AB Toxins: A Paradigm Switch from Deadly to Desirable

Abstract
:1. Introduction

{kind=link}
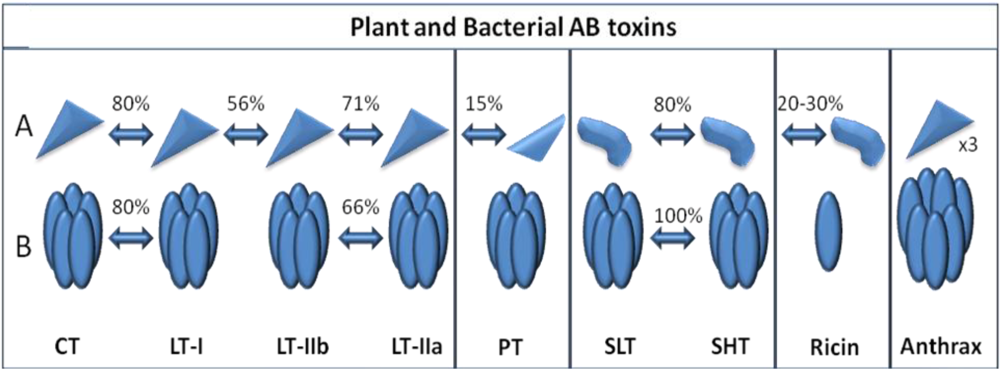{kind=link}
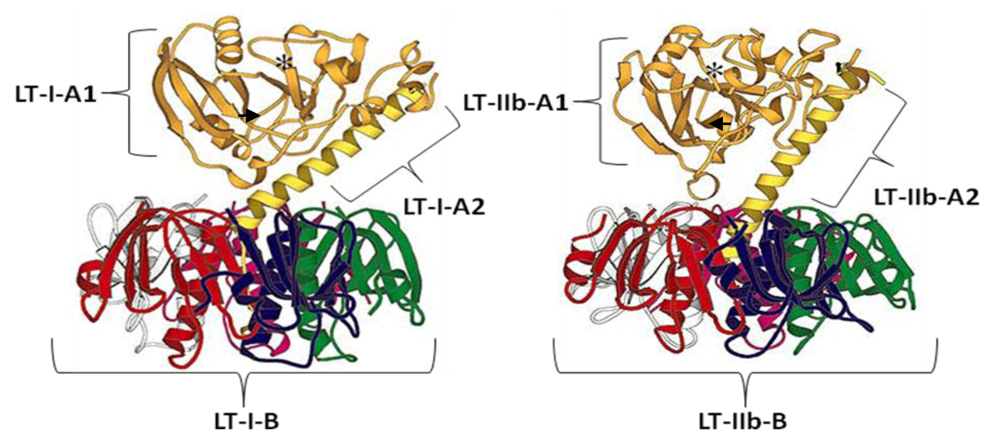{kind=link}
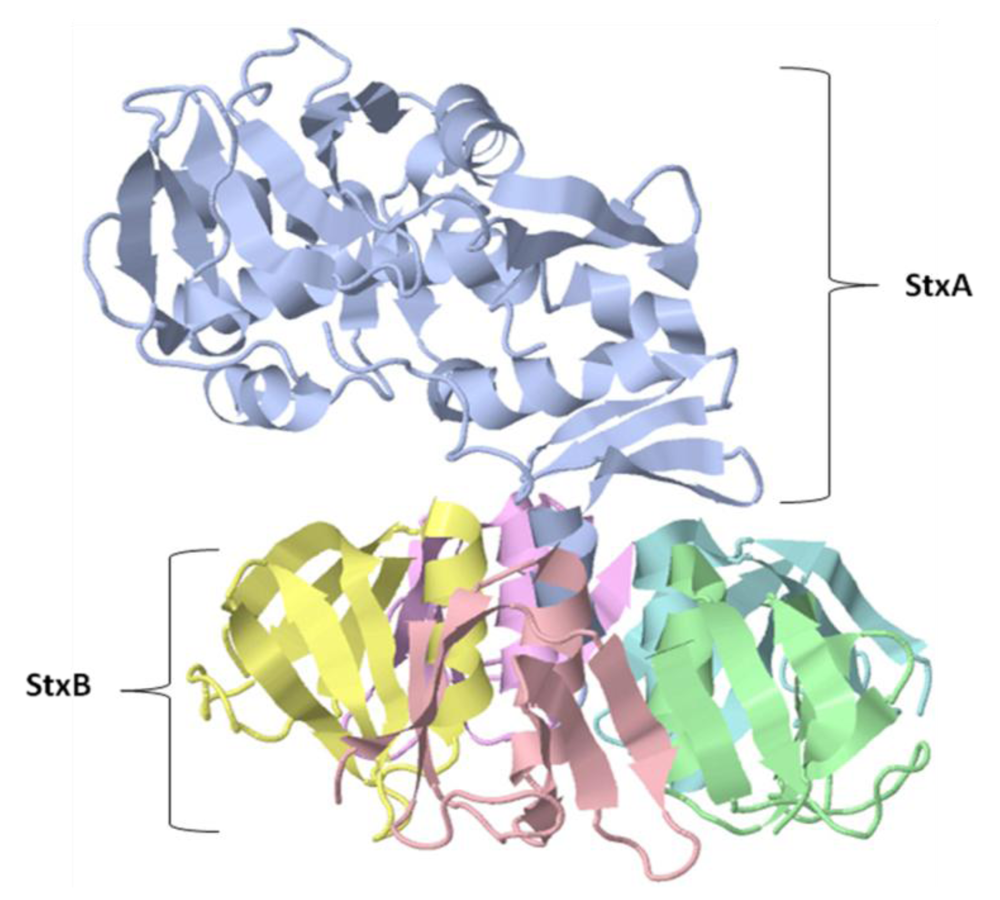{kind=link}
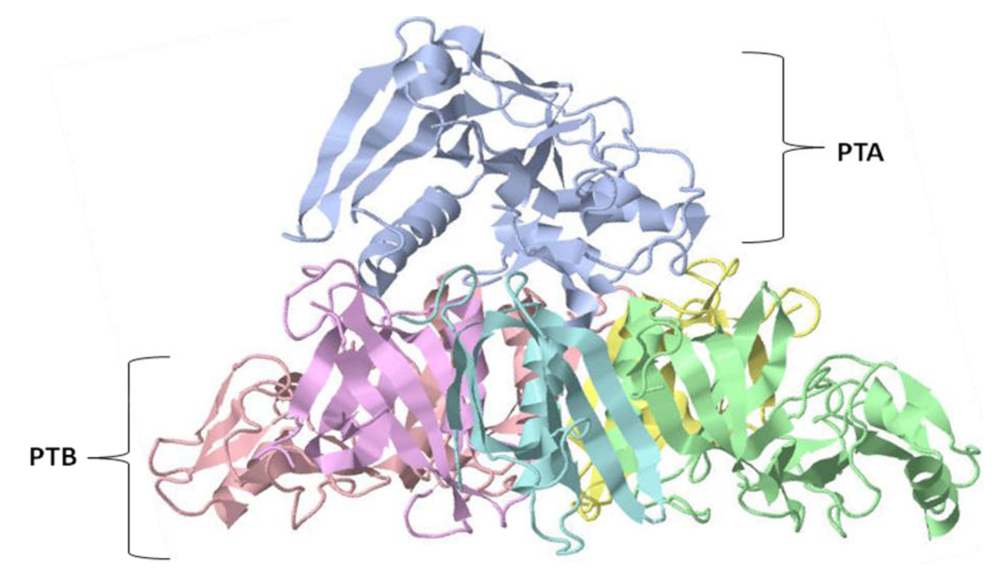{kind=link}
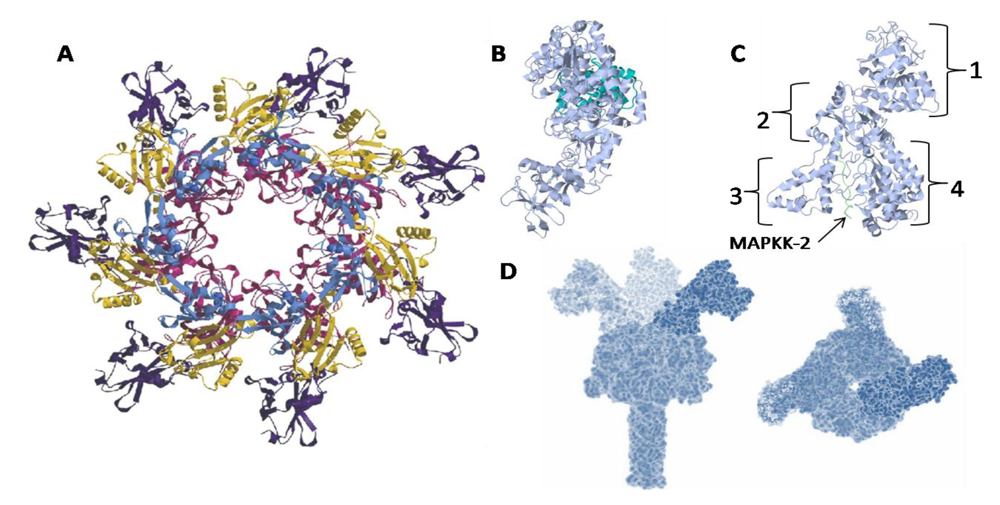{kind=link}
{kind=link}
| A Subunit(s) | B Subunit(s) | Enzymatic Activity | Target | Receptor(s) | |
|---|---|---|---|---|---|
| Cholera toxin | A1: 22 kDa | (×5) 10.6 kDa | ADP-ribosyl transferase | Adenylate cyclase | GM1 ganglioside |
| A2: 5 kDa | G-protein (Gsα) | ||||
| E. coli (LT) | A1: 22 kDa | (×5) 11.6 kDa | ADP-ribosyl transferase | G-protein (Gsα) | GM1 ganglioside |
| A2: 5 kDa | Asialoganglioside | ||||
| Shiga toxin | A1: 28 kDa | (×5) 7.7 kDa | N-glycosylase | rRNA (28S) | Gb3 glycolipid |
| A2: 4 kDa | (Cleaves adenine 4324) | ||||
| Pertussis toxin | S1: 28 kDa | S2: 23 kDa | ADP-ribosyl transferase | G-protein (Gsα) | GD1a ganglioside |
| S3: 22 kDa | |||||
| S4: (×2) 11.7 kDa | |||||
| S5: 9.3 kDa | |||||
| Anthrax | (LF): 90 kDa | (PA): (×7) 83 kDa | Zn metalloprotease | MAPKK | ANTXR 1 |
| (EF): 89 kDa | Adenylate cyclase | Protein kinases | ANTXR 2 | ||
| Ricin | 30 kDa | 29 kDa | N-glycosylase | rRNA (28S) | Glycoprotein |
| (Cleaves adenine 4324) | Glycolipid |
2. Cholera Toxin
2.1. Structure, Pathogenesis and Biological Function
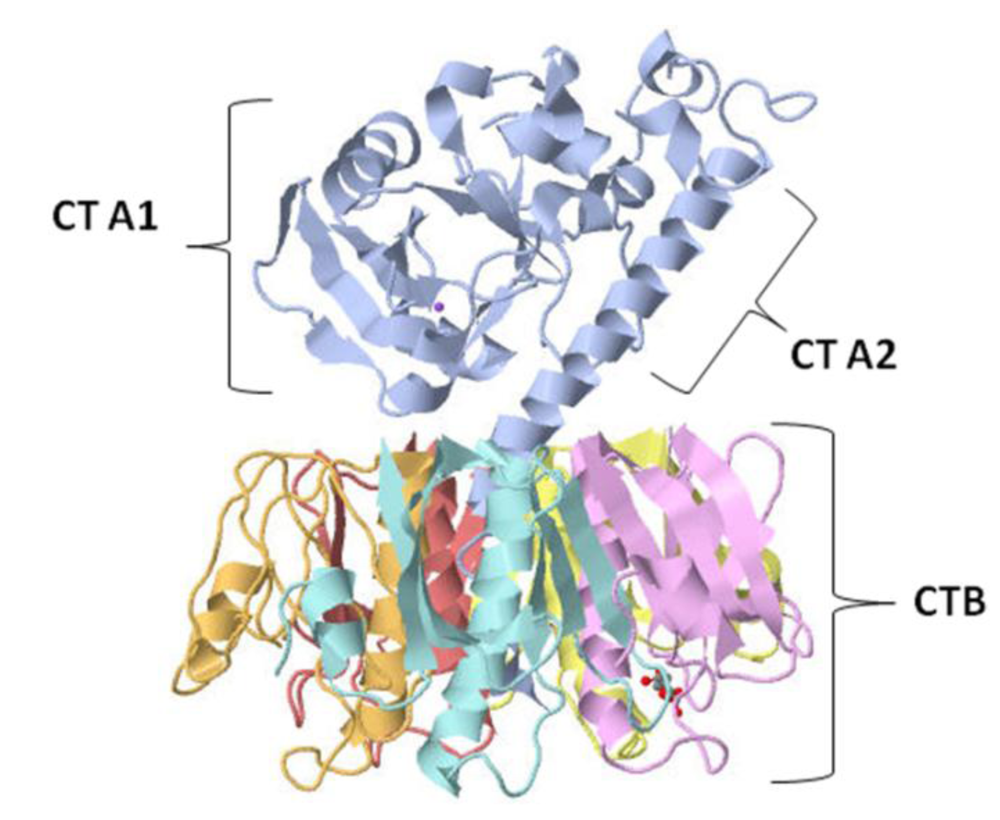
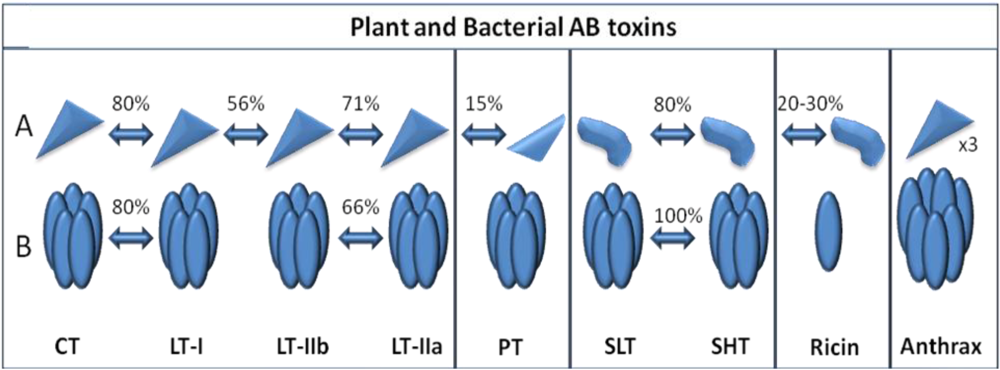
2.2. Immunological Activity and Clinical Applications of Cholera Toxin
3. Heat Labile Enterotoxin from Enterotoxigenic E. coli (LT)
3.1. Structure, Pathogenesis and Function

3.2. Immunological Activity and Clinical Applications of LT
4. Shiga and Shiga-like Toxins
4.1. Structure Pathogenesis and Function
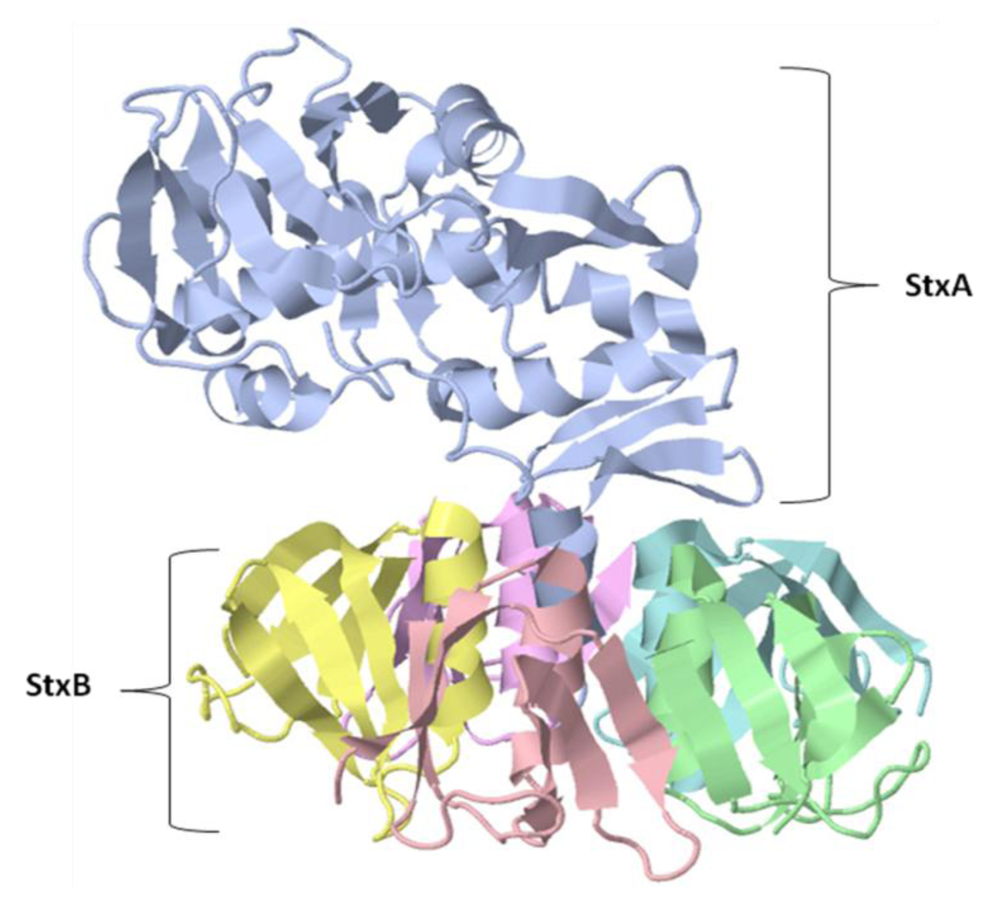
4.2. Immunological Activity and Clinical Applications of Shiga Toxin
5. Pertussis Toxin
5.1. Structure, Pathogenesis and Function

5.2. Immunological Activity and Clinical Applications of Pertussis Toxin
6. Anthrax Toxin
6.1. Structure, Pathogenesis and Function

6.2. Immunological Activity and Clinical Applications of Anthrax
7. Ricin Toxin
7.1. Structure, Pathogenesis and Function

7.2. Immunological and Clinical Applications of Ricin
8. Conclusions
References
- Finkelstein, R.A.; LoSpalluto, J.J. Pathogenesis of experimental cholera. Preparation and isolation of choleragen and choleragenoid. J. Exp. Med. 1969, 130, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Lonnroth, I.; Holmgren, J. Subunit structure of cholera toxin. J. Gen. Microbiol. 1973, 76, 417–427. [Google Scholar]
- Sattler, J.; Wiegandt, H. Studies of the subunit structure of choleragen. Eur. J. Biochem. 1975, 57, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Dauter, Z.; Kingma, J.; Witholt, B.; Hol, W.G. Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J. Mol. Biol. 1993, 230, 890–918. [Google Scholar]
- Merritt, E.A.; Sarfaty, S.; Chang, T.T.; Palmer, L.M.; Jobling, M.G.; Holmes, R.K.; Hol, W.G. Surprising leads for a cholera toxin receptor-binding antagonist: crystallographic studies of CTB mutants. Structure 1995, 3, 561–570. [Google Scholar]
- Holmgren, J.; Lonnroth, I.; Svennerholm, L. Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect. Immun. 1973, 8, 208–214. [Google Scholar]
- Holmgren, J.; Lonnroth, I.; Mansson, J.; Svennerholm, L. Interaction of cholera toxin and membrane GM1 ganglioside of small intestine. Proc. Natl. Acad. Sci. USA 1975, 72, 2520–2524. [Google Scholar]
- van Heyningen, W.E.; King, C.A. The role of gangliosides in the action of cholera toxin. Adv. Exp. Med. Biol. 1976, 71, 205–214. [Google Scholar]
- van Heyningen, S. The subunits of cholera toxin: structure, stoichiometry, and function. J. Infect. Dis. 1976, 133, 5–13. [Google Scholar]
- Holmgren, J.; Lindholm, L.; Lonnroth, I. Interaction of cholera toxin and toxin derivatives with lymphocytes. I. Binding properties and interference with lectin-induced cellular stimulation. J. Exp. Med. 1974, 139, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 2008, 65, 1347–1360. [Google Scholar]
- Camberg, J.L.; Johnson, T.L.; Patrick, M.; Abendroth, J.; Hol, W.G.; Sandkvist, M. Synergistic stimulation of EpsE ATP hydrolysis by EpsL and acidic phospholipids. EMBO J. 2007, 26, 19–27. [Google Scholar]
- Camberg, J.L.; Sandkvist, M. Molecular analysis of the Vibrio cholerae type II secretion ATPase EpsE. J. Bacteriol. 2005, 187, 249–256. [Google Scholar]
- Davis, B.M.; Lawson, E.H.; Sandkvist, M.; Ali, A.; Sozhamannan, S.; Waldor, M.K. Convergence of the secretory pathways for cholera toxin and the filamentous phage, CTXphi. Science 2000, 288, 333–335. [Google Scholar]
- Hirst, T.R.; Sanchez, J.; Kaper, J.B.; Hardy, S.J.; Holmgren, J. Mechanism of toxin secretion by Vibrio cholerae investigated in strains harboring plasmids that encode heat-labile enterotoxins of Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 7752–7756. [Google Scholar]
- Hardy, S.J.; Holmgren, J.; Johansson, S.; Sanchez, J.; Hirst, T.R. Coordinated assembly of multisubunit proteins: oligomerization of bacterial enterotoxins in vivo and in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 7109–7113. [Google Scholar]
