Scorpion Toxins Specific for Potassium (K+) Channels: A Historical Overview of Peptide Bioengineering

Abstract
:1. Introduction
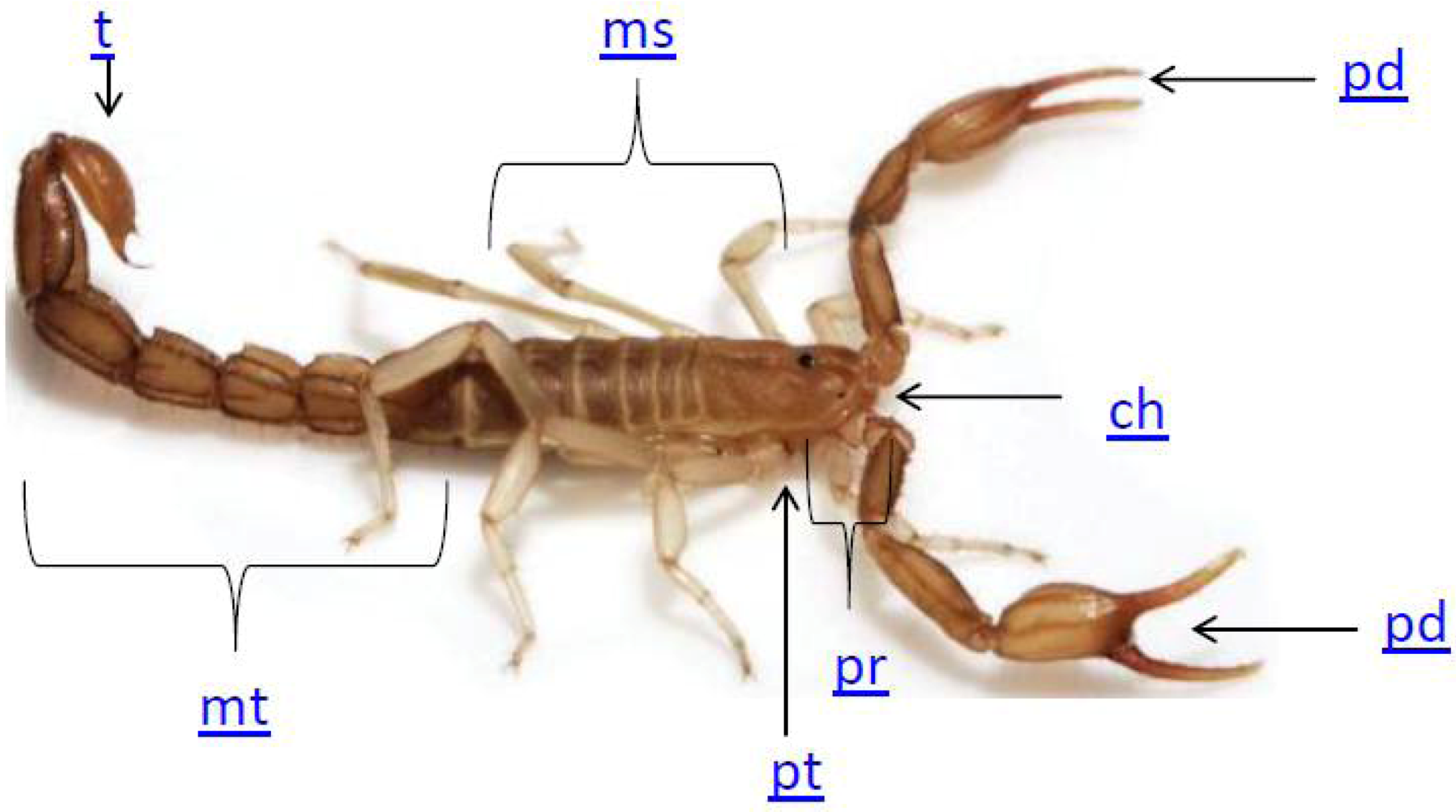
2. Scorpion Biology

3. The Use of Scorpion Toxins to Investigate Potassium Channels
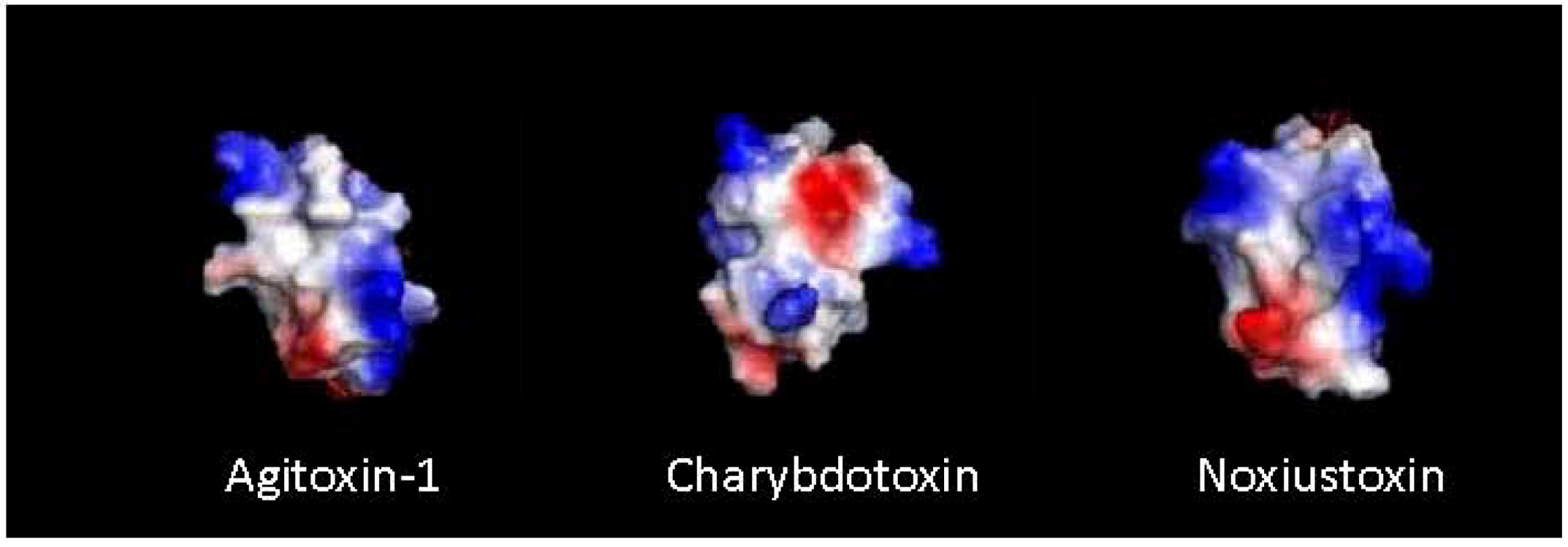
4. Peptide-Toxin Structure

5. Synthetic Toxin Production
6. Recombinant Toxin Production
7. Structure Activity Relationship

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Example | Disulfide connectivity | Target | Reference |
|---|---|---|---|---|
| I | KTX |  | KCa | [103] |
| II | CnERG1 |  | KV11 | [111] |
| III | MTX |  | KV1, KCa2, KCa3.1 | [46] |
| IV | HsTx1 |  | KV1.1, KV1.3 | [45] |
| V | TtBut-Tx |  | Shaker B | [112] |
8. Radiolabeled Scorpion Toxins and Receptor Localization
| Toxin | Modification | Name | Label | Target | Reference |
|---|---|---|---|---|---|
| ChTx | native | [125I]ChTX | [125I]-Y36 | KCa, KV | [113] |
| R19C | [3H]-ChTx-R19C | [3H]-C19 | KCa, KV | [118] | |
| ScyTx | F2Y | 125I-[Tyr2]ScyTx | [125I]-Y2 | KCa | [119] |
| MgTX | native | [125I]MgTX | [125I]-Y37 | KV1.2, 1.3 | [120] |
| IbTx | V5Y, Y36F | [mono-iodo-Tyr5, Phe36]-IbTx | [125I]-Y5 | KCa1.1(BK) | [121] |
| D19C | [3H]-IbTx-D19C | [3H]-C19 | KCa1.1(BK) | [116] | |
| D19Y/Y36F | [125I]IbTx-[D19Y/Y36F] | [125I]-Y19 | KCa1.1(BK) | [117] | |
| BeKm-1 | native | [127I]-BeKm-1 | [127I]-Y11 | KV11.1(hERG) | [122] |
| HgTx | A19Y/Y37F | 125I-HgTx1-A19Y/Y37F | [125I]-Y19 | KV1.1/2/3/6 | [123] |
| BmTX3 | native | 125I-sBmTX3 | [125I]-Y37 | A-type current | [124] |
| NTX | native | [125I]Noxiustoxin | [125I]-Y37 | KV1.1 | [125] |
| KTX | KTX(1-37) | 125I-KTX(1-37) | [125I]-H34 | KCa | [126] |
| Lqh III | native | [125I]Leiurutoxin III | [125I]-Y8 | KV1.1 | [125] |
9. Bioconjugation

| Toxin base sequence | Mutation | Bioconjugate | Target | Reference |
|---|---|---|---|---|
| ChTx | N-term | ChTx-biotin | KCa, KV | [68] |
| R19C | Rhodamine-ChTx-R19C | KCa, KV | [118] | |
| HgTx | A19C | HgTX1-A19C-Cy5 | KV1.1/2/3/6 | [69] |
| HgTX1-A19C-Cy3 | KV1.1/2/3/6 | [69] | ||
| HgTX1-A19C-Alex488 | KV1.1/2/3/6 | [69] | ||
| HgTX1-A19C-Alex546 | KV1.1/2/3/6 | [69] | ||
| HgTX1-A19C-Alex594 | KV1.1/2/3/6 | [69] | ||
| HgTX1-A19C-Alex660 | KV1.1/2/3/6 | [69] | ||
| HgTX1-A19C-Alex680 | KV1.1/2/3/6 | [69] | ||
| HgTX(1)-A19C-Cy5 | KV1.1/2/3/6 | [136] | ||
| IbTx | D19C | IbTx-D19C-Alexa488 | KCa1.1(BK) | [70] |
| D19K | IbTx-LC-biotin | KCa1.1(BK) | [5] | |
| IbTx-LC-FAM | KCa1.1(BK) | [138] |
10. Scorpion Toxin Chimeras
11. Native Chemical Ligation
12. Peptide Backbone Cyclization

13. Potassium Channels as Clinical Targets
13.1. Asthma
13.2. Cardiac Arrhythmia
13.3. T-Cell Mediated Autoimmune Diseases
13.4. Immune Response to Infection and Inflammation
13.5. Hypertension
14. Potassium Channel Toxins in the Clinic—Bioengineering Molecular Therapeutics
14.1. HIV/AIDS
14.1.1. Charybdotoxin
14.1.2. Scyllatoxin
14.2. T-Cell Mediated Autoimmune Disease
14.2.1. OSK-1[E16K, K20D]
14.2.2. ADWX-1
14.2.3. Mokatoxin-1
14.3. Malaria
15. Conclusions
Acknowledgements
Conflict of Interest
References
- Possani, L.D.; Becerril, B.; Delepierre, M.; Tytgat, J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 1999, 264, 287–300. [Google Scholar] [CrossRef]
- Bradding, P.; Wulff, H. The K+ channels KCa3.1 and Kv1.3 as novel targets for asthma therapy. Br. J. Pharmacol. 2009, 157, 1330–1339. [Google Scholar] [CrossRef]
- Jenkinson, D.H. Potassium channels-Multiplicity and challenges. Br. J. Pharmacol. 2006, 147, S63–S71. [Google Scholar] [CrossRef]
- Wickenden, A. K+ channels as therapeutic drug targets. Pharmacol. Ther. 2002, 94, 157–182. [Google Scholar] [CrossRef]
- Bingham, J.P.; Bian, S.; Tan, Z.Y.; Takacs, Z.; Moczydlowski, E. Synthesis of a biotin derivative of iberiotoxin: Binding interactions with streptavidin and the bk Ca2+-activated K+ channel expressed in a human cell line. Bioconjug. Chem. 2006, 17, 689–699. [Google Scholar] [CrossRef]
- Miller, C.; Moczydlowski, E.; Latorre, R.; Phillips, M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature 1985, 313, 316–318. [Google Scholar]
- Pimentel, C.; M’Barek, S.; Visan, V.; Grissmer, S.; Sampieri, F.; Sabatier, J.M.; Darbon, H.; Fajloun, Z. Chemical synthesis and 1h-nmr 3d structure determination of agtx2-mtx chimera, a new potential blocker for kv1.2 channel, derived from mtx and agtx2 scorpion toxins. Protein Sci. 2008, 17, 107–118. [Google Scholar]
- Giangiacomo, K.M.; Sugg, E.E.; Garcia-Calvo, M.; Leonard, R.J.; McManus, O.B.; Kaczorowski, G.J.; Garcia, M.L. Synthetic charybdotoxin-iberiotoxin chimeric peptides define toxin binding sites on calcium-activated and voltage-dependent potassium channels. Biochemistry 1993, 32, 2363–2370. [Google Scholar]
- Veiseh, M.; Gabikian, P.; Bahrami, S.B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin: Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar]
- Francke, O.F. Conspectus genericus scorpionum. Occas. Pap. Mus. 1985, 98, 1–32. [Google Scholar]
- Polis, G.A. The Biology of Scorpions; Stanford University Press: Palo Alto, CA, USA, 1990; pp. 1–679. [Google Scholar]
- Sissom, W.D. Systematics, Biogeography, and Paleontology. In The Biology of Scorpions; Polis, G.A., Ed.; Stanford University Press: Palo Alto, CA, USA, 1990; pp. 64–160. [Google Scholar]
- Ismail, M. The scorpion envenoming syndrome. Toxicon 1995, 33, 825–858. [Google Scholar] [CrossRef]
- Hjelle, J.T. Anatomy and Morphology. In The Biology of Scorpions; Polis, G.A., Ed.; Stanford University Press: Palo Alto, CA, USA, 1990; pp. 9–63. [Google Scholar]
- Webber, M.M.; Graham, M.R.; Jaeger, J.R. Wernerius inyoensis, an elusive new scorpion from the inyo mountains of california (scorpiones, vaejovidae). Zookeys 2012, 177, 1–13. [Google Scholar] [CrossRef]
