Cancer Cell Growth Inhibitory Effect of Bee Venom via Increase of Death Receptor 3 Expression and Inactivation of NF-kappa B in NSCLC Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
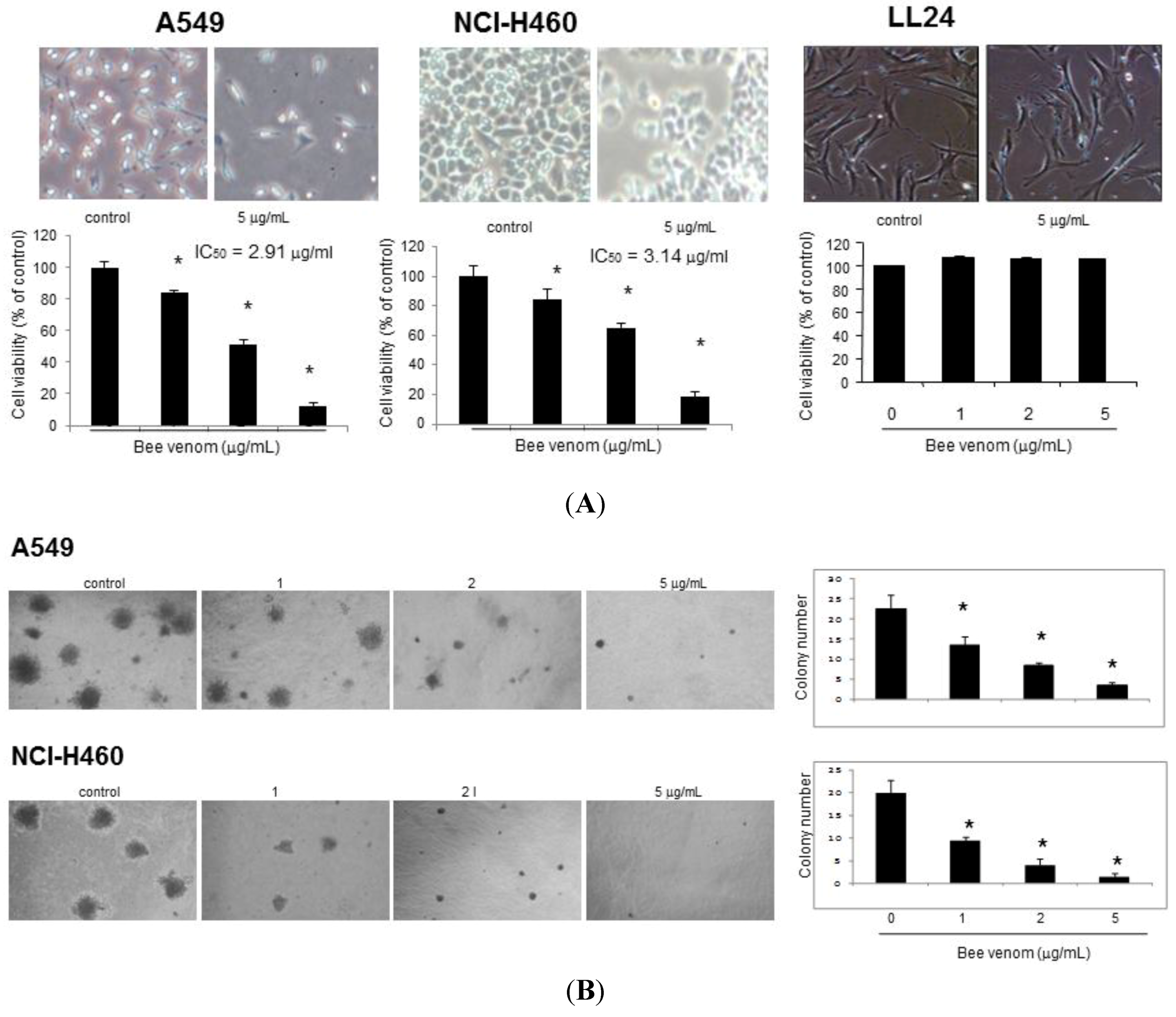
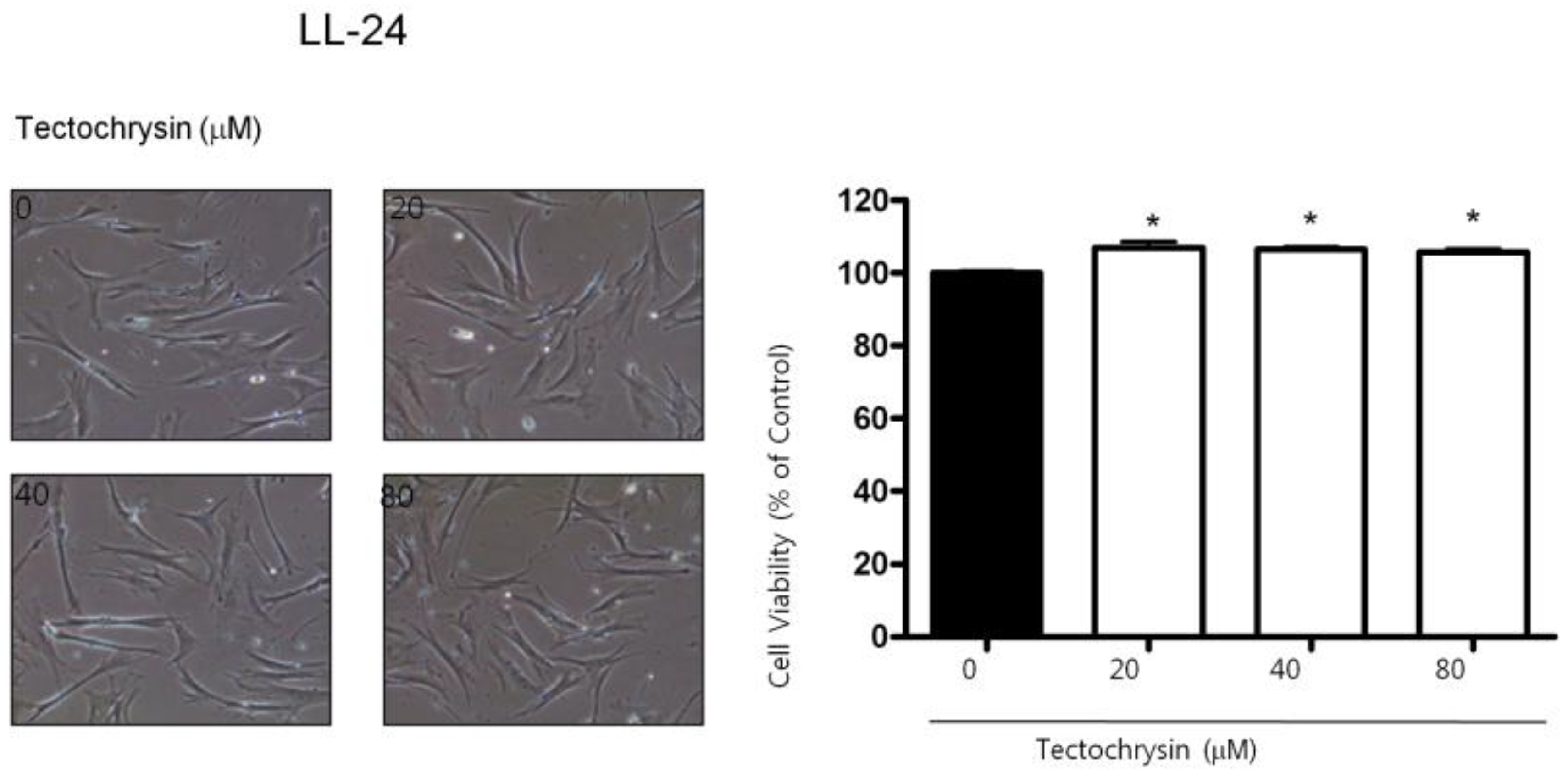
