Autoproteolytic Activation of Bacterial Toxins

Department of Pathology, Stanford School of Medicine, 300 Pasteur Drive, Stanford, California 94305, USA
Toxins 2010, 2(5), 963-977; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2050963
Submission received: 31 March 2010
/
Revised: 28 April 2010
/
Accepted: 5 May 2010
/
Published: 6 May 2010
(This article belongs to the Special Issue Protein Toxins as Proteases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Protease domains within toxins typically act as the primary effector domain within target cells. By contrast, the primary function of the cysteine protease domain (CPD) in Multifunctional Autoprocessing RTX-like (MARTX) and Clostridium sp. glucosylating toxin families is to proteolytically cleave the toxin and release its cognate effector domains. The CPD becomes activated upon binding to the eukaryotic-specific small molecule, inositol hexakisphosphate (InsP6), which is found abundantly in the eukaryotic cytosol. This property allows the CPD to spatially and temporally regulate toxin activation, making it a prime candidate for developing anti-toxin therapeutics. In this review, we summarize recent findings related to defining the regulation of toxin function by the CPD and the development of inhibitors to prevent CPD-mediated activation of bacterial toxins.
1. Autoproteolytic Activation of Bacterial Toxins
Many bacterial toxins whose targets are within host cells require proteolytic activation for their function. Whereas most toxins, such as cholera toxin and anthrax toxin, are cleaved by a eukaryotic cell protease, a subset of bacterial toxins is autoproteolytically activated by an internal cysteine protease domain (CPD). The CPD is found within two multidomain toxin families: the Multifunctional Autoprocessing RTX-like toxins (MARTX) toxins and the Clostridium sp. glucosylating toxins (GTs) (Figure 1a and b). Because the CPD is directly activated by the eukaryotic-specific small molecule inositol hexakisphosphate (InsP6), the CPD acts as a biosensor that appears to allow for temporal and spatial regulation of toxin activation. Once toxins translocate across eukaryotic cell membranes, the CPD binds to InsP6 within the cell (KD ~ 1-2 µM) [3,4,5] and induces toxin autoprocessing. Cleavage of the toxins by the CPD releases cognate effector domains and subsequently enhances toxin function [6,7,8,9]. Importantly, InsP6 is found at concentrations between 5-100 µM within the cytosol of mammalian cells, exhibits a long half-life in cells, and may be localized to cellular membranes and the nucleus [10,11,12].
1.1. MARTX toxins
The MARTX toxins are a newly discovered family of toxins that are encoded in the genomes of a number of Gram-negative bacteria [14]. Although only a few toxin members have been studied to date, MARTX toxins have been shown to modulate the virulence of Vibrio cholerae, Vibrio vulnificus, and Vibrio anguillarum. The MARTX toxin of V. cholerae, the causative agent of cholera, is produced by nearly all clinical and environmental isolates of V. cholerae [15,16,17]. Although it is not cytotoxic, it induces cell rounding and enhances colonization in a mouse model of infection [18,19,20,21]. By contrast, MARTX toxins of the marine pathogens V. vulnificus and V. anguillarum are cytotoxic and function as key virulence factors [22,23,24].
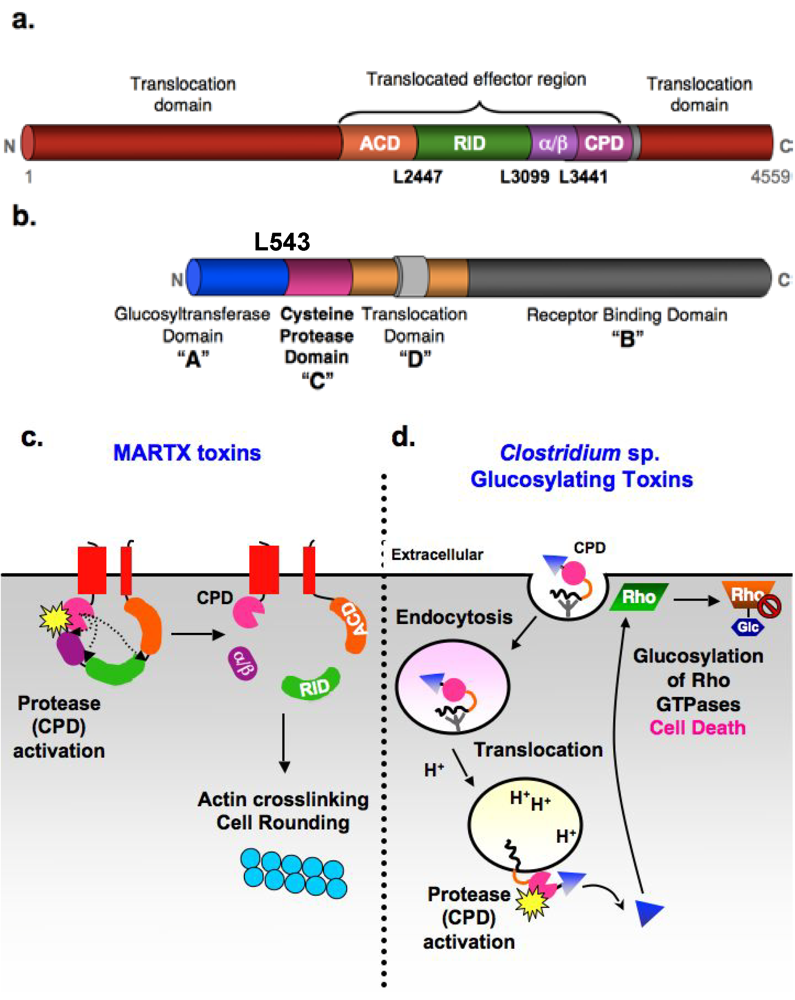
MARTX toxins are some of the largest bacterial proteins identified to date, with molecular weights often in excess of 450 kDa [14]. They are characterized by conserved N- and C-terminal repeat regions with similarity to RTX toxins that flank a central effector domain region (Figure 1a). These repeat regions have been proposed to form a pore that allows the central effector region to autotranslocate across host cell membranes [18]. While the composition of the central region varies across MARTX toxin family members, the most C-terminal effector of MARTX toxins is always the InsP6-sensing cysteine protease domain (CPD). In V. cholerae MARTX toxin, the best characterized family member, the CPD cleaves the toxin at multiple sites to release discrete effector domains (Figure 1c) [9,25].
Aside from the CPD, the activities of most MARTX effector domains remain uncharacterized. The functions of only two other effector domain functions have been identified to date; both these domains are found within V. cholerae MARTX toxins and disrupt host cell actin dynamics. The actin crosslinking domain (ACD) has homology to ATP-dependent ligases and covalently crosslinks actin monomers together through an atypical glutamate-lysine crosslink [26,27,28,29]. As a result, the ACD prevents growth of actin filaments and depletes the monomeric actin pool, which leads to cell rounding due to destruction of the actin cytoskeleton. The Rho Inactivating Domain (RID) domain inhibits small Rho GTPase activity through an as yet undefined mechanism [30].
Figure 1.