- Lourenco, W.R. The scorpion families and their geographical distribution. J. Venom. Anim. Toxins 2001, 7, 3–23. [Google Scholar]
- Sollod, B.L.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides 2005, 26, 131–139. [Google Scholar] [CrossRef]
- Bucherl, W.; Buckley, E.E. Venomous animals and their venoms. Venom. Invertabrates 1971, 3, 537. [Google Scholar]
- Miranda, F.; Lissitzky, S. Scorpamins: the Toxic Proteins of Scorpion Venoms. Nature. 1961, 190, 443–444. [Google Scholar] [CrossRef]
- Rochat, C.; Rochat, H.; Miranda, F.; Lissitzky, S. Purification and some properties of the neurotoxins of androctonus australis hector. Biochemistry 1967, 6, 578–585. [Google Scholar]
- Simard, J.M.; Watt, D.D. Venoms and toxins. In The Biology of Scorpions; Polis, G.A., Ed.; Stanford University Press: Palo Alto, CA, USA, 1990; pp. 414–444. [Google Scholar]
- Cole, K.S. Dynamic electrical characteristics of the squid axon membrane. Arch. Sci. Physiol. 1949, 3, 253–258. [Google Scholar]
- Hodgkin, A.L.; Huxley, K.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; Keating, M.T. Spectrum of mutations in long-qt syndrome genes. Kvlqt1, herg, scn5a, kcne1, and kcne2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef]
- Carbone, E.; Wanke, E.; Prestipino, G.; Possani, L.D.; Maelicke, A. Selective blockage of voltage-dependent K+ channels by a novel scorpion toxin. Nature 1982, 296, 90–91. [Google Scholar]
- Possani, L.D.; Martin, B.M.; Svendsen, I.B. The primary structure of noxiustoxin: A K+ channel blocking peptide, purified from the venom of the scorpion centruroides noxius hoffmann. Carlsberg Res. Commun. 1982, 47, 285–289. [Google Scholar] [CrossRef]
- Sugg, E.E.; Garcia, M.L.; Reuben, J.P.; Patchett, A.A.; Kaczorowski, G.J. Synthesis and structural characterization of charybdotoxin, a potent peptidyl inhibitor of the high conductance Ca2+-activated K+ channel. J. Biol. Chem. 1990, 265, 18745–18748. [Google Scholar]
- Garcia, M.L.; Galvez, A.; Garcia-Calvo, M.; King, V.F.; Vazquez, J.; Kaczorowski, G.J. Use of toxins to study potassium channels. J. Bioenerg. Biomembr. 1991, 23, 615–646. [Google Scholar] [CrossRef]
- Pedroso, E.; Grandas, A.; Amor, J.C.; Giralt, E. Reversed-phase high-performance liquid chromatography of protected peptide segments. J. Chromatogr. 1987, 409, 281–290. [Google Scholar] [CrossRef]
- Galvez, A.; Gimenez-Gallego, G.; Reuben, J.P.; Roy-Contancin, L.; Feigenbaum, P.; Kaczorowski, G.J.; Garcia, M.L. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion buthus tamulus. J. Biol. Chem. 1990, 265, 11083–11090. [Google Scholar]
- MacKinnon, R.; Heginbotham, L.; Abramson, T. Mapping the receptor site for charybdotoxin, a pore-blocking potassium channel inhibitor. Neuron 1990, 5, 767–771. [Google Scholar] [CrossRef]
- Legros, C.; Pollmann, V.; Knaus, H.G.; Farrell, A.M.; Darbon, H.; Bougis, P.E.; Martin-Eauclaire, M.F.; Pongs, O. Generating a high affinity scorpion toxin receptor in kcsa-kv1.3 chimeric potassium channels. J. Biol. Chem. 2000, 275, 16918–16924. [Google Scholar]
- Gross, A.; MacKinnon, R. Agitoxin footprinting the shaker potassium channel pore. Neuron 1996, 16, 399–406. [Google Scholar] [CrossRef]
- Mouhat, S.; Andreotti, N.; Jouirou, B.; Sabatier, J.M. Animal toxins acting on voltage-gated potassium channels. Curr. Pharm. Des. 2008, 14, 2503–2518. [Google Scholar] [CrossRef]
- Smith, J.J.; Hill, J.M.; Little, M.J.; Nicholson, G.M.; King, G.F.; Alewood, P.F. Unique scorpion toxin with a putative ancestral fold provides insight into evolution of the inhibitor cystine knot motif. Proc. Natl. Acad. Sci. USA 2011, 108, 10478–10483. [Google Scholar]
- Kopeyan, C.; Martinez, G.; Lissitzky, S.; Miranda, F.; Rochat, H. Disulfide bonds of toxin ii of the scorpion androctonus australis hector. Eur. J. Biochem. 1974, 47, 483–489. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Almassy, R.J.; Suddath, F.L.; Watt, D.D.; Bugg, C.E. Three-dimensional structure of a protein from scorpion venom: A new structural class of neurotoxins. Proc. Natl. Acad. Sci. USA 1980, 77, 6496–6500. [Google Scholar]
- Gimenez-Gallego, G.; Navia, M.A.; Reuben, J.P.; Katz, G.M.; Kaczorowski, G.J.; Garcia, M.L. Purification, sequence, and model structure of charybdotoxin, a potent selective inhibitor of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA 1988, 85, 3329–3333. [Google Scholar] [CrossRef]
- Pentelute, B.L.; Mandal, K.; Gates, Z.P.; Sawaya, M.R.; Yeates, T.O.; Kent, S.B. Total chemical synthesis and X-ray structure of kaliotoxin by racemic protein crystallography. Chem. Commun. 2010, 46, 8174–8176. [Google Scholar]
- Bystrov, V.F. Contribution of nmr spectroscopy to the study of structure-function relations of proteins and peptides. Bioorg. Khim. 1984, 10, 997–1043. [Google Scholar]
- Bontems, F.; Roumestand, C.; Boyot, P.; Gilquin, B.; Doljansky, Y.; Menez, A.; Toma, F. Three-dimensional structure of natural charybdotoxin in aqueous solution by 1h-nmr. Charybdotoxin possesses a structural motif found in other scorpion toxins. Eur. J. Biochem. 1991, 196, 19–28. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Vizzavona, J.; Neyton, J.; Aniort, V.; Bouet, F.; Virelizier, H.; Ménez, A.; Vita, C. Consequence of the removal of evolutionary conserved disulfide bridges on the structure and function of charybdotoxin and evidence that particular cysteine spacings govern specific disulfide bond formation. Biochemistry 1998, 37, 1292–1301. [Google Scholar]
- Johnson, B.A.; Sugg, E.E. Determination of the three-dimensional structure of iberiotoxin in solution by 1h nuclear magnetic resonance spectroscopy. Biochemistry 1992, 31, 8151–8159. [Google Scholar] [CrossRef]