- Merritt, E.A.; Sarfaty, S.; van den Akker, F.; L'Hoir, C.; Martial, J.A.; Hol, W.G. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994, 3, 166–175. [Google Scholar]
- Jobling, M.G.; Holmes, R.K. Mutational analysis of ganglioside GM(1)-binding ability, pentamer formation, and epitopes of cholera toxin B (CTB) subunits and CTB/heat-labile enterotoxin B subunit chimeras. Infect. Immun. 2002, 70, 1260–1271. [Google Scholar]
- Merritt, E.A.; Hol, W.G. AB5 toxins. Curr. Opin. Struct. Biol. 1995, 5, 165–171. [Google Scholar]
- Chinnapen, D.J.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar]
- Massol, R.H.; Larsen, J.E.; Fujinaga, Y.; Lencer, W.I.; Kirchhausen, T. Cholera toxin toxicity does not require functional Arf6- and dynamin-dependent endocytic pathways. Mol.Biol.Cell. 2004, 15, 3631–3641. [Google Scholar]
- Sandvig, K.; van Deurs, B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002, 529, 49–53. [Google Scholar]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar]
- O'Neal, C.J.; Jobling, M.G.; Holmes, R.K.; Hol, W.G. Structural basis for the activation of cholera toxin by human ARF6-GTP. Science 2005, 309, 1093–1096. [Google Scholar]
- Guidry, J.J.; Cardenas, L.; Cheng, E.; Clements, J.D. Role of receptor binding in toxicity, immunogenicity, and adjuvanticity of Escherichia coli heat-labile enterotoxin. Infect. Immun. 1997, 65, 4943–4950. [Google Scholar]
- Porgador, A.; Staats, H.F.; Itoh, Y.; Kelsall, B.L. Intranasal immunization with cytotoxic T-lymphocyte epitope peptide and mucosal adjuvant cholera toxin: selective augmentation of peptide-presenting dendritic cells in nasal mucosa-associated lymphoid tissue. Infect. Immun. 1998, 66, 5876–5881. [Google Scholar]
- Sun, J.B.; Holmgren, J.; Czerkinsky, C. Cholera toxin B subunit: an efficient transmucosal carrier-delivery system for induction of peripheral immunological tolerance. Proc. Natl. Acad. Sci. USA 1994, 91, 10795–10799. [Google Scholar]
- Czerkinsky, C.; Sun, J.B.; Lebens, M.; Li, B.L.; Rask, C.; Lindblad, M.; Holmgren, J. Cholera toxin B subunit as transmucosal carrier-delivery and immunomodulating system for induction of antiinfectious and antipathological immunity. Ann. N. Y. Acad. Sci. 1996, 778, 185–193. [Google Scholar]
- Sun, J.B.; Xiao, B.G.; Lindblad, M.; Li, B.L.; Link, H.; Czerkinsky, C.; Holmgren, J. Oral administration of cholera toxin B subunit conjugated to myelin basic protein protects against experimental autoimmune encephalomyelitis by inducing transforming growth factor-beta-secreting cells and suppressing chemokine expression. Int. Immunol. 2000, 12, 1449–1457. [Google Scholar]
- Eriksson, K.; Holmgren, J. Recent advances in mucosal vaccines and adjuvants. Curr. Opin. Immunol. 2002, 14, 666–672. [Google Scholar]
- Lucas, M.E.; Deen, J.L.; von Seidlein, L.; Wang, X.Y.; Ampuero, J.; Puri, M.; Ali, M.; Ansaruzzaman, M.; Amos, J.; Macuamule, A.; Cavailler, P.; Guerin, P.J.; Mahoudeau, C.; Kahozi-Sangwa, P.; Chaignat, C.L.; Barreto, A.; Songane, F.F.; Clemens, J.D. Effectiveness of mass oral cholera vaccination in Beira, Mozambique. N. Engl. J. Med. 2005, 352, 757–767. [Google Scholar]
- George-Chandy, A.; Eriksson, K.; Lebens, M.; Nordstrom, I.; Schon, E.; Holmgren, J. Cholera toxin B subunit as a carrier molecule promotes antigen presentation and increases CD40 and CD86 expression on antigen-presenting cells. Infect. Immun. 2001, 69, 5716–5725. [Google Scholar]
- D'Ambrosio, A.; Colucci, M.; Pugliese, O.; Quintieri, F.; Boirivant, M. Cholera toxin B subunit promotes the induction of regulatory T cells by preventing human dendritic cell maturation. J. Leukoc. Biol. 2008, 84, 661–668. [Google Scholar]
- Kim, N.; Cheng, K.C.; Kwon, S.S.; Mora, R.; Barbieri, M.; Yoo, T.J. Oral administration of collagen conjugated with cholera toxin induces tolerance to type II collagen and suppresses chondritis in an animal model of autoimmune ear disease. Ann. Otol. Rhinol. Laryngol. 2001, 110, 646–654. [Google Scholar]
- Phipps, P.A.; Stanford, M.R.; Sun, J.B.; Xiao, B.G.; Holmgren, J.; Shinnick, T.; Hasan, A.; Mizushima, Y.; Lehner, T. Prevention of mucosally induced uveitis with a HSP60-derived peptide linked to cholera toxin B subunit. Eur. J. Immunol. 2003, 33, 224–232. [Google Scholar]
- Bergerot, I.; Ploix, C.; Petersen, J.; Moulin, V.; Rask, C.; Fabien, N.; Lindblad, M.; Mayer, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1997, 94, 4610–4614. [Google Scholar]
- Arakawa, T.; Yu, J.; Chong, D.K.; Hough, J.; Engen, P.C.; Langridge, W.H. A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat. Biotechnol. 1998, 16, 934–938. [Google Scholar]
- Aspord, C.; Thivolet, C. Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 2002, 130, 204–211. [Google Scholar]
- Roncarolo, M.G.; Levings, M.K.; Traversari, C. Differentiation of T regulatory cells by immature dendritic cells. J. Exp. Med. 2001, 193, F5–F9. [Google Scholar]
- Arakawa, T.; Chong, D.K.; Langridge, W.H. Efficacy of a food plant-based oral cholera toxin B subunit vaccine. Nat. Biotechnol. 1998, 16, 292–297. [Google Scholar]