Autoproteolytic activation of MARTX and glucosylating toxin families by the CPD. (a) Domain organization of V. cholerae MARTX toxin. Conserved glycine-rich repeat regions in the N- and C-termini of MARTX toxins (MARTX conserved, red); actin crosslinking domain (ACD, orange); Rho-inactivating domain (RID, green); α/β hydrolase domain (α/β, purple), cysteine protease domain (CPD, pink). Cleavage sites are shown. (b) ABCD domain organization of C. difficile glucosylating toxins. A: Glucosyltransferase (Glc) “activity” domain; B: receptor “binding” domain; C: cysteine protease “cutting” domain; and D: hydrophobic translocation “delivery” domain. Cleavage site in TcdB is marked (c) Schematic model for CPD-mediated activation of V. cholerae MARTX toxin. Upon encountering a eukaryotic cell, the N- and C-terminal MARTXVc conserved regions insert into the plasma membrane and form a pore that permits translocation of the toxin central region across the cell membrane. Following toxin entry, the CPD binds inositol hexakisphosphate (InsP6, starburst), resulting in activation of its protease activity. The CPD first cleaves at Leu3441, the preferred autoprocessing site, then Leu2447, followed by Leu3099 (least preferred cleavage site) [9]. Processing of MARTXVc by the CPD releases the RID and (α/β) domains into the cytosol, while the ACD and CPD remain tethered to the membrane. Cleavage of MARTXVc, particularly at Leu2447, activates the actin crosslinking activity of the ACD. (d) Schematic model for CPD-mediated activation of Clostridium sp. glucosylating toxins. Binding of the toxins to unidentified receptors results in their uptake by receptor-mediated endocytosis. Acidification of the early endosome triggers toxin translocation of the Glc domain (triangle). This is the only part of the protein released into the cytosol, presumably due to CPD-mediated autoproteolysis [13]. Whether the CPD itself is translocated, and when and where it is activated by InsP6 (starburst) is unknown, although it likely occurs on the cytosolic face of the endosomal membrane.
Figure 1.
Autoproteolytic activation of MARTX and glucosylating toxin families by the CPD. (a) Domain organization of V. cholerae MARTX toxin. Conserved glycine-rich repeat regions in the N- and C-termini of MARTX toxins (MARTX conserved, red); actin crosslinking domain (ACD, orange); Rho-inactivating domain (RID, green); α/β hydrolase domain (α/β, purple), cysteine protease domain (CPD, pink). Cleavage sites are shown. (b) ABCD domain organization of C. difficile glucosylating toxins. A: Glucosyltransferase (Glc) “activity” domain; B: receptor “binding” domain; C: cysteine protease “cutting” domain; and D: hydrophobic translocation “delivery” domain. Cleavage site in TcdB is marked (c) Schematic model for CPD-mediated activation of V. cholerae MARTX toxin. Upon encountering a eukaryotic cell, the N- and C-terminal MARTXVc conserved regions insert into the plasma membrane and form a pore that permits translocation of the toxin central region across the cell membrane. Following toxin entry, the CPD binds inositol hexakisphosphate (InsP6, starburst), resulting in activation of its protease activity. The CPD first cleaves at Leu3441, the preferred autoprocessing site, then Leu2447, followed by Leu3099 (least preferred cleavage site) [9]. Processing of MARTXVc by the CPD releases the RID and (α/β) domains into the cytosol, while the ACD and CPD remain tethered to the membrane. Cleavage of MARTXVc, particularly at Leu2447, activates the actin crosslinking activity of the ACD. (d) Schematic model for CPD-mediated activation of Clostridium sp. glucosylating toxins. Binding of the toxins to unidentified receptors results in their uptake by receptor-mediated endocytosis. Acidification of the early endosome triggers toxin translocation of the Glc domain (triangle). This is the only part of the protein released into the cytosol, presumably due to CPD-mediated autoproteolysis [13]. Whether the CPD itself is translocated, and when and where it is activated by InsP6 (starburst) is unknown, although it likely occurs on the cytosolic face of the endosomal membrane.

1.2. Clostridium sp. glucosylating toxins
Similar to MARTX toxins, Clostridium sp. glucosylating toxins are multidomain toxins that alter target cell cytoskeletal dynamics in a manner dependent on CPD-mediated toxin autoprocessing (Figure 1b and d). Glucosylating toxins are large (~250 kDa) proteins with an ABCD toxin structure (A, biological activity; B, binding; C, cutting; D, delivery) [7,31,32]. The “biological activity” (A) is conferred by the N-terminal glucosyltransferase (Glc) domain, which glucosylates Rho GTPases (such as RhoA and Ras) at a conserved Thr residue [33,34]. This covalent modification prevents Rho GTP-GDP exchange and irreversibly inhibits Rho GTPase function, leading to disruption of epithelial barrier junctions, inhibition of Rho-mediated signaling, and ultimately cell death and inflammation [35]. The “binding activity” (B) of GTs is mediated by the C-terminal repeat region, which forms a solenoid structure that probably binds sugar moieties on cell surface receptors (the identity of which has not been determined). The toxins are then taken up by receptor-mediated endocytosis.
As “short-trip” toxins, acidification of the early endosomal compartment induces conformational changes in GTs that results in membrane insertion and toxin translocation. The central, hydrophobic region is thought to confer this “delivery activity” (D) [36]. While the mechanism by which this occurs remains unclear, the central region likely mediates translocation of the Glc domain across the endosomal membrane into the target cell cytosol. The CPD domain presumably is also transferred, but this has not been directly tested. Nevertheless, binding of InsP6 to the CPD (either in the endosome or in the host cytosol) results in CPD activation and cleavage of GTs at the Glc domain-CPD junction [3,37,38]. This “cutting” (C) event releases the Glc domain from the endosome, an event that presumably improves access of the Glc domain to its Rho GTPase substrates at the membrane.
The glucosylating toxins (GTs) are produced by select Clostridium sp. pathogens, which are Gram-positive, anaerobic, spore-forming bacteria. Members of the GT family include TcdL and TcdH of C. sordellii, TcnA of C. novyi, and C. perfringens types B and C, but the prototypical members are TcdA and TcdB of C. difficile. This latter bacterium is of particular interest as it is the leading cause of nosocomial diarrhea worldwide and is associated with significant morbidity and cost in health care-associated settings [39,40]. The glucosylating toxins TcdA and TcdB are the primary causes of C. difficile infection (CDI), with TcdB being the more active of the two toxins and essential for virulence [41]. The relevance of these toxins to disease is underscored by the observation that a strain that hyper-produces TcdA and TcdB causes more frequent and severe cases of CDIs [40,42]. Thus, the development of inhibitors that can inhibit TcdA and TcdB function or activation should have considerable therapeutic utility.