- Fernández, I.; Romi, R.; Szendeffy, S.; Martin-Eauclaire, M.F.; Rochat, H.; van Rietschoten, J.; Pons, M.; Giralt, E. Kaliotoxin (1-37) shows structural differences with related potassium channel blockers. Biochemistry 1994, 33, 14256–14263. [Google Scholar]
- Fajloun, Z.; Mosbah, A.; Carlier, E.; Mansuelle, P.; Sandoz, G.; Fathallah, M.; di Luccio, E.; Devaux, C.; Rochat, H.; Darbon, H.; et al. Maurotoxin versus pi1/hstx1 scorpion toxins. Toward new insights in the understanding of their distinct disulfide bridge patterns. J. Biol. Chem. 2000, 275, 39394–39402. [Google Scholar]
- M’Barek, S.; Lopez-Gonzalez, I.; Andreotti, N.; di Luccio, E.; Visan, V.; Grissmer, S.; Judge, S.; el Ayeb, M.; Darbon, H.; Rochat, H.; et al. A maurotoxin with constrained standard disulfide bridging: Innovative strategy of chemical synthesis, pharmacology, and docking on K+ channels. J. Biol. Chem. 2003, 278, 31095–31104. [Google Scholar]
- Lenffer, J.; Lai, P.; el Mejaber, W.; Khan, A.M.; Koh, J.L.; Tan, P.T.; Seah, S.H.; Brusic, V. Cysview: Protein classification based on cysteine pairing patterns. Nucleic Acids Res. 2004, 32, W350–W355. [Google Scholar]
- Lee, C.W.; Bae, C.; Lee, J.; Ryu, J.H.; Kim, H.H.; Kohno, T.; Swartz, K.J.; Kim, J.I. Solution structure of kurtoxin: A gating modifier selective for cav3 voltage-gated Ca2+ channels. Biochemistry 2012, 51, 1862–1873. [Google Scholar]
- Kumar, G.S.; Upadhyay, S.; Mathew, M.K.; Sarma, S.P. Solution structure of btk-2, a novel hk(v)1.1 inhibiting scorpion toxin, from the eastern indian scorpion mesobuthus tamulus. Biochim. Biophys. Acta 2011, 1814, 459–469. [Google Scholar] [CrossRef]
- Pardo-Lopez, L.; Zhang, M.; Liu, J.; Jiang, M.; Possani, L.D.; Tseng, G.N. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ergtx) to the channel’s outer vestibule. J. Biol. Chem. 2002, 277, 16403–16411. [Google Scholar]
- Korolkova, Y.V.; Tseng, G.N.; Grishin, E.V. Unique interaction of scorpion toxins with the herg channel. J. Mol. Recognit. 2004, 17, 209–217. [Google Scholar] [CrossRef]
- Yu, L.; Sun, C.; Song, D.; Shen, J.; Xu, N.; Gunasekera, A.; Hajduk, P.J.; Olejniczak, E.T. Nuclear magnetic resonance structural studies of a potassium channel-charybdotoxin complex. Biochemistry 2005, 44, 15834–15841. [Google Scholar] [CrossRef]
- Andersson, C.O. Mass spectrometric studies on amino acid and peptide derivatives. Acta Chem. Scand. 1958, 12, 1353. [Google Scholar]
- Biemann, K. Mass spectrometry. Annu. Rev. Biochem. 1963, 32, 755–780. [Google Scholar] [CrossRef]
- Barber, M.; Jolles, P.; Vilkas, E.; Lederer, E. Determination of amino acid sequences in oligopeptides by mass spectrometry I. The structure of fortuitine, an acylnonapeptide methyl ester. Biochem. Biophys. Res. Commun. 1965, 18, 469–473. [Google Scholar] [CrossRef]
- Vázquez, A.; Becerril, B.; Martin, B.M.; Zamudio, F.; Bolívar, F.; Possani, L.D. Primary structure determination and cloning of the cdna encoding toxin 4 of the scorpion centruroides noxius hoffmann. FEBS Lett. 1993, 320, 43–46. [Google Scholar] [CrossRef]
- Tayo, L.L.; Lu, B.; Cruz, L.J.; Yates, J.R. Proteomic analysis provides insights on venom processing in conus textile. J. Proteome Res. 2010, 9, 2292–2301. [Google Scholar] [CrossRef]
- Tamaoki, H.; Miura, R.; Kusunoki, M.; Kyogoku, Y.; Kobayashi, Y.; Moroder, L. Folding motifs induced and stabilized by distinct cystine frameworks. Protein Eng. 1998, 11, 649–659. [Google Scholar] [CrossRef]
- Hallgren, K.W.; Zhang, D.; Kinter, M.; Willard, B.; Berkner, K.L. Methylation of gamma-carboxylated glu (gla) allows detection by liquid chromatography-mass spectrometry and the identification of gla residues in the gamma-glutamyl carboxylase. J. Proteome Res. 2012. [Google Scholar] [CrossRef]
- Ramström, M.; Sandberg, H. Characterization of γ-carboxylated tryptic peptides by collision-induced dissociation and electron transfer dissociation mass spectrometry. Eur. J. Mass Spectrom. 2011, 17, 497–506. [Google Scholar] [CrossRef]
- Zhu, Q.; Liang, S.; Martin, L.; Gasparini, S.; Ménez, A.; Vita, C. Role of disulfide bonds in folding and activity of leiurotoxin i: Just two disulfides suffice. Biochemistry 2002, 41, 11488–11494. [Google Scholar] [CrossRef]
- Fajloun, Z.; Ferrat, G.; Carlier, E.; Fathallah, M.; Lecomte, C.; Sandoz, G.; di Luccio, E.; Mabrouk, K.; Legros, C.; Darbon, H.; et al. Synthesis, 1h nmr structure, and activity of a three-disulfide-bridged maurotoxin analog designed to restore the consensus motif of scorpion toxins. J. Biol. Chem. 2000, 275, 13605–13612. [Google Scholar]
- Gairí, M.; Romi, R.; Fernández, I.; Rochat, H.; Martin-Eauclaire, M.F.; van Rietschoten, J.; Pons, M.; Giralt, E. 3D structure of kaliotoxin: Is residue 34 a key for channel selectivity? J. Pept. Sci. 1997, 3, 314–319. [Google Scholar] [CrossRef]
- Harvey, A.L.; Vatanpour, H.; Rowan, E.G.; Pinkasfeld, S.; Vita, C.; Ménez, A.; Martin-Eauclaire, M.F. Structure-activity studies on scorpion toxins that block potassium channels. Toxicon 1995, 33, 425–436. [Google Scholar] [CrossRef]
- Gurrola, G.B.; Molinar-Rode, R.; Sitges, M.; Bayon, A.; Possani, L.D. Synthetic peptides corresponding to the sequence of noxiustoxin indicate that the active site of this K+ channel blocker is located on its amino-terminal portion. J. Neural. Transm. 1989, 77, 11–20. [Google Scholar]
- Lecomte, C.; Ferrat, G.; Fajloun, Z.; van Rietschoten, J.; Rochat, H.; Martin-Eauclaire, M.F.; Darbon, H.; Sabatier, J.M. Chemical synthesis and structure-activity relationships of ts kappa, a novel scorpion toxin acting on apamin-sensitive sk channel. J. Pept. Res. 1999, 54, 369–376. [Google Scholar] [CrossRef]
- Ferrat, G.; Bernard, C.; Fremont, V.; Mullmann, T.J.; Giangiacomo, K.M.; Darbon, H. Structural basis for alpha-K toxin specificity for K+ channels revealed through the solution 1h nmr structures of two noxiustoxin-iberiotoxin chimeras. Biochemistry 2001, 40, 10998–11006. [Google Scholar]