- Marinaro, M.; Staats, H.F.; Hiroi, T.; Jackson, R.J.; Coste, M.; Boyaka, P.N.; Okahashi, N.; Yamamoto, M.; Kiyono, H.; Bluethmann, H.; Fujihashi, K.; McGhee, J.R. Mucosal adjuvant effect of cholera toxin in mice results from induction of T helper 2 (Th2) cells and IL-4. J. Immunol. 1995, 155, 4621–4629. [Google Scholar]
- Lavelle, E.C.; Jarnicki, A.; McNeela, E.; Armstrong, M.E.; Higgins, S.C.; Leavy, O.; Mills, K.H. Effects of cholera toxin on innate and adaptive immunity and its application as an immunomodulatory agent. J.Leukoc.Biol. 2004, 75, 756–763. [Google Scholar]
- Lavelle, E.C.; McNeela, E.; Armstrong, M.E.; Leavy, O.; Higgins, S.C.; Mills, K.H. Cholera toxin promotes the induction of regulatory T cells specific for bystander antigens by modulating dendritic cell activation. J. Immunol. 2003, 171, 2384–2392. [Google Scholar]
- Isomura, I.; Yasuda, Y.; Tsujimura, K.; Takahashi, T.; Tochikubo, K.; Morita, A. Recombinant cholera toxin B subunit activates dendritic cells and enhances antitumor immunity. Microbiol. Immunol. 2005, 49, 79–87. [Google Scholar]
- Ploix, C.; Bergerot, I.; Durand, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes 1999, 48, 2150–2156. [Google Scholar]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand. J. Immunol. 2010, 71, 1–11. [Google Scholar]
- Denes, B.; Fodor, I.; Odumosu, O.; Langridge, W. Multi-factorial Vaccine Suppression of Diabetes Autoimmunity. 2010; Unpublished work. [Google Scholar]
- Sixma, T.K.; Pronk, S.E.; Kalk, K.H.; Wartna, E.S.; van Zanten, B.A.; Witholt, B.; Hol, W.G. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature 1991, 351, 371–377. [Google Scholar] [CrossRef] [PubMed]
- van den Akker, F.; Sarfaty, S.; Twiddy, E.; Connell, T.; Holmes, R.; Hol, W. Crystal structure of a new heat-labile enterotoxin, LT-IIb. Structure 1996, 4, 665–678. [Google Scholar]
- Alone, P.V.; Malik, G.; Krishnan, A.; Garg, L.C. Deletion mutations in N-terminal alpha1 helix render heat labile enterotoxin B subunit susceptible to degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 16056–16061. [Google Scholar]
- Alone, P.V.; Garg, L.C. Secretory and GM1 receptor binding role of N-terminal region of LTB in Vibrio cholerae. Biochem. Biophys. Res. Commun. 2008, 376, 770–774. [Google Scholar]
- Chung, W.Y.; Carter, R.; Hardy, T.; Sack, M.; Hirst, T.R.; James, R.F. Inhibition of Escherichia coli heat-labile enterotoxin B subunit pentamer (EtxB5) assembly in vitro using monoclonal antibodies. J. Biol. Chem. 2006, 281, 39465–39470. [Google Scholar]
- Schengrund, C.L.; Ringler, N.J. Binding of Vibrio cholera toxin and the heat-labile enterotoxin of Escherichia coli to GM1, derivatives of GM1, and nonlipid oligosaccharide polyvalent ligands. J. Biol. Chem. 1989, 264, 13233–13237. [Google Scholar]
- Sixma, T.K.; Pronk, S.E.; Kalk, K.H.; van Zanten, B.A.; Berghuis, A.M.; Hol, W.G. Lactose binding to heat-labile enterotoxin revealed by X-ray crystallography. Nature 1992, 355, 561–564. [Google Scholar]
- Karlsson, K.A.; Teneberg, S.; Angstrom, J.; Kjellberg, A.; Hirst, T.R.; Berstrom, J.; Miller-Podraza, H. Unexpected carbohydrate cross-binding by Escherichia coli heat-labile enterotoxin. Recognition of human and rabbit target cell glycoconjugates in comparison with cholera toxin. Bioorg. Med. Chem. 1996, 4, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R.; Magnani, J.L.; Fishman, P.H. Interaction of cholera toxin with rat intestinal brush border membranes. Relative roles of gangliosides and galactoproteins as toxin receptors. J. Biol. Chem. 1981, 256, 8724–8731. [Google Scholar] [PubMed]
- Orlandi, P.A.; Critchley, D.R.; Fishman, P.H. The heat-labile enterotoxin of Escherichia coli binds to polylactosaminoglycan-containing receptors in CaCo-2 human intestinal epithelial cells. Biochemistry 1994, 33, 12886–12895. [Google Scholar]
- Fukuta, S.; Magnani, J.L.; Twiddy, E.M.; Holmes, R.K.; Ginsburg, V. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect. Immun. 1988, 56, 1748–1753. [Google Scholar]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol. Biol. Cell. 2003, 14, 4783–4793. [Google Scholar]
- Spangler, B.D. Structure and Function of Cholera-Toxin and the Related Escherichia-Coli Heat-Labile Enterotoxin. Microbiol. Mol. Biol. Rev. 1992, 56, 622–647. [Google Scholar]
- Couch, R.B. Nasal vaccination, Escherichia coli enterotoxin, and Bell's palsy. N. Engl. J. Med. 2004, 350, 860–861. [Google Scholar]
- Lopes, L.M.; Maroof, A.; Dougan, G.; Chain, B.M. Inhibition of T-cell response by Escherichia coli heat-labile enterotoxin-treated epithelial cells. Infect. Immun. 2000, 68, 6891–6895. [Google Scholar]
- Negri, D.R.; Pinto, D.; Vendetti, S.; Patrizio, M.; Sanchez, M.; Riccomi, A.; Ruggiero, P.; Del Giudice, G.; De Magistris, M.T. Cholera toxin and Escherichia coli heat-labile enterotoxin, but not their nontoxic counterparts, improve the antigen-presenting cell function of human B lymphocytes. Infect. Immun. 2009, 77, 1924–1935. [Google Scholar]