2. Autoprocessing Cysteine Protease Domains
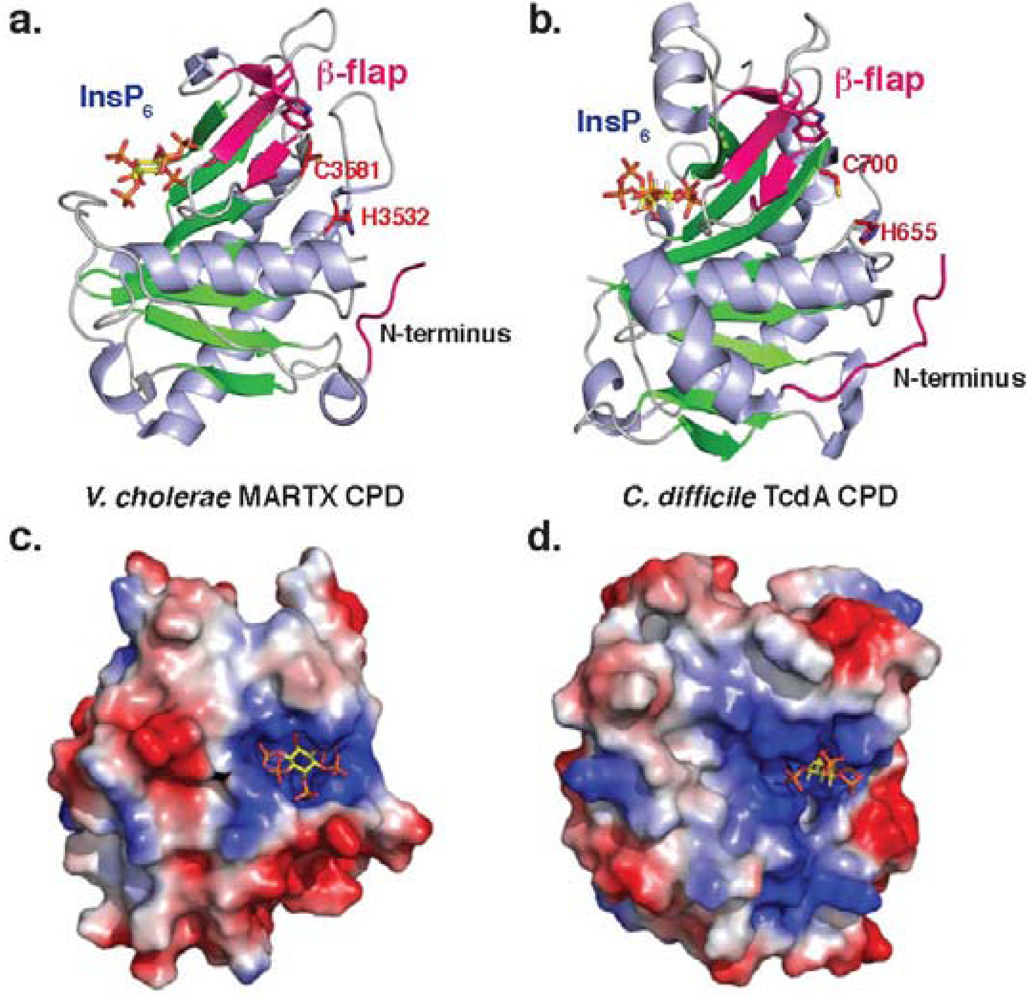
Although MARTX and GT toxin families differ in their activities and domain organization, they share a common cysteine protease domain (CPD) that proteolytically processes toxin family members. Despite differing in 60% of their amino acid residues, both MARTX and GT CPDs have a similar overall fold that resembles a canonical caspase-like fold (Figure 2a and b). As clan CD proteases (MEROPS classification) [43], both bacterial CPDs and caspases have a central β-sheet that is surrounded by α-helices [44]. Unlike the caspases, however, bacterial CPDs contain an InsP6 binding site and a region termed the β-flap (described below). Structures of both MARTX and TcdB CPDs reveal that the InsP6 binding site is comprised of numerous positively charged residues that trap a single negatively-charged InsP6 molecule [4,25,45]. Interestingly, the mechanism of InsP6 binding differs slightly between Tcd and MARTX CPDs, with InsP6 lying flat on the surface of the protease in V. cholerae MARTX CPD, and InsP6 being wedged into the side of TcdA CPD (Figure 2c and d) [45]. Both proteases, however, have similar low micromolar affinities for InsP6[4,5,37].
Figure 2.
Structure of bacterial CPDs. Ribbon structure of (a)V. cholerae MARTX CPD (PDB ID: 3EEB) and (b)C. difficile TcdA CPD (PDB ID: 3HO6). Electrostatic surface potential of (c)V. cholerae MARTX CPD and (d) C. difficile TcdB CPD as viewed from above the InsP6 binding site. Blue denotes positively charged surface; red denotes negatively charged surface. InsP6 is shown in the binding site as a stick model. Note that the orientation of the InsP6 molecule differs between the two structures.
Figure 2.
Structure of bacterial CPDs. Ribbon structure of (a)V. cholerae MARTX CPD (PDB ID: 3EEB) and (b)C. difficile TcdA CPD (PDB ID: 3HO6). Electrostatic surface potential of (c)V. cholerae MARTX CPD and (d) C. difficile TcdB CPD as viewed from above the InsP6 binding site. Blue denotes positively charged surface; red denotes negatively charged surface. InsP6 is shown in the binding site as a stick model. Note that the orientation of the InsP6 molecule differs between the two structures.

Notably, the InsP6 binding site is physically distinct from the active site, indicating that InsP6 likely functions as an allosteric activator of bacterial CPDs. Although a number of studies have shown that InsP6 binding improves access of the CPD to its substrates and inhibitors [3,4,25], the mechanism by which InsP6 binding is communicated to the active site has not been fully characterized. Current work suggests that the β-flap region - the second structural feature in which bacterial CPDs differ from other clan CD proteases - transduces the InsP6 binding to the active site. Mutation of key residues in the β-flap disrupt CPD function [4,5,37,45], and residues in this region have been shown to be resistant to limited proteolysis in the presence of InsP6 [25]. This latter result suggests that InsP6 binding induces stabilization of the β-flap in the activated protease. Interestingly, the β-flap region of TcdA CPD is larger than that of V. cholerae CPD, with TcdA CPD containing an additional alpha-helix [45]; the functional significance of this addition is unknown.