- Robitaille, R.; Garcia, M.L.; Kaczorowski, G.J.; Charlton, M.P. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 1993, 11, 635–655. [Google Scholar]
- Pragl, B.; Koschak, A.; Trieb, M.; Obermair, G.; Kaufmann, W.A.; Gerster, U.; Blanc, E.; Hahn, C.; Prinz, H.; Schütz, G.; et al. Synthesis, characterization, and application of cy-dye- and alexa-dye-labeled hongotoxin(1) analogues. The first high affinity fluorescence probes for voltage-gated K+ channels. Bioconjug. Chem. 2002, 13, 416–425. [Google Scholar] [CrossRef]
- Hafidi, A.; Beurg, M.; Dulon, D. Localization and developmental expression of bk channels in mammalian cochlear hair cells. Neuroscience 2005, 130, 475–484. [Google Scholar] [CrossRef]
- Wang, C.G.; Cai, Z.; Lu, W.; Wu, J.; Xu, Y.; Shi, Y.; Chi, C.W. A novel short-chain peptide bmkx from the chinese scorpion buthus martensi karsch, sequencing, gene cloning and structure determination. Toxicon 2005, 45, 309–319. [Google Scholar] [CrossRef]
- Takacs, Z.; Toups, M.; Kollewe, A.; Johnson, E.; Cuello, L.G.; Driessens, G.; Biancalana, M.; Koide, A.; Ponte, C.G.; Perozo, E.; et al. A designer ligand specific for kv1.3 channels from a scorpion neurotoxin-based library. Proc. Natl. Acad. Sci. USA 2009, 106, 22211–22216. [Google Scholar]
- Gao, B.; Peigneur, S.; Tytgat, J.; Zhu, S. A potent potassium channel blocker from mesobuthus eupeus scorpion venom. Biochimie 2010, 92, 1847–1853. [Google Scholar]
- Di Luccio, E.; Azulay, D.O.; Regaya, I.; Fajloun, Z.; Sandoz, G.; Mansuelle, P.; Kharrat, R.; Fathallah, M.; Carrega, L.; Estève, E.; et al. Parameters affecting in vitro oxidation/folding of maurotoxin, a four-disulphide-bridged scorpion toxin. Biochem. J. 2001, 358, 681–692. [Google Scholar] [CrossRef]
- Lecomte, C.; Sabatier, J.M.; van Rietschoten, J.; Rochat, H. Synthetic peptides as tools to investigate the structure and pharmacology of potassium channel-acting short-chain scorpion toxins. Biochimie 1998, 80, 151–154. [Google Scholar] [CrossRef]
- Merrifield, B.; Shaheen, J.C.; Hess, G.P. Concept and early development of solid-phase peptide synthesis. J. Am. Chem. Soc. 1955, 77, 1067. [Google Scholar] [CrossRef]
- Clark, R.J.; Craik, D.J. Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Pept. Sci. 2009, 94, 414–422. [Google Scholar]
- Lambert, P.; Kuroda, H.; Chino, N.; Watanabe, T.X.; Kimura, T.; Sakakibara, S. Solution synthesis of charybdotoxin (chtx), a K+ channel blocker. Biochem. Biophys. Res. Commun. 1990, 170, 684–690. [Google Scholar] [CrossRef]
- Sarin, V.K.; Kent, S.B.; Tam, J.P.; Merrifield, R.B. Quantitative monitoring of solid-phase peptide synthesis by the ninhydrin reaction. Anal. Biochem. 1981, 117, 147–157. [Google Scholar]
- Kharrat, R.; Mabrouk, K.; Crest, M.; Darbon, H.; Oughideni, R.; Martin-Eauclaire, M.F.; Jacquet, G.; el Ayeb, M.; van Rietschoten, J.; Rochat, H.; Sabatier, J.M. Chemical synthesis and characterization of maurotoxin, a short scorpion toxin with four disulfide bridges that acts on K+ channels. Eur. J. Biochem. 1996, 242, 491–498. [Google Scholar]
- Park, C.S.; Hausdorff, S.F.; Miller, C. Design, synthesis, and functional expression of a gene for charybdotoxin, a peptide blocker of K+ channels. Proc. Natl. Acad. Sci. USA 1991, 88, 2046–2050. [Google Scholar] [CrossRef]
- Sahdev, S.; Khattar, S.K.; Saini, K.S. Production of active eukaryotic proteins through bacterial expression systems: A review of the existing biotechnology strategies. Mol. Cell Biochem. 2008, 307, 249–264. [Google Scholar]
- Trundova, M.; Celer, V. Expression of porcine circovirus 2 orf2 gene requires codon optimized E.coli cells. Virus Genes 2007, 34, 199–204. [Google Scholar] [CrossRef]
- Winter, J.; Klappa, P.; Freedman, R.B.; Lilie, H.; Rudolph, R. Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem. 2002, 277, 310–317. [Google Scholar]
- Chun, J.B.; Baker, M.R.; Kim, D.H.; Leroy, M.; Toribo, P.; Bingham, J.P. Cone snail milked venom dynamics-A quantitative study of conus purpurascens. Toxicon 2012, 60, 83–94. [Google Scholar] [CrossRef]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Post-translationally modified neuropeptides from conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar] [CrossRef]
- Wang, L.; Xie, J.; Schultz, P.G. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 225–249. [Google Scholar] [CrossRef]
- Xie, J.; Schultz, P.G. A chemical toolkit for proteins-An expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. [Google Scholar] [CrossRef]
- Xie, J.; Schultz, P.G. Adding amino acids to the genetic repertoire. Curr. Opin. Chem. Biol. 2005, 9, 548–554. [Google Scholar] [CrossRef]
- Kent, S. Novel forms of chemical protein diversity-In nature and in the laboratory. Curr. Opin. Biotechnol. 2004, 15, 607–614. [Google Scholar] [CrossRef]
- Wang, L.; Schultz, P.G. Expanding the genetic code. Angew. Chem. Int. Ed. 2004, 44, 34–66. [Google Scholar]
- Alfonta, L.; Zhang, Z.; Uryu, S.; Loo, J.A.; Schultz, P.G. Site-specific incorporation of a redox-active amino acid into proteins. J. Am. Chem. Soc. 2003, 125, 14662–14663. [Google Scholar]
- Hooker, J.M.; Kovacs, E.W.; Francis, M.B. Interior surface modification of bacteriophage ms2. J. Am. Chem. Soc. 2004, 126, 3718–3719. [Google Scholar]
- Datta, D.; Wang, P.; Carrico, I.S.; Mayo, S.L.; Tirrell, D.A. A designed phenylalanyl-trna synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins. J. Am. Chem. Soc. 2002, 124, 5652–5653. [Google Scholar]
- Kiick, K.L.; Saxon, E.; Tirrell, D.A.; Bertozzi, C.R. Incorporation of azides into recombinant proteins for chemoselective modification by the staudinger ligation. Proc. Natl. Acad. Sci. USA 2002, 99, 19–24. [Google Scholar]