- Hajishengallis, G.; Nawar, H.; Tapping, R.I.; Russell, M.W.; Connell, T.D. The Type II heat-labile enterotoxins LT-IIa and LT-IIb and their respective B pentamers differentially induce and regulate cytokine production in human monocytic cells. Infect. Immun. 2004, 72, 6351–6358. [Google Scholar]
- Domingos, M.O.; Andrade, R.G.; Barbaro, K.C.; Borges, M.M.; Lewis, D.J.; New, R.R. Influence of the A and B subunits of cholera toxin (CT) and Escherichia coli toxin (LT) on TNF-alpha release from macrophages. Toxicon 2009, 53, 570–577. [Google Scholar]
- Lee, C.H.; Nawar, H.F.; Mandell, L.; Liang, S.; Hajishengallis, G.; Connell, T.D. Enhanced antigen uptake by dendritic cells induced by the B pentamer of the type II heat-labile enterotoxin LT-IIa requires engagement of TLR2. Vaccine 2010, 28, 3696–3705. [Google Scholar]
- Liang, S.; Hosur, K.B.; Nawar, H.F.; Russell, M.W.; Connell, T.D.; Hajishengallis, G. In vivo and in vitro adjuvant activities of the B subunit of Type IIb heat-labile enterotoxin (LT-IIb-B5) from Escherichia coli. Vaccine 2009, 27, 4302–4308. [Google Scholar]
- Anosova, N.G.; Chabot, S.; Shreedhar, V.; Borawski, J.A.; Dickinson, B.L.; Neutra, M.R. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal. Immunol. 2008, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Glenn, G.M.; Thomas, D.N.; Poffenberger, K.L.; Flyer, D.C.; Ellingsworth, L.R.; Andersen, B.H.; Frech, S.A. Safety and immunogenicity of an influenza vaccine A/H5N1 (A/Vietnam/1194/2004) when coadministered with a heat-labile enterotoxin (LT) adjuvant patch. Vaccine 2009, 27, G60–G66. [Google Scholar] [PubMed]
- Alfano, M.; Rizzi, C.; Corti, D.; Adduce, L.; Poli, G. Bacterial toxins: potential weapons against HIV infection. Curr. Pharm. Des. 2005, 11, 2909–2926. [Google Scholar]
- Zhou, W.Y.; Shi, Y.; Wu, C.; Zhang, W.J.; Mao, X.H.; Guo, G.; Li, H.X.; Zou, Q.M. Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine 2009, 27, 5013–5019. [Google Scholar]
- Facciabene, A.; Aurisicchio, L.; Elia, L.; Palombo, F.; Mennuni, C.; Ciliberto, G.; La Monica, N. Vectors encoding carcinoembryonic antigen fused to the B subunit of heat-labile enterotoxin elicit antigen-specific immune responses and antitumor effects. Vaccine 2007, 26, 47–58. [Google Scholar]
- Lemere, C.A. Developing novel immunogens for a safe and effective Alzheimer's disease vaccine. Prog. Brain Res. 2009, 175, 83–93. [Google Scholar]
- Ryan, E.J.; McNeela, E.; Pizza, M.; Rappuoli, R.; O'Neill, L.; Mills, K.H. Modulation of innate and acquired immune responses by Escherichia coli heat-labile toxin: distinct pro- and anti-inflammatory effects of the nontoxic AB complex and the enzyme activity. J. Immunol. 2000, 165, 5750–5759. [Google Scholar]
- Raveney, B.J.; Richards, C.; Aknin, M.L.; Copland, D.A.; Burton, B.R.; Kerr, E.; Nicholson, L.B.; Williams, N.A.; Dick, A.D. The B subunit of Escherichia coli heat-labile enterotoxin inhibits Th1 but not Th17 cell responses in established experimental autoimmune uveoretinitis. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4008–4017. [Google Scholar]
- Scerbo, M.J.; Rupil, L.L.; Bibolini, M.J.; Roth, G.A.; Monferran, C.G. Protective effect of a synapsin peptide genetically fused to the B subunit of Escherichia coli heat-labile enterotoxin in rat autoimmune encephalomyelitis. J. Neurosci. Res. 2009, 87, 2273–2281. [Google Scholar]
- Carter, J.E., III; Yu, J.; Choi, N.W.; Hough, J.; Henderson, D.; He, D.; Langridge, W.H. Bacterial and plant enterotoxin B subunit-autoantigen fusion proteins suppress diabetes insulitis. Mol. Biotechnol. 2006, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ravin, N.V.; Kuprianov, V.V.; Zamchuk, L.A.; Kochetov, A.V.; Dorokhov, Y.L.; Atabekov, J.G.; Skryabin, K.G. Highly efficient expression of Escherichia coli heat-labile enterotoxin B subunit in plants using potato virus X-based vector. Biochemistry (Mosc) 2008, 73, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Chikwamba, R.; Cunnick, J.; Hathaway, D.; McMurray, J.; Mason, H.; Wang, K. A functional antigen in a practical crop: LT-B producing maize protects mice against Escherichia coli heat labile enterotoxin (LT) and cholera toxin (CT). Transgenic Res. 2002, 11, 479–493. [Google Scholar]
- Kim, T.G.; Kim, M.Y.; Kim, B.G.; Kang, T.J.; Kim, Y.S.; Jang, Y.S.; Arntzen, C.J.; Yang, M.S. Synthesis and assembly of Escherichia coli heat-labile enterotoxin B subunit in transgenic lettuce (Lactuca sativa). Protein Expr. Purif. 2007, 51, 22–27. [Google Scholar]
- Rosales-Mendoza, S.; Soria-Guerra, R.E.; Lopez-Revilla, R.; Moreno-Fierros, L.; Alpuche-Solis, A.G. Ingestion of transgenic carrots expressing the Escherichia coli heat-labile enterotoxin B subunit protects mice against cholera toxin challenge. Plant Cell Rep. 2008, 27, 79–84. [Google Scholar]
- Zhang, X.; Yuan, Z.; Duan, Q.; Zhu, H.; Yu, H.; Wang, Q. Mucosal immunity in mice induced by orally administered transgenic rice. Vaccine 2009, 27, 1596–1600. [Google Scholar]
- Trofa, A.F.; Ueno-Olsen, H.; Oiwa, R.; Yoshikawa, M. Dr. Kiyoshi Shiga: discoverer of the dysentery bacillus. Clin. Infect. Dis. 1999, 29, 1303–1306. [Google Scholar]
- Strelkauskas, A.J.; Stelkauskas, J.E.; Moszyk-Strelkauskas, D. Microbiology: A Clinical Approach; Garland Science: New York, NY, USA, 2010. [Google Scholar]