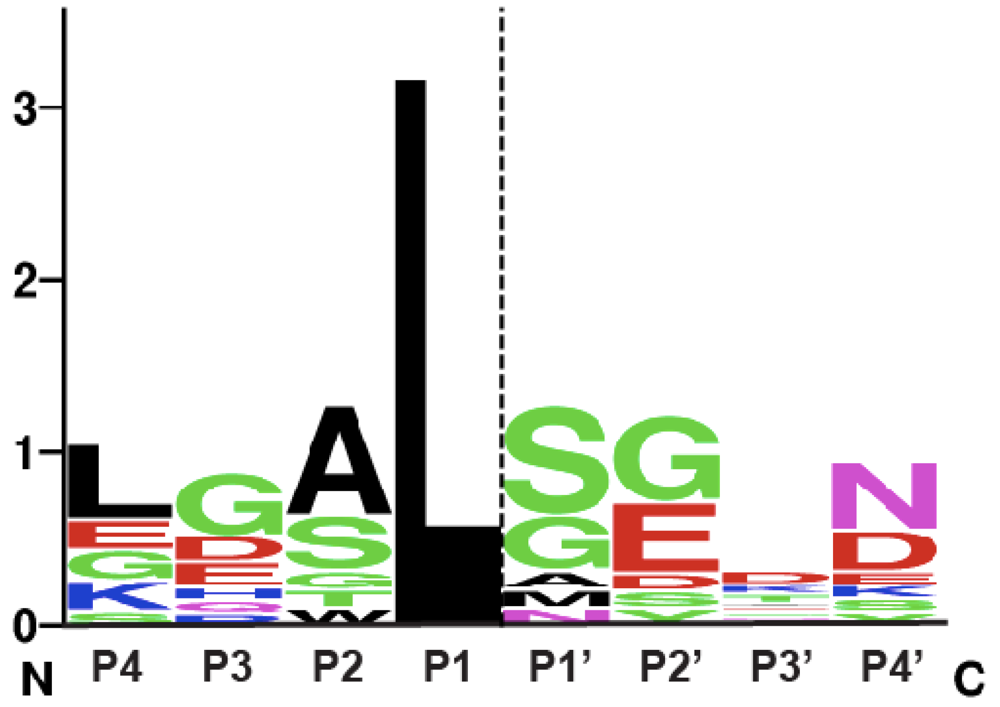
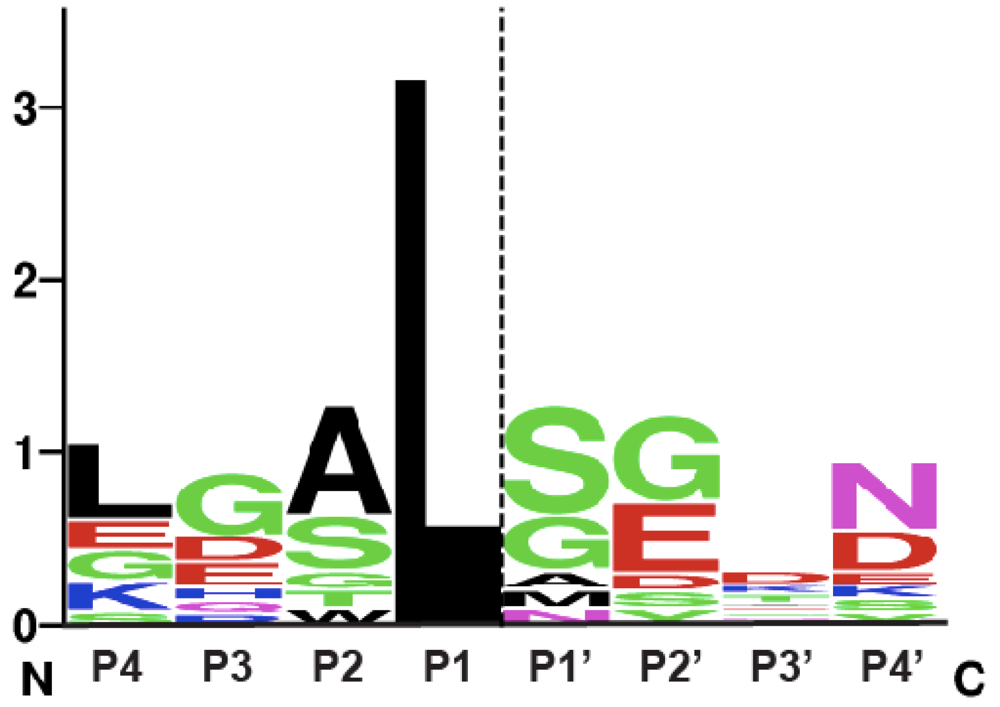
This mechanism of allosteric regulation by a small metabolite is unique among proteases. Nevertheless, bacterial CPDs exhibit many similarities in substrate recognition to clan CD proteases. In particular, like the caspases, the substrate specificity of MARTX CPDs is governed by the P1 residue, the amino acid immediately N-terminal to the scissile bond [9,25,46,47,48]. Just as caspases cleave almost exclusively after P1 Asp residues, bacterial CPDs appear to cleave exclusively after P1 Leu residues: all known MARTX and GT toxin autoprocessing sites occur after Leu residues [3,9,25,49]. In addition, like the caspases, MARTX CPDs appear to exhibit a preference for a small amino acid in the P1’ position, the residue immediately C-terminal to the scissile bond. A comparison of known and inferred cleavage sites within MARTX and GT toxins suggests that small residues in the P1’ position are favored (Figure 3). Furthermore, the CPD has been shown to cleave more efficiently when the P1’ residue is an Ala versus a Ser residue, while the presence of a bulky P1’ Leu residue is sufficient to abrogate CPD-mediated processing [9].
Figure 3.
Sequence logo representation of MARTX and GT CPD consensus cleavage site. The sequence logo was created using natural MARTX CPD cleavage sites mapped by intact mass spectrometry [9] and cleavage sites mapped and/or inferred in GT CPDs [38,49], see http://weblogo.berkeley.edu. The dashed line indicates the scissile bond.
Figure 3.
Sequence logo representation of MARTX and GT CPD consensus cleavage site. The sequence logo was created using natural MARTX CPD cleavage sites mapped by intact mass spectrometry [9] and cleavage sites mapped and/or inferred in GT CPDs [38,49], see http://weblogo.berkeley.edu. The dashed line indicates the scissile bond.
3. Development of Inhibitors of Bacterial CPD Protease Activity
The restraints on substrate specificity determined by cleavage site comparisons are also observed in studies with MARTX CPD inhibitors [9]. Inhibitors of V. cholerae MARTX CPD processing were recently identified in a screen of a focused library of cysteine protease inhibitors. All CPD inhibitors derived from the screen contain a Leu in the P1 position, the residue immediately N-terminal to the scissile bond. They were also all functionalized with an aza-epoxide group, which reacts with the caspases [50]. The screen produced a small structure-activity relationship (SAR) series that shows remarkable similarities to an SAR series previously performed on the caspases. In particular, the regio- and stereochemistry at the epoxide moiety influences inhibitor potency for both V. cholerae CPD and caspase-3, with S,S > trans > R,R [9,51].