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915. [Google Scholar] [CrossRef]
- Mendel, D.; Ellman, J.; Schultz, P.G. Construction of a light-activated protein by site directed unnatural amino acid mutagenesis. J. Am. Chem. Soc. 1991, 113, 2758–2760. [Google Scholar]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.; Schultz, P.G. An expanded eukaryotic genetic code. Science 2003, 301, 964–967. [Google Scholar]
- Wang, J.; Xie, J.; Schultz, P.G. A genetically encoded fluorescent amino acid. J. Am. Chem. Soc. 2006, 128, 8738–8739. [Google Scholar] [CrossRef]
- Charbon, G.; Brustad, E.; Scott, K.A.; Wang, J.; Løbner-Olesen, A.; Schultz, P.G.; Jacobs-Wagner, C.; Chapman, E. Subcellular protein localization by using a genetically encoded fluorescent amino acid. Chembiochem 2011, 12, 1818–1821. [Google Scholar]
- Zhang, Z.; Smith, B.A.; Wang, L.; Brock, A.; Cho, C.; Schultz, P.G. A new strategy for the site-specific modification of proteins in vivo. Biochemistry 2003, 42, 6735–6746. [Google Scholar]
- Zhang, Z.; Alfonta, L.; Tian, F.; Bursulaya, B.; Uryu, S.; King, D.S.; Schultz, P.G. Selective incorporation of 5-hydroxytryptophan into proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 8882–8887. [Google Scholar]
- Pimenta, A.M.; Legros, C.; Almeida Fde, M.; Mansuelle, P.; de Lima, M.E.; Bougis, P.E.; Martin-Eauclaire, M.F. Novel structural class of four disulfide-bridged peptides from tityus serrulatus venom. Biochem. Biophys. Res. Commun. 2003, 301, 1086–1092. [Google Scholar] [CrossRef]
- Park, C.S.; Miller, C. Mapping function to structure in a channel-blocking peptide: Electrostatic mutants of charybdotoxin. Biochemistry 1992, 31, 7749–7755. [Google Scholar] [CrossRef]
- Lopatin, A.N.; Nichols, C.G. Ion Channel Localization; Methods and Protocols; Humana Press: Totowa, NJ, USA, 2011. [Google Scholar]
- Garcia, M.L.; Hanner, M.; Knaus, H.G.; Koch, R.; Schmalhofer, W.; Slaughter, R.S.; Kaczorowski, G.J. Pharmacology of potassium channels. Adv. Pharmacol. 1997, 39, 425–471. [Google Scholar] [CrossRef]
- Martínez, F.; Muñoz-Garay, C.; Gurrola, G.; Darszon, A.; Possani, L.D.; Becerril, B. Site directed mutants of noxiustoxin reveal specific interactions with potassium channels. FEBS Lett. 1998, 429, 381–384. [Google Scholar] [CrossRef]
- Yu, K.; Fu, W.; Liu, H.; Luo, X.; Chen, K.X.; Ding, J.; Shen, J.; Jiang, H. Computational simulations of interactions of scorpion toxins with the voltage-gated potassium ion channel. Biophys. J. 2004, 86, 3542–3555. [Google Scholar] [CrossRef]
- Stehling, E.G.; Sforça, M.L.; Zanchin, N.I.; Oyama, S., Jr.; Pignatelli, A.; Belluzzi, O.; Polverini, E.; Corsini, R.; Spisni, A.; Pertinhez, T.A. Looking over toxin-K+ channel interactions. Clues from the structural and functional characterization of α-ktx toxin tc32, a kv1.3 channel blocker. Biochemistry 2012, 51, 1885–1894. [Google Scholar]
- Bingham, J.P.; Andrews, A.E.; Kiyabu, S.M.; Cabalteja, C.C. Drugs from slugs, part II-Conopeptide bioengineering. Chem. Biol. Interact. 2012. [Google Scholar] [CrossRef]
- Torres, A.M.; Bansal, P.; Alewood, P.F.; Bursill, J.A.; Kuchel, P.W.; Vandenberg, J.I. Solution structure of cnerg1 (ergtoxin), a herg specific scorpion toxin. FEBS Lett. 2003, 539, 138–142. [Google Scholar] [CrossRef]
- Coronas, F.V.; de Roodt, A.R.; Portugal, T.O.; Zamudio, F.Z.; Batista, C.V.; Gómez-Lagunas, F.; Possani, L.D. Disulfide bridges and blockage of shaker b K+-channels by another butantoxin peptide purified from the argentinean scorpion tityus trivittatus. Toxicon 2003, 41, 173–179. [Google Scholar] [CrossRef]
- Vázquez, J.; Feigenbaum, P.; Katz, G.; King, V.F.; Reuben, J.P.; Roy-Contancin, L.; Slaughter, R.S.; Kaczorowski, G.J.; Garcia, M.L. Characterization of high affinity binding sites for charybdotoxin in sarcolemmal membranes from bovine aortic smooth muscle. Evidence for a direct association with the high conductance calcium-activated potassium channel. J. Biol. Chem. 1989, 264, 20902–20909. [Google Scholar]
- Salacinski, P.; Hope, J.; McLean, C.; Clement-Jones, V.; Sykes, J.; Price, J.; Lowry, P.J. A new simple method which allows theoretical incorporation of radio-iodine into proteins and peptides without damage. J. Endocrinol. 1979, 81, 131. [Google Scholar] [CrossRef]
- Auguste, P.; Hugues, M.; Mourre, C.; Moinier, D.; Tartar, A.; Lazdunski, M. Scyllatoxin, a blocker of Ca2+-activated K+ channels: Structure-function relationships and brain localization of the binding sites. Biochemistry 1991, 31, 648–654. [Google Scholar]
- Knaus, H.G.; Schwarzer, C.; Koch, R.O.; Eberhart, A.; Kaczorowski, G.J.; Glossmann, H.; Wunder, F.; Pongs, O.; Garcia, M.L.; Sperk, G. Distribution of high-conductance Ca2+-activated K+ channels in rat brain: Targeting to axons and nerve terminals. J. Neurosci. 1996, 16, 955–963. [Google Scholar]
- Koschak, A.; Koch, R.O.; Liu, J.; Kaczorowski, G.J.; Reinhart, P.H.; Garcia, M.L.; Knaus, H.G. [125I]Iberiotoxin-d19y/y36f, the first selective, high specific activity radioligand for high-conductance calcium-activated potassium channels. Biochemistry 1997, 36, 1943–1952. [Google Scholar] [CrossRef]
- Shimony, E.; Sun, T.; Kolmakova-Partensky, L.; Miller, C. Engineering a uniquely reactive thiol into a cysteine-rich peptide. Protein Eng. 1994, 7, 503–507. [Google Scholar] [CrossRef]
- Auguste, P.; Hugues, M.; Gravé, B.; Gesquière, J.C.; Maes, P.; Tartar, A.; Romey, G.; Schweitz, H.; Lazdunski, M. Leiurotoxin i (scyllatoxin), a peptide ligand for Ca2+-activated K+ channels. Chemical synthesis, radiolabeling, and receptor characterization. J. Biol. Chem. 1990, 265, 4753–4759. [Google Scholar]
- Knaus, H.G.; Koch, R.O.; Eberhart, A.; Kaczorowski, G.J.; Garcia, M.L.; Slaughter, R.S. [125I]Margatoxin, an extraordinarily high affinity ligand for voltage-gated potassium channels in mammalian brain. Biochemistry 1995, 34, 13627–13634. [Google Scholar]
