y.; Matsuda, M. The enterotoxin of Clostridium perfringens type A binds to the presynaptic nerve endings in neuromuscular junctions of mouse phrenic nerve-diaphragm. Toxicon 1995, 33, 499–506. [Google Scholar]
- Rolfe, V.E.; Levin, R.J. Vagotomy inhibits the jejunal fluid secretion activated by luminal ileal Escherichia coli STa in the rat in vivo. Gut 1999, 44, 615–619. [Google Scholar]
- Mourad, F.H.; Nassar, C.F. Effect of vasoactive intestinal polypeptide (VIP) antagonism on rat jejunal fluid and electrolyte secretion induced by cholera and Escherichia coli enterotoxins. Gut 2000, 47, 382–386. [Google Scholar]
- Rolfe, V.; Levin, R.J. Enterotoxin Escherichia coli STa activates a nitric oxide-dependent myenteric plexus secretory reflex in the rat ileum. J. Physiol. 1994, 475, 531–537. [Google Scholar]
- Eklund, S.; Jodal, M.; Lundgren, O. The enteric nervous system participates in the secretory response to the heat stable enterotoxins of Escherichia coli in rats and cats. Neuroscience 1985, 14, 673–681. [Google Scholar]
- Stenfors Arnesen, L.P.; Fagerlund, A.; Granum, P.E. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 2008, 32, 579–606. [Google Scholar]
- Toh, M.; Moffitt, M.C.; Henrichsen, L.; Raftery, M.; Barrow, K.; Cox, J.M.; Marquis, C.P.; Neilan, B.A. Cereulide, the emetic toxin of Bacillus cereus, is putatively a product of nonribosomal peptide synthesis. J. Appl. Microbiol. 2004, 97, 992–1000. [Google Scholar]
- Agata, N.; Ohta, M.; Mori, M.; Isobe, M. A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus cereus. FEMS Microbiol. Lett. 1995, 129, 17–20. [Google Scholar]
- Horwood, P.F.; Burgess, G.W.; Oakey, H.J. Evidence for non-ribosomal peptide synthetase production of cereulide (the emetic toxin) in Bacillus cereus. FEMS Microbiol. Lett. 2004, 236, 319–324. [Google Scholar]
- Agata, N.; Ohta, M.; Mori, M. Production of an emetic toxin, cereulide, is associated with a specific class of Bacillus cereus. Curr. Microbiol. 1996, 33, 67–69. [Google Scholar]
- Shinagawa, K.; Konuma, H.; Sekita, H.; Sugii, S. Emesis of rhesus monkeys induced by intragastric administration with the HEp-2 vacuolation factor (cereulide) produced by Bacillus cereus. FEMS Microbiol. Lett. 1995, 130, 87–90. [Google Scholar]
- Agata, N.; Mori, M.; Ohta, M.; Suwan, S.; Ohtani, I.; Isobe, M. A novel dodecadepsipeptide, cereulide, isolated from Bacillus cereus causes vacuole formation in HEp-2 cells. FEMS Microbiol. Lett. 1994, 121, 31–34. [Google Scholar]
- Yokoyama, K.; Ito, M.; Agata, N.; Isobe, M.; Shibayama, K.; Horii, T.; Ohta, M. Pathological effect of synthetic cereulide, an emetic toxin of Bacillus cereus, is reversible in mice. FEMS Immunol. Med. Microbiol. 1999, 24, 115–120. [Google Scholar]
- Virtanen, S.M.; Roivainen, M.; Andersson, M.A.; Ylipaasto, P.; Hoornstra, D.; Mikkola, R.; Salkinoja-Salonen, M.S. In vitro toxicity of cereulide on porcine pancreatic Langerhans islets. Toxicon 2008, 51, 1029–1037. [Google Scholar]
- Andersson, M.A.; Hakulinen, P.; Honkalampi-Hamalainen, U.; Hoornstra, D.; Lhuguenot, J.C.; Maki-Paakkanen, J.; Savolainen, M.; Severin, I.; Stammati, A.L.; Turco, L.; Weber, A.; von Wright, A.; Zucco, F.; Salkinoja-Salonen, M. Toxicological profile of cereulide, the Bacillus cereus emetic toxin, in functional assays with human, animal and bacterial cells. Toxicon 2007, 49, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, R.; Saris, N.E.; Grigoriev, P.A.; Andersson, M.A.; Salkinoja-Salonen, M.S. Ionophoretic properties and mitochondrial effects of cereulide: the emetic toxin of B. cereus. Eur. J. Biochem. 1999, 263, 112–117. [Google Scholar]
- Teplova, V.V.; Mikkola, R.; Tonshin, A.A.; Saris, N.E.; Salkinoja-Salonen, M.S. The higher toxicity of cereulide relative to valinomycin is due to its higher affinity for potassium at physiological plasma concentration. Toxicol. Appl. Pharmacol. 2006, 210, 39–46. [Google Scholar]
- Saris, N.E.; Andersson, M.A.; Mikkola, R.; Andersson, L.C.; Teplova, V.V.; Grigoriev, P.A.; Salkinoja-Salonen, M.S. Microbial toxin's effect on mitochondrial survival by increasing K+ uptake. Toxicol. Ind. Health. 2009, 25, 441–446. [Google Scholar]
- Krakauer, T.; Stiles, B.G. Staphylococcal enterotoxins, toxic-shock syndrome toxin-1, and streptococcal pyrogenic exotoxins: Some basic biology of bacterial superantigens. Rec. Res. Dev. Infect. Immun. 2003, 1, 21–27. [Google Scholar]
- Jett, M.; Brinkley, W.; Neill, R.; Gemski, P.; Hunt, R. Staphylococcus aureus enterotoxin B challenge of monkeys: Correlation of plasma levels of arachidonic acid cascade products with occurrence of illness. Infect. Immun. 1990, 58, 3494–3499. [Google Scholar]
- Alber, G.; Scheuber, P.H.; Reck, B.; Sailer-Kramer, B.; Hartmann, A.; Hammer, D.K. Role of substance P in immediate-type skin reactions induced by staphylococcal enterotoxin B in unsensitized monkeys. J. Allergy Clin. Immunol. 1989, 84, 880–885. [Google Scholar]
- Tiegs, G.; Bang, R.; Neuhuber, W.L. Requirement of peptidergic sensory innervation for disease activity in murine models of immune hepatitis and protection by beta-adrenergic stimulation. J. Neuroimmunol. 1999, 96, 131–143. [Google Scholar]
- Wang, X.; Wang, B.R.; Zhang, X.J.; Duan, X.L.; Guo, X.; Ju, G. Fos expression in the rat brain after intraperitoneal injection of Staphylococcus enterotoxin B and the effect of vagotomy. Neurochem. Res. 2004, 29, 1667–1674. [Google Scholar]
- Hu, D.L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell Microbiol. 2007, 9, 2267–2277. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Popoff, M.R.; Poulain, B. Bacterial Toxins and the Nervous System: Neurotoxins and Multipotential Toxins Interacting with Neuronal Cells. Toxins 2010, 2, 683-737. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040683
Popoff MR, Poulain B. Bacterial Toxins and the Nervous System: Neurotoxins and Multipotential Toxins Interacting with Neuronal Cells. Toxins. 2010; 2(4):683-737. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040683
Chicago/Turabian StylePopoff, Michel R., and Bernard Poulain. 2010. "Bacterial Toxins and the Nervous System: Neurotoxins and Multipotential Toxins Interacting with Neuronal Cells" Toxins 2, no. 4: 683-737. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040683