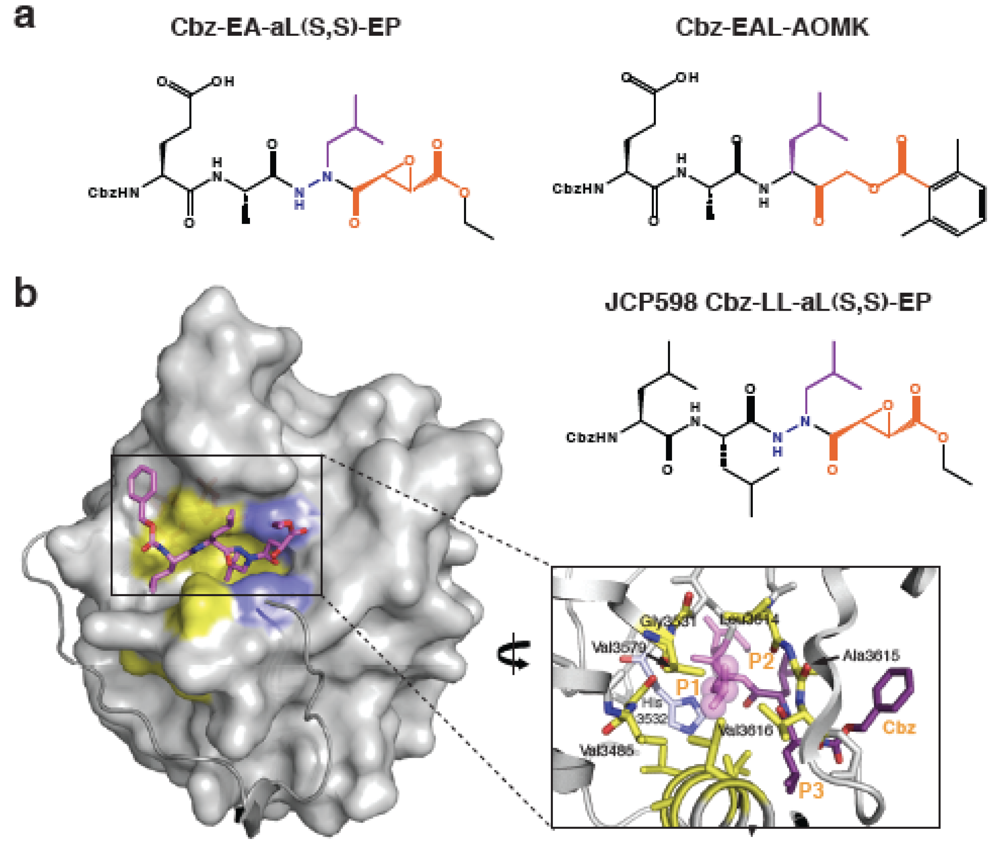
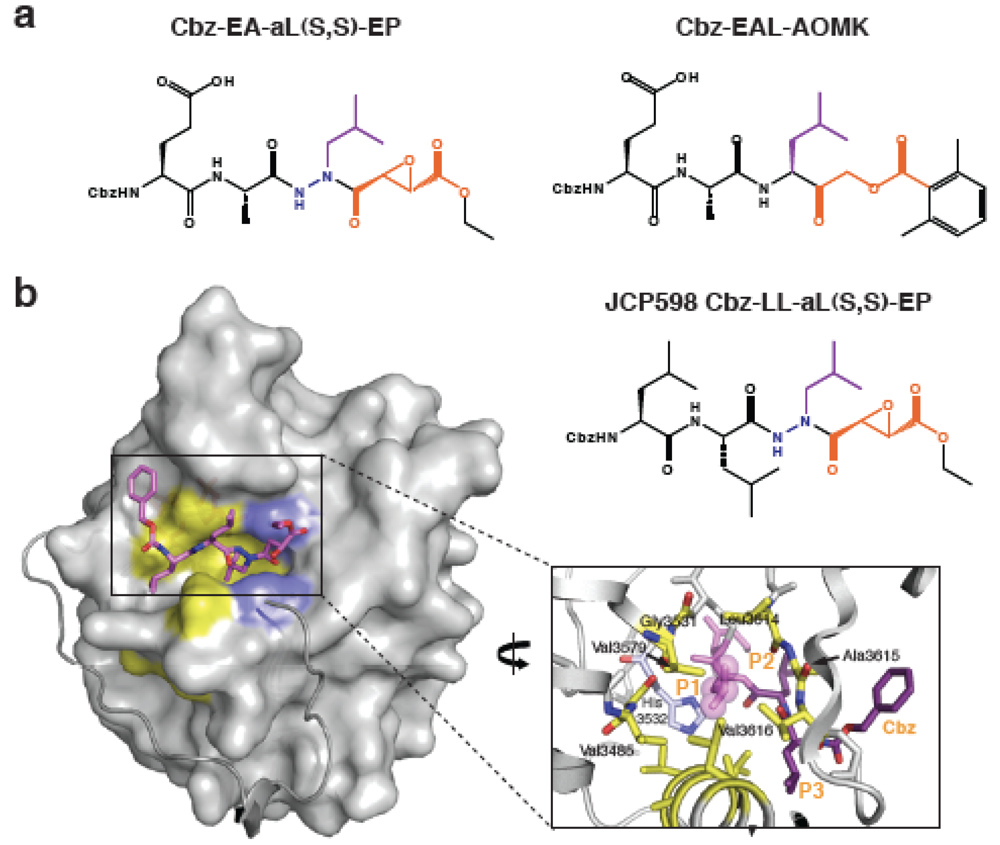
The crystal structure of V. cholerae CPD bound to the most potent inhibitor from the screen (JCP598, Z-LL-azaL-EP, Figure 4a) provided the most convincing evidence that MARTX CPDs and caspases share similar mechanisms of substrate recognition and catalysis [9]. In both the V. cholerae CPD and caspase-3 structures bound to aza-epoxide inhibitors, the catalytic Cys of V. cholerae CPD attacks on the C3 position of the expoxide rather than the C2. Furthermore, the catalytic residues of V. cholerae MARTX CPD and caspase-3 exhibit similar geometries around the P1-P1’ peptide bond, and the S1 binding pockets (that recognize the P1 residue) are similarly aligned. Indeed, the S1 pocket of V. cholerae MARTX CPD forms a deep, hydrophobic cavity that perfectly accommodates a Leu residue (Figure 4b), and residues that form this S1 pocket are conserved across MARTX and GT CPDs. Notably, the structure of aza-epoxide inhibited V. cholerae MARTX CPD is essentially superimposable with the recently solved structure of the uncleaved form of the protease. This uncleaved form carries the native P1 Leu and non-native TEV protease cleavage site [25]. The P1 Leu residues of both MARTX CPD structures are similarly aligned, while mutational analyses of the P2 site also support the conclusion that the primary substrate specificity determinant is a P1 Leu [25].
As observed with the natural cleavage substrates of MARTX toxins, the P2 and P3 positions of the inhibitors do not strongly contribute to substrate recognition [9]. For example, no difference in inhibitor potency was observed between a Cbz-Leu-Leu-azaLeu (Z-LLaL-EP) epoxide inhibitor and Cbz-Asp-Ala-azaLeu (Z-EAaL-EP) epoxide inhibitor. Furthermore, the S2 and S3 sub-sites in the crystal structure of inhibitor-bound MARTX CPD do not make contacts with the P2 and P3 side chains of the inhibitor; rather, the protease interacts with the peptide backbone of the inhibitor. These conclusions are consistent with SAR series results, indicating that inhibitors carrying P2 and P3 residues were more potent than inhibitors with only a single P1 Leu.
The functional group of the P1 Leu inhibitors also affects inhibitor potency. Whereas inhibitors with a P1 Leu and acyloxymethylketone (AOMK) functional group, which reacts with caspases [52], block the activity of V. cholerae MARTX CPD, inhibitors with the same peptide sequence as JCP598 (but different reactive groups) do not inhibit CPD function [9]. Specifically, MG132, a common proteasome inhibitor (Cbz-LLL-aldehyde) and Z-L3VS (Cbz-LLL-vinyl sulfone) do not block V. cholerae CPD autocleavage. The lack of inhibitory activity of Z-L3VS may reflect the preference of MARTX CPDs for small P1’ residues in the consensus cleavage site sequence. Indeed, the S1’ subsite (which interacts with the P1’ residue) is flat and featureless and may not accommodate large constituents in the P1’ position. Nevertheless, making contacts with the S1’ subsite would appear to be important for inhibitor potency given the lack on inhibitor activity of MG132.