- Kozlowski, E.S.; Johnson, G.; Dischino, D.D.; Dworetzky, S.I.; Boissard, C.G.; Gribkoff, V.K. Synthesis and biological evaluation of an iodinated iberiotoxin analogue, [mono-iodo-tyr5, phe36]-iberiotoxin. Int. J. Pept. Protein Res. 1996, 48, 194–199. [Google Scholar]
- Angelo, K.; Korolkova, Y.V.; Grunnet, M.; Grishin, E.V.; Pluzhnikov, K.A.; Klaerke, D.A.; Knaus, H.G.; Møller, M.; Olesen, S.P. A radiolabeled peptide ligand of the herg channel, [125I]-bekm-1. Pflugers Arch. 2003, 447, 55–63. [Google Scholar] [CrossRef]
- Grunnet, M.; Rasmussen, H.B.; Hay-Schmidt, A.; Klaerke, D.A. The voltage-gated potassium channel subunit, kv1.3, is expressed in epithelia. Biochim. Biophys. Acta 2003, 1616, 85–94. [Google Scholar] [CrossRef]
- Vacher, H.; Romi-Lebrun, R.; Mourre, C.; Lebrun, B.; Kourrich, S.; Masméjean, F.; Nakajima, T.; Legros, C.; Crest, M.; Bougis, P.E.; Martin-Eauclaire, M.F. A new class of scorpion toxin binding sites related to an a-type K+ channel: Pharmacological characterization and localization in rat brain. FEBS Lett. 2001, 501, 31–36. [Google Scholar] [CrossRef]
- Valdivia, H.H.; Martin, B.M.; Escobar, L.; Possani, L.D. Noxiustoxin and leiurutoxin iii, two homologous peptide toxins with binding properties to synaptosomal membrane K+ channels. Biochem. Int. 1992, 27, 953–962. [Google Scholar]
- Romi, R.; Crest, M.; Gola, M.; Sampieri, F.; Jacquet, G.; Zerrouk, H.; Mansuelle, P.; Sorokine, O.; van Dorsselaer, A.; Rochat, H.; et al. Synthesis and characterization of kaliotoxin. Is the 26-32 sequence essential for potassium channel recognition? J. Biol. Chem. 1993, 268, 26302–26309. [Google Scholar]
- Kantchev, E.A.; Chang, C.C.; Cheng, S.F.; Roche, A.C.; Chang, D.K. Direct solid-phase synthesis and fluorescence labeling of large, monodisperse mannosylated dendrons in a peptide synthesizer. Org. Biomol. Chem. 2008, 6, 1377–1385. [Google Scholar] [CrossRef]
- Johnson, D.A.; Yguerabide, J. Solute accessibility to N epsilon-fluorescein isothiocyanate-lysine-23 cobra alpha-toxin bound to the acetylcholine receptor. A consideration of the effect of rotational diffusion and orientation constraints on fluorescence quenching. Biophys. J. 1985, 48, 949–955. [Google Scholar] [CrossRef]
- Akcan, M.; Stroud, M.R.; Hansen, S.J.; Clark, R.J.; Daly, N.L.; Craik, D.J.; Olson, J.M. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 2011, 54, 782–787. [Google Scholar] [CrossRef]
- Shi, Y.; Xiang, R.; Horváth, C.; Wilkins, J.A. Design and synthesis of a solid-phase fluorescent mass tag. J. Sep. Sci. 2005, 28, 1812–1817. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W.; Nielsen, T.E.; Diness, F.; le Quement, S.T.; Christensen, C.A.; Jensen, J.F.; Worm-Leonhard, K.; Groth, T.; Bouakaz, L.; et al. Hirschmann award address 2009: Merger of organic chemistry with peptide diversity. Biopolymers 2010, 94, 161–182. [Google Scholar] [CrossRef]
- Kamaruddin, M.A.; Ung, P.; Hossain, M.I.; Jarasrassamee, B.; O’Malley, W.; Thompson, P.; Scanlon, D.; Cheng, H.C.; Graham, B. A facile, click chemistry-based approach to assembling fluorescent chemosensors for protein tyrosine kinases. Bioorg. Med. Chem. Lett. 2011, 21, 329–331. [Google Scholar]
- Beal, D.M.; Albrow, V.E.; Burslem, G.; Hitchen, L.; Fernandes, C.; Lapthorn, C.; Roberts, L.R.; Selby, M.D.; Jones, L.H. Click-enabled heterotrifunctional template for sequential bioconjugations. Org. Biomol. Chem. 2012, 10, 548–554. [Google Scholar]
- Robitaille, R.; Adler, E.M.; Charlton, M.P. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 1990, 5, 773–779. [Google Scholar] [CrossRef]
- Jones, O.T.; Kunze, D.L.; Angelides, K.J. Localization and mobility of omega-conotoxin-sensitive Ca2+ channels in hippocampal ca1 neurons. Science 1989, 244, 1189–1193. [Google Scholar]
- Freudenthaler, G.; Axmann, M.; Schindler, H.; Pragl, B.; Knaus, H.G.; Schütz, G.J. Ultrasensitive pharmacological characterisation of the voltage-gated potassium channel K(v)1.3 studied by single-molecule fluorescence microscopy. Histochem. Cell Biol. 2002, 117, 97–102. [Google Scholar]
- Bergeron, Z.L. The Molecular Engineering of Potassium Channel Probes: A Stepwise Approach to Cellular Imaging of BK and HERG Ion Channels; Clarkson University: Potsdam, NY, USA, 2007. [Google Scholar]
- Bergeron, Z.L. Peptide Toxin Bioengineering-Advancement of Fluorescent Probe Design for Targeting Human K+ Channels. Ph.D. Thesis, University of Hawaii at Manoa, 2012. [Google Scholar]
- Vita, C.; Roumestand, C.; Toma, F.; Ménez, A. Scorpion toxins as natural scaffolds for protein engineering. Proc. Natl. Acad. Sci. USA 1995, 92, 6404–6408. [Google Scholar] [CrossRef]
- Tassonyi, E.; Charpantier, E.; Muller, D.; Dumont, L.; Bertrand, D. The role of nicotinic acetylcholine receptors in the mechanisms of anesthesia. Brain Res. Bull. 2002, 57, 133–150. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Zinn-Justin, S.; Guenneugues, M.; Gilqin, B.; Ménez, A.; Vita, C. Changing the structural context of a functional beta-hairpin. Synthesis and characterization of a chimera containing the curaremimetic loop of a snake toxin in the scorpion alpha/beta scaffold. J. Biol. Chem. 1996, 271, 11979–11987. [Google Scholar]
- Fajloun, Z.; Ferrat, G.; Carlier, E.; M’Barek, S.; Regaya, I.; Fathallah, M.; Rochat, H.; Darbon, H.; de Waard, M.; Sabatier, J.M. Synthesis, 3-D structure, and pharmacology of a reticulated chimeric peptide derived from maurotoxin and tsk scorpion toxins. Biochem. Biophys. Res. Commun. 2002, 291, 640–648. [Google Scholar] [CrossRef]