- Johannes, L.; Romer, W. Shiga toxins—from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar]
- Wilson, C.B.; Rowell, E.; Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 91–105. [Google Scholar]
- Melton-Celsa, A.R.; Darnell, S.C.; O'Brien, A.D. Activation of Shiga-like toxins by mouse and human intestinal mucus correlates with virulence of enterohemorrhagic Escherichia coli O91:H21 isolates in orally infected, streptomycin-treated mice. Infect. Immun. 1996, 64, 1569–1576. [Google Scholar]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Entry of ricin and Shiga toxin into cells: molecular mechanisms and medical perspectives. EMBO J. 2000, 19, 5943–5950. [Google Scholar]
- Sandvig, K.; Grimmer, S.; Iversen, T.G.; Rodal, K.; Torgersen, M.L.; Nicoziani, P.; van Deurs, B. Ricin transport into cells: studies of endocytosis and intracellular transport. Int. J. Med. Microbiol. 2000, 290, 415–420. [Google Scholar]
- Haicheur, N.; Benchetrit, F.; Amessou, M.; Leclerc, C.; Falguieres, T.; Fayolle, C.; Bismuth, E.; Fridman, W.H.; Johannes, L.; Tartour, E. The B subunit of Shiga toxin coupled to full-size antigenic protein elicits humoral and cell-mediated immune responses associated with a Th1-dominant polarization. Int. Immunol. 2003, 15, 1161–1171. [Google Scholar]
- Ohmura, M.; Yamamoto, M.; Tomiyama-Miyaji, C.; Yuki, Y.; Takeda, Y.; Kiyono, H. Nontoxic Shiga toxin derivatives from Escherichia coli possess adjuvant activity for the augmentation of antigen-specific immune responses via dendritic cell activation. Infect. Immun. 2005, 73, 4088–4097. [Google Scholar]
- Adotevi, O.; Vingert, B.; Freyburger, L.; Shrikant, P.; Lone, Y.C.; Quintin-Colonna, F.; Haicheur, N.; Amessou, M.; Herbelin, A.; Langlade-Demoyen, P.; Fridman, W.H.; Lemonnier, F.; Johannes, L.; Tartour, E. B subunit of Shiga toxin-based vaccines synergize with alpha-galactosylceramide to break tolerance against self antigen and elicit antiviral immunity. J. Immunol. 2007, 179, 3371–3379. [Google Scholar]
- Oloomi, M.; Bouzari, S.; Emami, S. A recombinant hybrid peptide composed of AAF adhesin of enteroaggregative Escherichia coli and Shiga toxin B subunit elicits protective immune response in mice. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1311–1316. [Google Scholar]
- al-Jaufy, A.Y.; Haddad, J.E.; King, S.R.; McPhee, R.A.; Jackson, M.P. Cytotoxicity of a shiga toxin A subunit-CD4 fusion protein to human immunodeficiency virus-infected cells. Infect. Immun. 1994, 62, 956–960. [Google Scholar]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol. 2008, 10, 1130–1139. [Google Scholar]
- Oakley, C.L. Jules Jean Baptiste Vincent Bordet. 1870–1961. Biogr. Mem. Fellows R. Soc. 1962, 19–25. [Google Scholar]
- Center for Diseases Control and Prevention (CDC). Pertussis (Whooping Cough) Vaccination. Available online: http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm (Accessed on 21 June 2010).
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J. Leukoc. Biol. 2002, 72, 962–969. [Google Scholar]
- Worthington, Z.E.; Carbonetti, N.H. Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect. Immun. 2007, 75, 2946–2953. [Google Scholar]
- Hausman, S.Z.; Burns, D.L. Binding of pertussis toxin to lipid vesicles containing glycolipids. Infect. Immun. 1993, 61, 335–337. [Google Scholar]
- el Baya, A.; Linnemann, R.; von Olleschik-Elbheim, L.; Robenek, H.; Schmidt, M.A. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar]
- Xu, Y.; Barbieri, J.T. Pertussis toxin-mediated ADP-ribosylation of target proteins in Chinese hamster ovary cells involves a vesicle trafficking mechanism. Infect. Immun. 1995, 63, 825–832. [Google Scholar]
- Wang, Z.Y.; Yang, D.; Chen, Q.; Leifer, C.A.; Segal, D.M.; Su, S.B.; Caspi, R.R.; Howard, Z.O.; Oppenheim, J.J. Induction of dendritic cell maturation by pertussis toxin and its B subunit differentially initiate Toll-like receptor 4-dependent signal transduction pathways. Exp. Hematol. 2006, 34, 1115–1124. [Google Scholar]
- Fujimoto, C.; Yu, C.R.; Shi, G.; Vistica, B.P.; Wawrousek, E.F.; Klinman, D.M.; Chan, C.C.; Egwuagu, C.E.; Gery, I. Pertussis toxin is superior to TLR ligands in enhancing pathogenic autoimmunity, targeted at a neo-self antigen, by triggering robust expansion of Th1 cells and their cytokine production. J. Immunol. 2006, 177, 6896–6903. [Google Scholar]
- Hou, W.; Wu, Y.; Sun, S.; Shi, M.; Sun, Y.; Yang, C.; Pei, G.; Gu, Y.; Zhong, C.; Sun, B. Pertussis toxin enhances Th1 responses by stimulation of dendritic cells. J. Immunol. 2003, 170, 1728–1736. [Google Scholar]
- Chen, X.; Winkler-Pickett, R.T.; Carbonetti, N.H.; Ortaldo, J.R.; Oppenheim, J.J.; Howard, O.M. Pertussis toxin as an adjuvant suppresses the number and function of CD4+CD25+ T regulatory cells. Eur. J. Immunol. 2006, 36, 671–680. [Google Scholar]
- Alfano, M.; Schmidtmayerova, H.; Amella, C.A.; Pushkarsky, T.; Bukrinsky, M. The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J. Exp. Med. 1999, 190, 597–605. [Google Scholar]
- Alfano, M.; Pushkarsky, T.; Poli, G.; Bukrinsky, M. The B-oligomer of pertussis toxin inhibits human immunodeficiency virus type 1 replication at multiple stages. J. Virol. 2000, 74, 8767–8770. [Google Scholar]