Although the aza-epoxide inhibitors identified in the screen are designed to covalently inhibit cysteine proteases, they failed to irreversibly react with V. cholerae MARTX CPD under standard assay conditions (A. Shen, unpublished data). The apparent slow reactivity of V. cholerae MARTX CPD with the aza-epoxide inhibitors may reflect the rapid rate with which InsP6 likely binds to and dissociates from the CPD relative to inhibitor binding. In contrast, when the aza-epoxide inhibitor was co-crystallized with V. cholerae CPD in the presence of InsP6, the inhibitor likely had sufficient time to react with the enzyme constrained in the crystal lattice. The lack of reactivity of the aza-epoxide inhibitors may also reflect the need for optimization of substrate binding or electrophile reactivity to V. cholerae CPD. To the latter point, functionalized alkylating agents have been shown to react with the sole catalytic Cys of V. cholerae CPD in a manner that is enhanced by the presence of InsP6[9,25].
Figure 4.
Chemical inhibition of MARTX CPD activity. (a) Structures of the most potent MARTX CPD inhibitors. Navy blue indicates aza-linkage; purple highlights P1 Leu, and orange denotes electrophile warhead. (b) Surface topology of activated MARTXVc CPD bound to an aza-peptide epoxide inhibitor. Hydrophobic residues in the substrate binding cleft are in yellow. The aza-peptide epoxide inhibitor (JCP598) is shown as a stick model bound in the substrate binding pocket. The N-terminus is shown as a grey ribbon, terminating at Ile3437 and highlighting the threading of this region along the surface of the core domain.
Figure 4.
Chemical inhibition of MARTX CPD activity. (a) Structures of the most potent MARTX CPD inhibitors. Navy blue indicates aza-linkage; purple highlights P1 Leu, and orange denotes electrophile warhead. (b) Surface topology of activated MARTXVc CPD bound to an aza-peptide epoxide inhibitor. Hydrophobic residues in the substrate binding cleft are in yellow. The aza-peptide epoxide inhibitor (JCP598) is shown as a stick model bound in the substrate binding pocket. The N-terminus is shown as a grey ribbon, terminating at Ile3437 and highlighting the threading of this region along the surface of the core domain.
Nevertheless, MARTX CPD inhibitors are capable of reducing CPD-mediated processing of MARTX toxin during growth of V. cholerae in broth culture [9]. While the inhibitors are not effective at preventing CPD autocleavage in the native toxin, they do prevent processing of the toxin by the CPD at distal sites. Although they have limited potency on the native toxin, the CPD inhibitors were useful in determining the relative affinity of the CPD for its different cleavage sites, which correlated the relative affinities determined using in vitro cleavage reactions. Notably, the inhibitors were also capable of preventing MARTX toxin activity (specifically actin crosslinking activity) during intoxication of target cells. The aza-epoxide inhibitors were the most potent, since they were more cell permeable than the AOMK inhibitors; Cbz-LLaL-EP was the most cell permeable inhibitor. While the specificity of these inhibitors in complex mixtures still needs to be assessed, these results strongly suggest that CPD inhibitors can prevent the autoproteolytic activation of MARTX toxins inside target cells. Thus, the MARTX CPD inhibitors provide a starting point for the development of active site inhibitors of MARTX CPDs and potentially GT CPDs (given that S1 residues in MARTX and GT CPDs appear to be conserved).
4. Requirement of CPD-Mediated Processing for Toxin Activity
Although CPD-mediated processing of V. cholerae MARTX and C. difficile Tcd toxins is necessary for optimal toxin function, it is not absolutely required. Toxins carrying mutations in the catalytic Cys of both V. cholerae MARTX and C. difficile TcdB CPDs can still exert cytopathic effects on cells, but the toxin activity is significantly decreased [3,4,6,8]. Why proteolytic cleavage enhances effector domain function has not been characterized. The known substrates of both V. cholerae MARTX and Tcd toxins are often localized to the host cell plasma membrane, so it has been hypothesized that CPD-mediated processing improves access of toxin effector domains to their substrates.
C. difficile glucosylating toxins are cleaved by the CPD after a single conserved Leu at the glucosyltransferase (Glc) domain-CPD junction [3,49]. This cleavage event releases the Glc domain from the endosomal membrane into the cytosol, while the remainder of the toxin remains membrane-associated (Figure 1b, [13,49]). This cleavage event likely enhances access of the Glc domain to its Rho GTPase substrates at the plasma membrane given that a membrane localization domain (MLD) was recently identified in its N-terminus [53]. This MLD was recently shown to be sufficient to mediate plasma membrane localization, with enrichment observed at cell-cell junctions in particular. An analysis of the localization of the Glc domain and its activity in toxins carrying mutations of the CPD cleavage site and/or CPD catalytic residues will be important for determining the precise function of the CPD in regulating Clostridium sp. glucosylating toxin activation. Such analyses will be greatly facilitated with the advent of new tools for manipulating C. difficile genetically [54].
In contrast with the glucosylating toxins, the precise mechanism by which CPD-mediated processing increases effector domain activity in MARTX toxins is less apparent. Whereas the CPD cleaves Clostridium sp. glucosylating toxins at a single site, the CPD processes MARTX toxins at multiple sites. The cleavage sites all occur after a Leu residue and are found at interdomain boundaries predicted to be disordered [9]. The identity of cleavage sites was confirmed by mutational analyses with both recombinant and native MARTX toxin. These analyses indicate that these three sites are the primary cleavage sites in the toxin, although a small amount of second site processing of these toxins can be observed in a mutant in which all three P1 Leu cleavage sites have been mutated to Ala residues [9]. It should be noted that a fourth cleavage site was mapped by Prochazkova et al. [25] using N-terminal sequencing of in vitro cleavage reactions of recombinant V. cholerae MARTX toxin. However, extensive Western blot analyses of wildtype and cleavage site mutants indicate that this site is not physiologically relevant in the native toxin [9].
CPD-mediated processing separates the actin crosslinking domain (ACD), Rho-inactivating domain (RID), and α/β hydrolase domains (Figure 1a). While these cleavage events should liberate the RID and α/β hydrolase domains from the membrane, both the ACD and CPDs are predicted to remain tethered to the membrane. The membrane localization of these domains may be important to their function, perhaps by optimizing access of the ACD and CPD to their substrates. Indeed, the CPD is always found adjacent to the C-terminal MARTX conserved region, while the ACD in other MARTX toxins is always found just downstream of the N-terminal MARTX conserved region. Tethering of the CPD to the membrane likely facilitates processing of the toxin at distal sites, by keeping the substrates in close proximity. The dependence of the ACD domain on CPD-mediated processing remains less clear. Studies of V. cholerae cleavage site mutants indicate that processing of the ACD-RID junction is necessary for optimal ACD activity [9]. Nevertheless, cleavage at any site would appear to stimulate ACD activity, although why this is observed is unknown. Further studies of the ACD and its interaction with its substrate will likely provide important insight into the functional requirements of this domain and the role of CPD-mediated processing in stimulating its activity.
The dependence of the remaining V. cholerae effector domains on CPD-mediated processing has not yet been tested. Further analysis of the biochemical functions of the RID and α/β hydrolase domains will likely provide insight into any role that the CPD may play in activating the function of these effector domains. These studies will require that a function for the α/β hydrolase domain be determined, since there is no information on the activity or localization of this domain. Likewise, a more thorough characterization of the function of the RID domain would contribute to an understanding of the role of proteolysis in activating RID function. Although the RID domain has been shown to disrupt Rho GTPase function [30], the mechanism by which this occurs is unknown. CPD-mediated processing should liberate the RID domain from MARTX toxin at the membrane. However, a recent study suggests that the RID should remain associated with the membrane through its N-terminal MLD domain [53], which preferentially localizes to cell-cell junctions and is necessary for optimal RID activity. If these sites are where the putative RID substrate is enriched, CPD-mediated cleavage may similarly enhance access of the RID domain to its substrates. Indeed, localization of the RID to the membrane has been shown to significantly increase its ability to induce cell rounding [53]. Alternatively, CPD-mediated processing may function to produce free N- and C-termini that are necessary for optimal RID enzymatic activity.