- Blanc, E.; Lecomte, C.; Rietschoten, J.V.; Sabatier, J.M.; Darbon, H. Solution structure of tskapa, a charybdotoxin-like scorpion toxin from tityus serrulatus with high affinity for apamin-sensitive Ca2+-activated K+ channels. Proteins 1997, 29, 359–369. [Google Scholar] [CrossRef]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar]
- Huys, I.; Waelkens, E.; Tytgat, J. Structure-function study of a chlorotoxin-chimer and its activity on kv1.3 channels. J. Chromatogr. B 2004, 803, 67–73. [Google Scholar] [CrossRef]
- Holaday, S.K.J.; Martin, B.M.; Fletcher, P.L., Jr.; Krishna, N.R. Nmr solution structure of butantoxin. Arch. Biochem. Biophys. 2000, 379, 18–27. [Google Scholar] [CrossRef]
- Miranda, L.P.; Alewood, P.F. Accelerated chemical synthesis of peptides and small proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 1181–1186. [Google Scholar]
- Valiyaveetil, F.I.; MacKinnon, R.; Muir, T.W. Semisynthesis and folding of the potassium channel kcsa. J. Am. Chem. Soc. 2002, 124, 9113–9120. [Google Scholar] [CrossRef]
- Komarov, A.G.; Linn, K.M.; Devereaux, J.J.; Valiyaveetil, F.I. A modular strategy for the semisynthesis of a K+ channel: Investigating interactions of the pore helix. ACS Chem. Biol. 2009, 4, 1029–1038. [Google Scholar] [CrossRef]
- Yu, H.H.; Nakase, I.; Pujals, S.; Hirose, H.; Tanaka, G.; Katayama, S.; Imanishi, M.; Futaki, S. Expressed protein ligation for the preparation of fusion proteins with cell penetrating peptides for endotoxin removal and intracellular delivery. Biochim. Biophys. Acta 2010, 1798, 2249–2257. [Google Scholar] [CrossRef]
- Clippingdale, A.B.; Barrow, C.J.; Wade, J.D. Peptide thioester preparation by fmoc solid phase peptide synthesis for use in native chemical ligation. J. Pept. Sci. 2000, 6, 225–234. [Google Scholar] [CrossRef]
- Li, X.; Kawakami, T.; Aimoto, S. Direct preparation of peptide thioesters using an fmoc solid-phase method. Tetrahedron Lett. 1998, 39, 8669–8672. [Google Scholar] [CrossRef]
- Bingham, J.P.; Chun, J.B.; Ruzicka, M.R.; Li, Q.X.; Tan, Z.Y.; Kaulin, Y.A.; Englebretsen, D.R.; Moczydlowski, E.G. Synthesis of an iberiotoxin derivative by chemical ligation: A method for improved yields of cysteine-rich scorpion toxin peptides. Peptides 2009, 30, 1049–1057. [Google Scholar] [CrossRef]
- Clark, R.J.; Craik, D.J. Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Biopolymers 2010, 94, 414–422. [Google Scholar] [CrossRef]
- Craik, D.J.; Clark, R.J.; Daly, N.L. Potential therapeutic applications of the cyclotides and related cystine knot mini-proteins. Expert Opin. Investig. Drugs 2007, 16, 595–604. [Google Scholar] [CrossRef]
- Cemazar, M.; Kwon, S.; Mahatmanto, T.; Ravipati, A.S.; Craik, D.J. Discovery and applications of disulfide-rich cyclic peptides. Curr. Top Med. Chem. 2012, 12, 1534–1545. [Google Scholar] [CrossRef]
- Ireland, D.C.; Colgrave, M.L.; Nguyencong, P.; Daly, N.L.; Craik, D.J. Discovery and characterization of a linear cyclotide from viola odorata: Implications for the processing of circular proteins. J. Mol. Biol. 2006, 357, 1522–1535. [Google Scholar] [CrossRef]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef]
- Craik, D.J.; Swedberg, J.E.; Mylne, J.S.; Cemazar, M. Cyclotides as a basis for drug design. Expert Opin. Drug Discov. 2012, 7, 179–194. [Google Scholar] [CrossRef]
- Clark, R.J.; Daly, N.L.; Craik, D.J. Structural plasticity of the cyclic-cystine-knot framework: Implications for biological activity and drug design. Biochem. J. 2006, 394, 85–93. [Google Scholar] [CrossRef]
- Carstens, B.B.; Clark, R.J.; Daly, N.L.; Harvey, P.J.; Kaas, Q.; Craik, D.J. Engineering of conotoxins for the treatment of pain. Curr. Pharm. Des. 2011, 17, 4242–4253. [Google Scholar] [CrossRef]
- Daly, N.L.; Clark, R.J.; Craik, D.J. Disulfide folding pathways of cystine knot proteins. Tying the knot within the circular backbone of the cyclotides. J. Biol. Chem. 2003, 278, 6314–6322. [Google Scholar]
- Craik, D.J.; Cemazar, M.; Daly, N.L. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr. Opin. Drug Discov. Devel. 2006, 9, 251–260. [Google Scholar]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Sharpe, I.A.; Palant, E.; Schroeder, C.I.; Kaye, D.M.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Inhibition of the norepinephrine transporter by the venom peptide chi-mria. Site of action, Na+ dependence, and structure-activity relationship. J. Biol. Chem. 2003, 278, 40317–40323. [Google Scholar]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic mria: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar]
- Villalonga, N.; David, M.; Bielanska, J.; Vicente, R.; Comes, N.; Valenzuela, C.; Felipe, A. Immunomodulation of voltage-dependent K+ channels in macrophages: Molecular and biophysical consequences. J. Gen. Physiol. 2010, 135, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Leonetti, M.D.; Hsiung, Y.; MacKinnon, R. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 2011, 481, 94–97. [Google Scholar]
- Ataga, K.I.; Stocker, J. Senicapoc (ica-17043): A potential therapy for the prevention and treatment of hemolysis-associated complications in sickle cell anemia. Expert Opin. Investig. Drugs 2009, 18, 231–239. [Google Scholar] [CrossRef]
- Wettwer, E.; Hála, O.; Christ, T.; Heubach, J.F.; Dobrev, D.; Knaut, M.; Varró, A.; Ravens, U. Role of ikur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004, 110, 2299–2306. [Google Scholar]
- Peroz, D.; Rodriguez, N.; Choveau, F.; Baró, I.; Mérot, J.; Loussouarn, G. Kv7.1 (kcnq1) properties and channelopathies. J. Physiol. 2008, 586, 1785–1789. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Tristani-Firouzi, M. Herg potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar]
- Center for Disease Control and Prevention. State-specific mortality from sudden cardiac death-United States, 1999. MMWR Morb. Mortal. Wkly. Rep. 2002, 51, 123–126.