- Rizzi, C.; Crippa, M.P.; Jeeninga, R.E.; Berkhout, B.; Blasi, F.; Poli, G.; Alfano, M. Pertussis toxin B-oligomer suppresses IL-6 induced HIV-1 and chemokine expression in chronically infected U1 cells via inhibition of activator protein 1. J. Immunol. 2006, 176, 999–1006. [Google Scholar]
- van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar]
- Remacle, A.G.; Gawlik, K.; Golubkov, V.S.; Cadwell, G.W.; Liddington, R.C.; Cieplak, P.; Millis, S.Z.; Desjardins, R.; Routhier, S.; Yuan, X.W.; Neugebauer, W.A.; Day, R.; Strongin, A.Y. Selective and potent furin inhibitors protect cells from anthrax without significant toxicity. Int. J. Biochem. Cell Biol. 2010, 42, 987–995. [Google Scholar]
- Lacy, D.B.; Wigelsworth, D.J.; Melnyk, R.A.; Harrison, S.C.; Collier, R.J. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA 2004, 101, 13147–13151. [Google Scholar]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar]
- Mogridge, J.; Cunningham, K.; Collier, R.J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–1082. [Google Scholar]
- Abrami, L.; Kunz, B.; van der Goot, F.G. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 1420–1424. [Google Scholar]
- Tama, F.; Ren, G.; Brooks, C.L., III; Mitra, A.K. Model of the toxic complex of anthrax: responsive conformational changes in both the lethal factor and the protective antigen heptamer. Protein Sci. 2006, 15, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar]
- Gao, M.; Schulten, K. Onset of anthrax toxin pore formation. Biophys. J. 2006, 90, 3267–3279. [Google Scholar]
- Basilio, D.; Juris, S.J.; Collier, R.J.; Finkelstein, A. Evidence for a proton-protein symport mechanism in the anthrax toxin channel. J. Gen. Physiol. 2009, 133, 307–314. [Google Scholar]
- Wesche, J.; Elliott, J.L.; Falnes, P.O.; Olsnes, S.; Collier, R.J. Characterization of membrane translocation by anthrax protective antigen. Biochemistry 1998, 37, 15737–15746. [Google Scholar]
- Thoren, K.L.; Worden, E.J.; Yassif, J.M.; Krantz, B.A. Lethal factor unfolding is the most force-dependent step of anthrax toxin translocation. Proc. Natl. Acad. Sci. USA 2009, 106, 21555–21560. [Google Scholar]
- Janowiak, B.E.; Fischer, A.; Collier, R.J. Effects of introducing a single charged residue into the phenylalanine clamp of multimeric anthrax protective antigen. J. Biol. Chem. 2010, 285, 8130–8137. [Google Scholar]
- Zhang, S.; Finkelstein, A.; Collier, R.J. Evidence that translocation of anthrax toxin's lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc. Natl. Acad. Sci. USA 2004, 101, 16756–16761. [Google Scholar]
- Halverson, K.M.; Panchal, R.G.; Nguyen, T.L.; Gussio, R.; Little, S.F.; Misakian, M.; Bavari, S.; Kasianowicz, J.J. Anthrax biosensor, protective antigen ion channel asymmetric blockade. J. Biol. Chem. 2005, 280, 34056–34062. [Google Scholar]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; Vande Woude, G.F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacy, D.B.; Collier, R.J.; Park, S.; Leppla, S.H.; Hanna, P.; Liddington, R.C. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar]
- Leppla, S.H. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar]
- Rossi Paccani, S.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell Microbiol. 2007, 9, 924–929. [Google Scholar]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar]
- Kim, J.S.; Bokoch, G.M. Anthrax edema toxin inhibits Nox1-mediated formation of reactive oxygen species by colon epithelial cells. J. Innate Immun. 2009, 1, 145–152. [Google Scholar]
- Ono, K.; Han, J. The p38 signal transduction pathway: activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar]
- Wu, W.; Mehta, H.; Chakrabarty, K.; Booth, J.L.; Duggan, E.S.; Patel, K.B.; Ballard, J.D.; Coggeshall, K.M.; Metcalf, J.P. Resistance of human alveolar macrophages to Bacillus anthracis lethal toxin. J. Immunol. 2009, 183, 5799–5806. [Google Scholar]
- Khan, M.A.; Gallo, R.M.; Brutkiewicz, R.R. Anthrax Lethal Toxin Impairs CD1d-Mediated Antigen Presentation by Targeting the ERK1/2 MAPK Pathway. Infect. Immun. 2010. [Google Scholar]
- Yang, J.; Woo, S.S.; Ryu, Y.H.; Yun, C.H.; Cho, M.H.; Rhie, G.E.; Kim, B.S.; Oh, H.B.; Han, S.H. Bacillus anthracis lethal toxin attenuates lipoteichoic acid-induced maturation and activation of dendritic cells through a unique mechanism. Mol. Immunol. 2009, 46, 3261–3268. [Google Scholar]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar]
- Maldonado-Arocho, F.J.; Bradley, K.A. Anthrax edema toxin induces maturation of dendritic cells and enhances chemotaxis towards macrophage inflammatory protein 3beta. Infect. Immun. 2009, 77, 2036–2042. [Google Scholar]
- Maldonado-Arocho, F.J.; Fulcher, J.A.; Lee, B.; Bradley, K.A. Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol. Microbiol. 2006, 61, 324–337. [Google Scholar]
- Alileche, A.; Serfass, E.R.; Muehlbauer, S.M.; Porcelli, S.A.; Brojatsch, J. Anthrax lethal toxin-mediated killing of human and murine dendritic cells impairs the adaptive immune response. PLoS Pathog. 2005, 1, e19. [Google Scholar]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar]