5. Conclusions and Perspectives
Analyses of V. cholerae MARTX toxin will likely be transferable to other MARTX toxins, which typically contain RID, ACD, and/or α/β domains. Studies of other MARTX toxins will be greatly enhanced by mapping CPD cleavage sites, since these will define effector domain boundaries and contribute to a more global understanding of the role of this protease domain in toxin regulation. In general, the CPD appears to function analogously to viral polyprotein processing proteases such as those produced by picornaviruses (such as poliovirus and Hepatitis C). Viruses often use a conserved internal protease domain to proteolytically process the single polyprotein precursor into discrete functional domains that mediate viral replication and assembly [55]. Notably, both viral and CPD autoprocessing proteases exhibit a high degree of sequence specificity but poor transcleavage activity [56]. As a result, these proteases do not appear to cleave substrates in their target host cells. Consistent with this proposal, a MARTX toxin that carries only the CPD in the central translocated region does not overtly affect the morphology of target cells exposed to this mutant toxin (A. Shen, unpublished data), strongly suggesting that the CPD does not have many substrates within target cells aside from the MARTX toxins.
Nevertheless, just as viral polyprotein processing enzymes have been prime targets for therapeutic intervention [57], the CPDs of both MARTX and Clostridium sp. glucosylating toxins are excellent targets for drug design. The C. difficile glucosylating toxin CPDs are particularly good candidates because TcdA and TcdB toxins are the primary factors responsible for C. difficile-associated disease [40]. Given that C. difficile infections are often promoted by antibiotic administration, targeting virulence rather than viability will likely be the most effective strategy for controlling C. difficile-associated disease. Likewise, given that MARTX toxins are comprised of multiple effector domains that vary among toxin family members [14], targeting the conserved CPD domain will likely be a more effective strategy at inhibiting MARTX toxin activation that developing inhibitors against each domain. MARTX CPD inhibitors would have particular applicability in aquaculture settings where infection by V. anguillarum and V. vulnificus infection of fish and bivalves is particularly costly to the seafood industry.
The identification of the first active site inhibitors of bacterial CPDs has provided an excellent starting point for the optimization of inhibitors with greater potency, selectivity, and stability, and the structure of inhibitor-bound CPD may serve as a platform for inhibitor modeling. An alternative strategy to targeting the active sites of these proteases may be to prevent the activation of bacterial CPDs by restraining their conformational mobility and thus allosteric activation [58]. Thus, a better understanding of the mechanism of CPD activation will likely lead to more effective strategies for preventing toxin activation.
Acknowledgements
A.S. is funded by the National Institute of Health Awards K99 GM092934-01.
References and Notes
- Gordon, V.M.; Leppla, S.H. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infect. Immun. 1994, 62, 333–340. [Google Scholar]
- Lencer, W.I.; Constable, C.; Moe, S.; Rufo, P.A.; Wolf, A.; Jobling, M.G.; Ruston, S.P.; Madara, J.L.; Holmes, R.K.; Hirst, T.R. Proteolytic activation of cholera toxin and Escherichia coli labile toxin by entry into host epithelial cells. Signal transduction by a protease-resistant toxin variant. J. Biol. Chem. 1997, 272, 15562–15568. [Google Scholar] [PubMed]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar]
- Lupardus, P.J.; Shen, A.; Bogyo, M.; Garcia, K.C. Small molecule-induced allosteric activation of the vibrio cholerae rtx cysteine protease domain. Science 2008, 322, 265–268. [Google Scholar]
- Prochazkova, K.; Satchell, K.J. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing of the Vibrio cholerae multifunctional autoprocessing RTX toxin. J. Biol. Chem. 2008, 283, 23656–23664. [Google Scholar] [CrossRef]
- Barroso, L.A.; Moncrief, J.S.; Lyerly, D.M.; Wilkins, T.D. Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microb. Pathog. 1994, 16, 297–303. [Google Scholar]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef]
- Sheahan, K.L.; Cordero, C.L.; Satchell, K.J. Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. Embo. J. 2007, 26, 2552–2561. [Google Scholar] [CrossRef]
- Shen, A.; Lupardus, P.J.; Albrow, V.E.; Guzzetta, A.; Powers, J.C.; Garcia, K.C.; Bogyo, M. Mechanistic and structural insights into the proteolytic activation of Vibrio cholerae MARTX toxin. Nat. Chem. Biol. 2009, 5, 469–478. [Google Scholar] [CrossRef]
- Irvine, R.F.; Schell, M.J. Back in the water: the return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar]
- Michell, R.H. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell. Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef]
- Shears, S.B. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal. 2001, 13, 151–158. [Google Scholar] [CrossRef]
- Pfeifer, G.; Schirmer, J.; Leemhuis, J.; Busch, C.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003, 278, 44535–44541. [Google Scholar] [PubMed]
- Satchell, K.J. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect. Immun. 2007, 75, 5079–5084. [Google Scholar] [CrossRef]
- Rahman, M.H.; Biswas, K.; Hossain, M.A.; Sack, R.B.; Mekalanos, J.J.; Faruque, S.M. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol. 2008, 27, 347–355. [Google Scholar] [CrossRef]
- Chow, K.H.; Ng, T.K.; Yuen, K.Y.; Yam, W.C. Detection of RTX toxin gene in Vibrio cholerae by PCR. J. Clin. Microbiol. 2001, 39, 2594–2597. [Google Scholar]
- Cordero, C.L.; Sozhamannan, S.; Satchell, K.J. RTX toxin actin cross-linking activity in clinical and environmental isolates of Vibrio cholerae. J. Clin. Microbiol. 2007, 45, 2289–2292. [Google Scholar] [CrossRef]
- Lin, W.; Fullner, K.J.; Clayton, R.; Sexton, J.A.; Rogers, M.B.; Calia, K.E.; Calderwood, S.B.; Fraser, C.; Mekalanos, J.J. Identification of a vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl. Acad. Sci. USA 1999, 96, 1071–1076. [Google Scholar]
- Olivier, V.; Haines, G.K.; Satchell, K.J. Hemolysin and the multifunctional autoprocessing RTX toxin are virulence factors during intestinal infection of mice with Vibrio cholerae El Tor O1 strains. Infect. Immun. 2007, 75, 5035–5042. [Google Scholar] [CrossRef] [PubMed]
- Olivier, V.; Queen, J.; Satchell, K.J. Successful small intestine colonization of adult mice by Vibrio cholerae requires ketamine anesthesia and accessory toxins. PLoS One 2009, 4, e7352. [Google Scholar]