- Roy, M.; Dumaine, R.; Brown, A.M. Herg, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation 1996, 94, 817–823. [Google Scholar] [CrossRef]
- Friedrichs, G.S.; Patmore, L.; Bass, A. Non-clinical evaluation of ventricular repolarization (ich s7b): Results of an interim survey of international pharmaceutical companies. J. Pharmacol. Toxicol Methods 2005, 52, 6–11. [Google Scholar] [CrossRef]
- Korolkova, Y.V.; Kozlov, S.A.; Lipkin, A.V.; Pluzhnikov, K.A.; Hadley, J.K.; Filippov, A.K.; Brown, D.A.; Angelo, K.; Strøbaek, D.; Jespersen, T.; et al. An erg channel inhibitor from the scorpion buthus eupeus. J. Biol. Chem. 2001, 276, 9868–9876. [Google Scholar]
- Li, Y.; Wang, P.; Xu, J.; Desir, G.V. Voltage-gated potassium channel kv1.3 regulates glut4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am. J. Physiol. Cell Physiol. 2006, 290, C345–C351. [Google Scholar]
- Villalonga, N.; Escalada, A.; Vicente, R.; Sánchez-Tilló, E.; Celada, A.; Solsona, C.; Felipe, A. Kv1.3/kv1.5 heteromeric channels compromise pharmacological responses in macrophages. Biochem. Biophys. Res. Commun. 2007, 352, 913–918. [Google Scholar] [CrossRef]
- Varga, Z.; Gurrola-Briones, G.; Papp, F.; Rodríguez de la Vega, R.C.; Pedraza-Alva, G.; Tajhya, R.B.; Gaspar, R.; Cardenas, L.; Rosenstein, Y.; Beeton, C.; Possani, L.D.; Panyi, G. Vm24, a natural immunosuppressive peptide, potently and selectively blocks kv1.3 potassium channels of human t cells. Mol. Pharmacol. 2012, 82, 372–382. [Google Scholar] [CrossRef]
- Eichhorn, B.; Dobrev, D. Vascular large conductance calcium-activated potassium channels: Functional role and therapeutic potential. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 376, 145–155. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Zhang, N.; Wu, G.; Wu, H. The solution structure of bmtx3b, a member of the scorpion toxin subfamily alpha-ktx 16. Proteins 2005, 58, 489–497. [Google Scholar]
- Yao, J.; Chen, X.; Li, H.; Zhou, Y.; Yao, L.; Wu, G.; Chen, X.; Zhang, N.; Zhou, Z.; Xu, T.; Wu, H.; Ding, J. Bmp09, a “long chain” scorpion peptide blocker of bk channels. J. Biol. Chem. 2005, 280, 14819–14828. [Google Scholar]
- Zuberi, S.M.; Eunson, L.H.; Spauschus, A.; de Silva, R.; Tolmie, J.; Wood, N.W.; McWilliam, R.C.; Stephenson, J.B.; Kullmann, D.M.; Hanna, M.G. A novel mutation in the human voltage-gated potassium channel gene (kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 1999, 122, 817–825. [Google Scholar] [CrossRef]
- Angulo, E.; Noé, V.; Casadó, V.; Mallol, J.; Gomez-Isla, T.; Lluis, C.; Ferrer, I.; Ciudad, C.J.; Franco, R. Up-regulation of the kv3.4 potassium channel subunit in early stages of alzheimer’s disease. J. Neurochem. 2004, 91, 547–557. [Google Scholar] [CrossRef]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef]
- Wickenden, A.D.; McNaughton-Smith, G. Kv7 channels as targets for the treatment of pain. Curr. Pharm. Des. 2009, 15, 1773–1798. [Google Scholar] [CrossRef]
- Abdel-Mottaleb, Y.; Clynen, E.; Jalali, A.; Bosmans, F.; Vatanpour, H.; Schoofs, L.; Tytgat, J. The first potassium channel toxin from the venom of the iranian scorpion odonthobuthus doriae. FEBS Lett. 2006, 580, 6254–6258. [Google Scholar]
- Vacher, H.; Diochot, S.; Bougis, P.E.; Martin-Eauclaire, M.F.; Mourre, C. Kv4 channels sensitive to bmtx3 in rat nervous system: Autoradiographic analysis of their distribution during brain ontogenesis. Eur. J. Neurosci. 2006, 24, 1325–1340. [Google Scholar] [CrossRef]
- Unaids Data Tables 2011, United Nations: Geneva, Switzerland, 2011.
- Kripke, C. Antiretroviral prophylaxis for occupational exposure to hiv. Am. Fam. Physician. 2007, 76, 375–376. [Google Scholar]
- Li, C.; Dowd, C.S.; Zhang, W.; Chaiken, I.M. Phage randomization in a charybdotoxin scaffold leads to cd4-mimetic recognition motifs that bind hiv-1 envelope through non-aromatic sequences. J. Pept. Res. 2001, 57, 507–518. [Google Scholar] [CrossRef]
- Vita, C.; Drakopoulou, E.; Vizzavona, J.; Rochette, S.; Martin, L.; Ménez, A.; Roumestand, C.; Yang, Y.S.; Ylisastigui, L.; Benjouad, A.; Gluckman, J.C. Rational engineering of a miniprotein that reproduces the core of the cd4 site interacting with hiv-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 13091–13096. [Google Scholar]
- Mouhat, S.; Visan, V.; Ananthakrishnan, S.; Wulff, H.; Andreotti, N.; Grissmer, S.; Darbon, H.; de Waard, M.; Sabatier, J.M. K+ channel types targeted by synthetic osk1, a toxin from orthochirus scrobiculosus scorpion venom. Biochem. J. 2005, 385, 95–104. [Google Scholar] [CrossRef]
- Han, S.; Yi, H.; Yin, S.J.; Chen, Z.Y.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Structural basis of a potent peptide inhibitor designed for kv1.3 channel, a therapeutic target of autoimmune disease. J. Biol. Chem. 2008, 283, 19058–19065. [Google Scholar]
- Renisio, J.G.; Romi-Lebrun, R.; Blanc, E.; Bornet, O.; Nakajima, T.; Darbon, H. Solution structure of bmktx, a K+ blocker toxin from the chinese scorpion buthus martensi. Proteins 2000, 38, 70–78. [Google Scholar] [CrossRef]
- Filler, S.J.; MacArthur, J.R.; Parise, M.; Wirtz, R.; Eliades, M.J.; Dasilva, A.; Steketee, R. Centers for disease control and prevention locally Acquired mosquito-transmitted malaria: A guide for investigations in the United States. Morb. Mortal. Wkly. Rep. 2006, 55, 1–9. [Google Scholar]
- Zhu, S.; Gao, B.; Aumelas, A.; del Carmen Rodríguez, M.; Lanz-Mendoza, H.; Peigneur, S.; Diego-Garcia, E.; Martin-Eauclaire, M.F.; Tytgat, J.; Possani, L.D. Meutxkbeta1, a scorpion venom-derived two-domain potassium channel toxin-like peptide with cytolytic activity. Biochim. Biophys. Acta 2010, 1804, 872–873. [Google Scholar] [CrossRef]
- Gao, B.; Xu, J.; Rodriguez Mdel, C.; Lanz-Mendoza, H.; Hernández-Rivas, R.; Du, W.; Zhu, S. Characterization of two linear cationic antimalarial peptides in the scorpion mesobuthus eupeus. Biochimie 2010, 92, 350–359. [Google Scholar] [CrossRef]
- He, Q.Y.; He, Q.Z.; Deng, X.C.; Yao, L.; Meng, E.; Liu, Z.H.; Liang, S.P. Atdb: A uni-database platform for animal toxins. Nucleic Acids Res. 2008, 36, D293–D297. [Google Scholar]
- Lim, E.; Pon, A.; Djoumbou, Y.; Knox, C.; Shrivastava, S.; Guo, A.C.; Neveu, V.; Wishart, D.S. T3db: A comprehensively annotated database of common toxins and their targets. Nucleic Acids Res. 2010, 38, D781–D786. [Google Scholar] [CrossRef]
- Wood, D.L.; Miljenović, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. Arachnoserver: A database of protein toxins from spiders. BMC Genomics 2009, 10, 375. [Google Scholar] [CrossRef]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. Conoserver, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Srinivasan, K.N.; Gopalakrishnakone, P.; Tan, P.T.; Chew, K.C.; Cheng, B.; Kini, R.M.; Koh, J.L.; Seah, S.H.; Brusic, V. Scorpion, a molecular database of scorpion toxins. Toxicon 2002, 40, 23–31. [Google Scholar]
- Tan, P.T.; Veeramani, A.; Srinivasan, K.N.; Ranganathan, S.; Brusic, V. Scorpion2: A database for structure-function analysis of scorpion toxins. Toxicon 2006, 47, 356–363. [Google Scholar] [CrossRef]
Appendix
Structural Databases
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bergeron, Z.L.; Bingham, J.-P. Scorpion Toxins Specific for Potassium (K+) Channels: A Historical Overview of Peptide Bioengineering. Toxins 2012, 4, 1082-1119. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins4111082
Bergeron ZL, Bingham J-P. Scorpion Toxins Specific for Potassium (K+) Channels: A Historical Overview of Peptide Bioengineering. Toxins. 2012; 4(11):1082-1119. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins4111082
Chicago/Turabian StyleBergeron, Zachary L., and Jon-Paul Bingham. 2012. "Scorpion Toxins Specific for Potassium (K+) Channels: A Historical Overview of Peptide Bioengineering" Toxins 4, no. 11: 1082-1119. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins4111082