- Fang, H.; Cordoba-Rodriguez, R.; Lankford, C.S.; Frucht, D.M. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J. Immunol. 2005, 174, 4966–4971. [Google Scholar]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D'Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar]
- Lee, D.Y.; Chun, J.H.; Ha, H.J.; Park, J.; Kim, B.S.; Oh, H.B.; Rhie, G.E. Poly-gamma-d-glutamic acid and protective antigen conjugate vaccines induce functional antibodies against the protective antigen and capsule of Bacillus anthracis in guinea-pigs and rabbits. FEMS Immunol. Med. Microbiol. 2009, 57, 165–172. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: progress and problems. Cell. Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar]
- Lubelli, C.; Chatgilialoglu, A.; Bolognesi, A.; Strocchi, P.; Colombatti, M.; Stirpe, F. Detection of ricin and other ribosome-inactivating proteins by an immuno-polymerase chain reaction assay. Anal. Biochem. 2006, 355, 102–109. [Google Scholar]
- O'Hare, M.; Roberts, L.M.; Lord, J.M. Biological activity of recombinant Ricinus communis agglutinin A chain produced in Escherichia coli. FEBS Lett. 1992, 299, 209–212. [Google Scholar]
- Furstenberg, C. Computer genereated model of Ricin toxin; Lund University: Lund, Sweden, 2008. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Chiou, J.C.; Li, X.P.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 2008, 70, 1441–1452. [Google Scholar]
- Ogasawara, T.; Sawasaki, T.; Morishita, R.; Ozawa, A.; Madin, K.; Endo, Y. A new class of enzyme acting on damaged ribosomes: ribosomal RNA apurinic site specific lyase found in wheat germ. EMBO J. 1999, 18, 6522–6531. [Google Scholar]
- Eiklid, K.; Olsnes, S.; Pihl, A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp. Cell Res. 1980, 126, 321–326. [Google Scholar]
- Marsden, C.J.; Fulop, V.; Day, P.J.; Lord, J.M. The effect of mutations surrounding and within the active site on the catalytic activity of ricin A chain. Eur. J. Biochem. 2004, 271, 153–162. [Google Scholar]
- Schlossman, D.; Withers, D.; Welsh, P.; Alexander, A.; Robertus, J.; Frankel, A. Role of glutamic acid 177 of the ricin toxin A chain in enzymatic inactivation of ribosomes. Mol. Cell Biol. 1989, 9, 5012–5021. [Google Scholar]
- Spilsberg, B.; Van Meer, G.; Sandvig, K. Role of lipids in the retrograde pathway of ricin intoxication. Traffic 2003, 4, 544–552. [Google Scholar]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Rothberg, K.G.; Ying, Y.S.; Kamen, B.A.; Anderson, R.G. Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J. Cell. Biol. 1990, 111, 2931–2938. [Google Scholar]
- Rodal, S.K.; Skretting, G.; Garred, O.; Vilhardt, F.; van Deurs, B.; Sandvig, K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell. 1999, 10, 961–974. [Google Scholar]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar]
- Iversen, T.G.; Skretting, G.; Llorente, A.; Nicoziani, P.; van Deurs, B.; Sandvig, K. Endosome to Golgi transport of ricin is independent of clathrin and of the Rab9- and Rab11-GTPases. Mol. Biol. Cell. 2001, 12, 2099–2107. [Google Scholar]
- Grimmer, S.; Iversen, T.G.; van Deurs, B.; Sandvig, K. Endosome to Golgi transport of ricin is regulated by cholesterol. Mol. Biol. Cell. 2000, 11, 4205–4216. [Google Scholar]
- Moisenovich, M.; Tonevitsky, A.; Maljuchenko, N.; Kozlovskaya, N.; Agapov, I.; Volknandt, W.; Bereiter-Hahn, J. Endosomal ricin transport: involvement of Rab4- and Rab5-positive compartments. Histochem. Cell. Biol. 2004, 121, 429–439. [Google Scholar]
- Day, P.J.; Owens, S.R.; Wesche, J.; Olsnes, S.; Roberts, L.M.; Lord, J.M. An interaction between ricin and calreticulin that may have implications for toxin trafficking. J. Biol. Chem. 2001, 276, 7202–7208. [Google Scholar]
- Richardson, P.T.; Westby, M.; Roberts, L.M.; Gould, J.H.; Colman, A.; Lord, J.M. Recombinant proricin binds galactose but does not depurinate 28 S ribosomal RNA. FEBS Lett. 1989, 255, 15–20. [Google Scholar]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosen, A.; Solda, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004, 67, 1721–1731. [Google Scholar]
- Day, P.J.; Pinheiro, T.J.; Roberts, L.M.; Lord, J.M. Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 2002, 41, 2836–2843. [Google Scholar]
- Griffiths, G.D.; Lindsay, C.D.; Allenby, A.C.; Bailey, S.C.; Scawin, J.W.; Rice, P.; Upshall, D.G. Protection against inhalation toxicity of ricin and abrin by immunisation. Hum. Exp. Toxicol. 1995, 14, 155–164. [Google Scholar]
- Hewetson, J.F.; Rivera, V.R.; Creasia, D.A.; Lemley, P.V.; Rippy, M.K.; Poli, M.A. Protection of mice from inhaled ricin by vaccination with ricin or by passive treatment with heterologous antibody. Vaccine 1993, 11, 743–746. [Google Scholar]
- Godal, A.; Fodstad, O.; Pihl, A. Antibody formation against the cytotoxic proteins abrin and ricin in humans and mice. Int. J. Cancer 1983, 32, 515–521. [Google Scholar]
- Foxwell, B.M.; Donovan, T.A.; Thorpe, P.E.; Wilson, G. The removal of carbohydrates from ricin with endoglycosidases H, F and D and alpha-mannosidase. Biochim. Biophys. Acta 1985, 840, 193–203. [Google Scholar]