- Olivier, V.; Salzman, N.H.; Satchell, K.J. Prolonged colonization of mice by Vibrio cholerae El Tor O1 depends on accessory toxins. Infect. Immun. 2007, 75, 5043–5051. [Google Scholar]
- Kim, Y.R.; Lee, S.E.; Kook, H.; Yeom, J.A.; Na, H.S.; Kim, S.Y.; Chung, S.S.; Choy, H.E.; Rhee, J.H. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell Microbiol. 2008, 10, 848–862. [Google Scholar]
- Lee, J.H.; Kim, M.W.; Kim, B.S.; Kim, S.M.; Lee, B.C.; Kim, T.S.; Choi, S.H. Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J. Microbiol. 2007, 45, 146–152. [Google Scholar] [PubMed]
- Li, L.; Rock, J.L.; Nelson, D.R. Identification and characterization of a repeat-in-toxin gene cluster in Vibrio anguillarum. Infect. Immun. 2008, 76, 2620–2632. [Google Scholar]
- Prochazkova, K.; Shuvalova, L.A.; Minasov, G.; Voburka, Z.; Anderson, W.F.; Satchell, K.J. Structural and molecular mechanism for autoprocessing of MARTX toxin of Vibrio cholerae at multiple sites. J. Biol. Chem. 2009, 284, 26557–26568. [Google Scholar]
- Cordero, C.L.; Kudryashov, D.S.; Reisler, E.; Satchell, K.J. The Actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J. Biol. Chem. 2006, 281, 32366–32374. [Google Scholar]
- Geissler, B.; Bonebrake, A.; Sheahan, K.L.; Walker, M.E.; Satchell, K.J. Genetic determination of essential residues of the Vibrio cholerae actin cross-linking domain reveals functional similarity with glutamine synthetases. Mol. Microbiol. 2009, 73, 858–868. [Google Scholar]
- Kudryashov, D.S.; Cordero, C.L.; Reisler, E.; Satchell, K.J. Characterization of the enzymatic activity of the actin cross-linking domain from the Vibrio cholerae MARTX Vc toxin. J. Biol. Chem. 2008, 283, 445–452. [Google Scholar]
- Kudryashov, D.S.; Durer, Z.A.; Ytterberg, A.J.; Sawaya, M.R.; Pashkov, I.; Prochazkova, K.; Yeates, T.O.; Loo, R.R.; Loo, J.A.; Satchell, K.J.; Reisler, E. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc. Natl. Acad. Sci. USA 2008, 105, 18537–18542. [Google Scholar]
- Sheahan, K.L.; Satchell, K.J. Inactivation of small Rho GTPases by the multifunctional RTX toxin from Vibrio cholerae. Cell Microbiol. 2007, 9, 1324–1335. [Google Scholar]
- Albesa-Jove, D.; Bertrand, T.; Carpenter, E.P.; Swain, G.V.; Lim, J.; Zhang, J.; Haire, L.F.; Vasisht, N.; Braun, V.; Lange, A.; von Eichel-Streiber, C.; Svergun, D.I.; Fairweather, N.F.; Brown, K.A. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J. Mol. Biol. 2010, 396, 1260–1270. [Google Scholar]
- Giesemann, T.; Egerer, M.; Jank, T.; Aktories, K. Processing of Clostridium difficile toxins. J. Med. Microbiol. 2008, 57, 690–696. [Google Scholar]
- Genth, H.; Aktories, K.; Just, I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J. Biol. Chem. 1999, 274, 29050–29056. [Google Scholar]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar]
- Barth, H.; Pfeifer, G.; Hofmann, F.; Maier, E.; Benz, R.; Aktories, K. Low pH-induced formation of ion channels by clostridium difficile toxin B in target cells. J. Biol. Chem. 2001, 276, 10670–10676. [Google Scholar]
- Egerer, M.; Giesemann, T.; Herrmann, C.; Aktories, K. Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J. Biol. Chem. 2009, 284, 3389–3395. [Google Scholar] [PubMed]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile--more difficult than ever. N. Engl. J. Med. 2008, 359, 1932–1940. [Google Scholar]
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526–536. [Google Scholar] [CrossRef]
- Lyras, D.; O'Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; Gerding, D.N.; Rood, J.I. Toxin B is essential for virulence of Clostridium difficile. Nature 2009, 458, 1176–1179. [Google Scholar]
- Stabler, R.A.; Gerding, D.N.; Songer, J.G.; Drudy, D.; Brazier, J.S.; Trinh, H.T.; Witney, A.A.; Hinds, J.; Wren, B.W. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J. Bacteriol. 2006, 188, 7297–7305. [Google Scholar]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: the peptidase database. Nucleic Acids Res. 2008, 36, D320–D325. [Google Scholar]
- Barrett, A.J.; Rawlings, N.D. Evolutionary lines of cysteine peptidases. Biol. Chem. 2001, 382, 727–733. [Google Scholar]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar]
- Pop, C.; Salvesen, G.S. Human caspases: activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [PubMed]
- Schilling, O.; Overall, C.M. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 2008, 26, 685–694. [Google Scholar]
- Stennicke, H.R.; Renatus, M.; Meldal, M.; Salvesen, G.S. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 2000, 350 (Pt 2), 563–568. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, M.; Pabst, S.; Rupnik, M.; von Eichel-Streiber, C.; Urlaub, H.; Soling, H.D. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 2005, 151, 199–208. [Google Scholar]
- Asgian, J.L.; James, K.E.; Li, Z.Z.; Carter, W.; Barrett, A.J.; Mikolajczyk, J.; Salvesen, G.S.; Powers, J.C. Aza-peptide epoxides: A new class of inhibitors selective for clan CD cysteine proteases. J. Med. Chem. 2002, 45, 4958–4960. [Google Scholar]
- Ganesan, R.; Jelakovic, S.; Campbell, A. J.; Li, Z.Z.; Asgian, J.L.; Powers, J.C.; Grutter, M.G. Exploring the S4 and S1 prime subsite specificities in caspase-3 with aza-peptide epoxide inhibitors. Biochemistry 2006, 45, 9059–9067. [Google Scholar]
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38. [Google Scholar] [CrossRef]
- Geissler, B.; Tungekar, R.; Satchell, K.J. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Pro. Nat. Acad Sci. USA 2010, 107, 5581–5586. [Google Scholar]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The ClosTron: Mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar]
- Blair, W.S.; Semler, B.L. Self-cleaving proteases. Curr. Opin. Cell Biol. 1991, 3, 1039–1045. [Google Scholar] [CrossRef]
- Bedard, K.M.; Semler, B.L. Regulation of picornavirus gene expression. Microbes. Infect. 2004, 6, 702–713. [Google Scholar] [CrossRef]
- Lall, M.S.; Jain, R.P.; Vederas, J.C. Inhibitors of 3C cysteine proteinases from Picornaviridae. Curr. Top Med. Chem. 2004, 4, 1239–1253. [Google Scholar]
- Lee, G.M.; Craik, C.S. Trapping moving targets with small molecules. Science 2009, 324, 213–215. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Shen, A. Autoproteolytic Activation of Bacterial Toxins. Toxins 2010, 2, 963-977. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2050963
AMA Style
Shen A. Autoproteolytic Activation of Bacterial Toxins. Toxins. 2010; 2(5):963-977. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2050963
Chicago/Turabian StyleShen, Aimee. 2010. "Autoproteolytic Activation of Bacterial Toxins" Toxins 2, no. 5: 963-977. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2050963