- Foxwell, B.M.; Detre, S.I.; Donovan, T.A.; Thorpe, P.E. The use of anti-ricin antibodies to protect mice intoxicated with ricin. Toxicology 1985, 34, 79–88. [Google Scholar]
- Ready, M.P.; Kim, Y.; Robertus, J.D. Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins 1991, 10, 270–278. [Google Scholar]
- Lemley, P.V.; Amanatides, P.; Wright, D.C. Identification and characterization of a monoclonal antibody that neutralizes ricin toxicity in vitro and in vivo. Hybridoma 1994, 13, 417–421. [Google Scholar]
- Colombatti, M.; Johnson, V.G.; Skopicki, H.A.; Fendley, B.; Lewis, M.S.; Youle, R.J. Identification and characterization of a monoclonal antibody recognizing a galactose-binding domain of the toxin ricin. J. Immunol. 1987, 138, 3339–3344. [Google Scholar]
- Lebeda, F.J.; Olson, M.A. Prediction of a conserved, neutralizing epitope in ribosome-inactivating proteins. Int. J. Biol. Macromol. 1999, 24, 19–26. [Google Scholar]
- Maddaloni, M.; Cooke, C.; Wilkinson, R.; Stout, A.V.; Eng, L.; Pincus, S.H. Immunological characteristics associated with the protective efficacy of antibodies to ricin. J. Immunol. 2004, 172, 6221–6228. [Google Scholar]
- Mantis, N.J. Vaccines against the category B toxins: Staphylococcal enterotoxin B, epsilon toxin and ricin. Adv. Drug Deliv. Rev. 2005, 57, 1424–1439. [Google Scholar]
- Thorpe, P.E.; Detre, S.I.; Foxwell, B.M.; Brown, A.N.; Skilleter, D.N.; Wilson, G.; Forrester, J.A.; Stirpe, F. Modification of the carbohydrate in ricin with metaperiodate-cyanoborohydride mixtures. Effects on toxicity and in vivo distribution. Eur. J. Biochem. 1985, 147, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.D.; Bailey, S.C.; Hambrook, J.L.; Keyte, M.; Jayasekera, P.; Miles, J.; Williamson, E. Liposomally-encapsulated ricin toxoid vaccine delivered intratracheally elicits a good immune response and protects against a lethal pulmonary dose of ricin toxin. Vaccine 1997, 15, 1933–1939. [Google Scholar]
- Yan, C.; Rill, W.L.; Malli, R.; Hewetson, J.; Tammariello, R.; Kende, M. Dependence of ricin toxoid vaccine efficacy on the structure of poly(lactide-co-glycolide) microparticle carriers. Vaccine 1995, 13, 645–651. [Google Scholar]
- Kende, M.; Yan, C.; Hewetson, J.; Frick, M.A.; Rill, W.L.; Tammariello, R. Oral immunization of mice with ricin toxoid vaccine encapsulated in polymeric microspheres against aerosol challenge. Vaccine 2002, 20, 1681–1691. [Google Scholar]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar]
- Piascik, P. FDA approves fusion protein for treatment of lymphoma. J. Am. Pharm. Assoc. (Wash.) 1999, 39, 571–572. [Google Scholar] [PubMed]
- Carter, J.E., III; Odumosu, O.; Langridge, W.H. Expression of a ricin toxin B subunit: insulin fusion protein in edible plant tissues. Mol. Biotechnol. 2009, 44, 90–100. [Google Scholar]
- Kobayashi, M.; Abiru, N.; Arakawa, T.; Fukushima, K.; Zhou, H.; Kawasaki, E.; Yamasaki, H.; Liu, E.; Miao, D.; Wong, F.S.; Eisenbarth, G.S.; Eguchi, K. Altered B:9-23 insulin, when administered intranasally with cholera toxin adjuvant, suppresses the expression of insulin autoantibodies and prevents diabetes. J. Immunol. 2007, 179, 2082–2088. [Google Scholar]
- Arakawa, T.; Yu, J.; Langridge, W.H. Food plant-delivered cholera toxin B subunit for vaccination and immunotolerization. Adv. Exp. Med. Biol. 1999, 464, 161–178. [Google Scholar]
- Denes, B.; Fodor, I.; Langridge, W. Autoantigen plus interleukin-10 suppress diabetes autoimmunity. Diabetes Technol. Ther. 2010. submitted for publication.. [Google Scholar]
- Fensterle, J.; Bergmann, B.; Yone, C.L.; Hotz, C.; Meyer, S.R.; Spreng, S.; Goebel, W.; Rapp, U.R.; Gentschev, I. Cancer immunotherapy based on recombinant Salmonella enterica serovar Typhimurium aroA strains secreting prostate-specific antigen and cholera toxin subunit B. Cancer Gene Ther. 2008, 15, 85–93. [Google Scholar]
- Peppoloni, S.; Ruggiero, P.; Contorni, M.; Morandi, M.; Pizza, M.; Rappuoli, R.; Podda, A.; Del Giudice, G. Mutants of the Escherichia coli heat-labile enterotoxin as safe and strong adjuvants for intranasal delivery of vaccines. Expert Rev. Vaccines 2003, 2, 285–293. [Google Scholar]
- Lewis, D.J.; Huo, Z.; Barnett, S.; Kromann, I.; Giemza, R.; Galiza, E.; Woodrow, M.; Thierry-Carstensen, B.; Andersen, P.; Novicki, D.; Del Giudice, G.; Rappuoli, R. Transient facial nerve paralysis (Bell's palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS One 2009, 4, e6999. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Odumosu, O.; Nicholas, D.; Yano, H.; Langridge, W. AB Toxins: A Paradigm Switch from Deadly to Desirable. Toxins 2010, 2, 1612-1645. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2071612
Odumosu O, Nicholas D, Yano H, Langridge W. AB Toxins: A Paradigm Switch from Deadly to Desirable. Toxins. 2010; 2(7):1612-1645. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2071612
Chicago/Turabian StyleOdumosu, Oludare, Dequina Nicholas, Hiroshi Yano, and William Langridge. 2010. "AB Toxins: A Paradigm Switch from Deadly to Desirable" Toxins 2, no. 7: 1612-1645. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2071612