2.1. Effect of BV on Cell Growth in Lung Cancer Cells

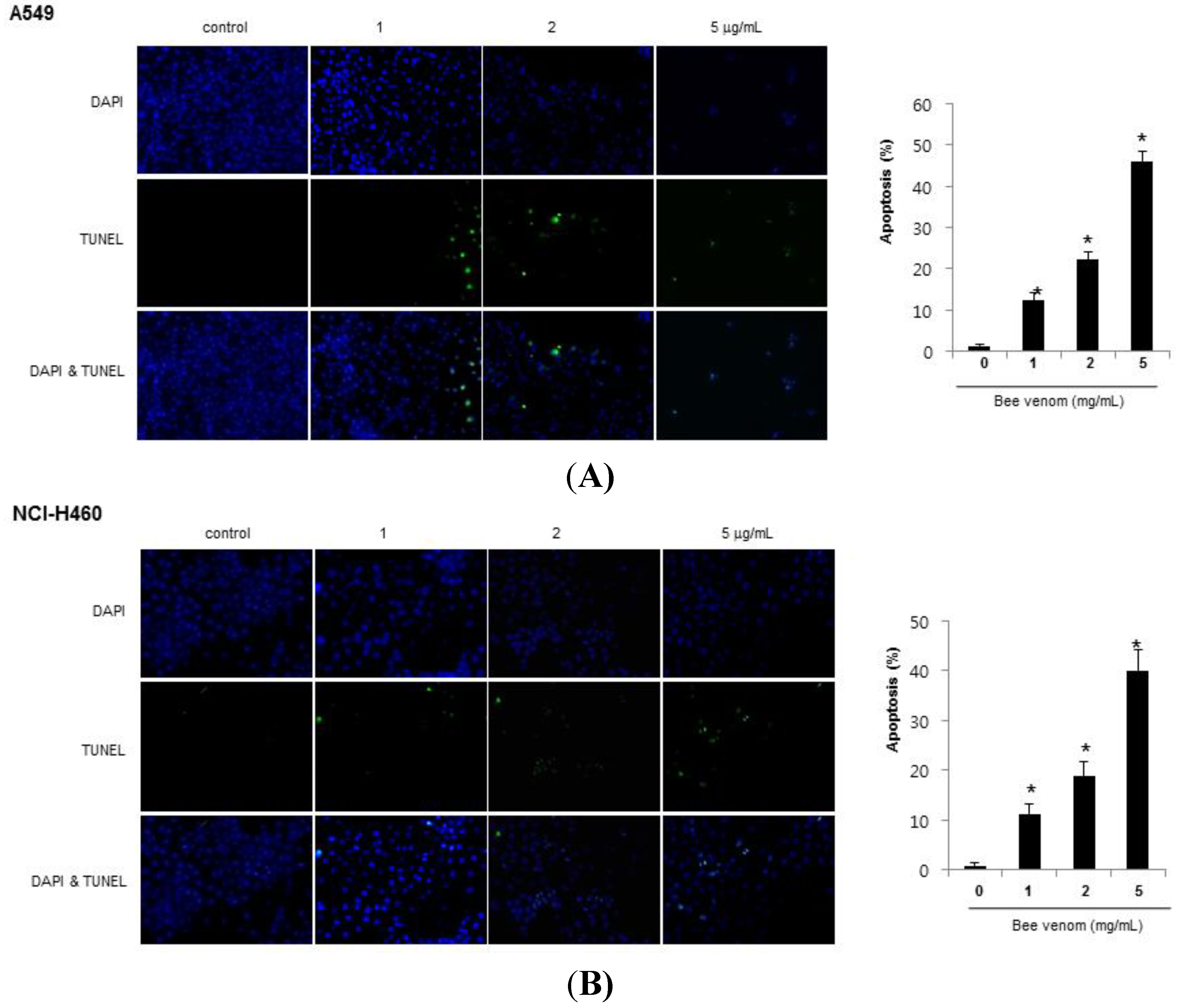
2.2. Apoptotic Cell Death by BV

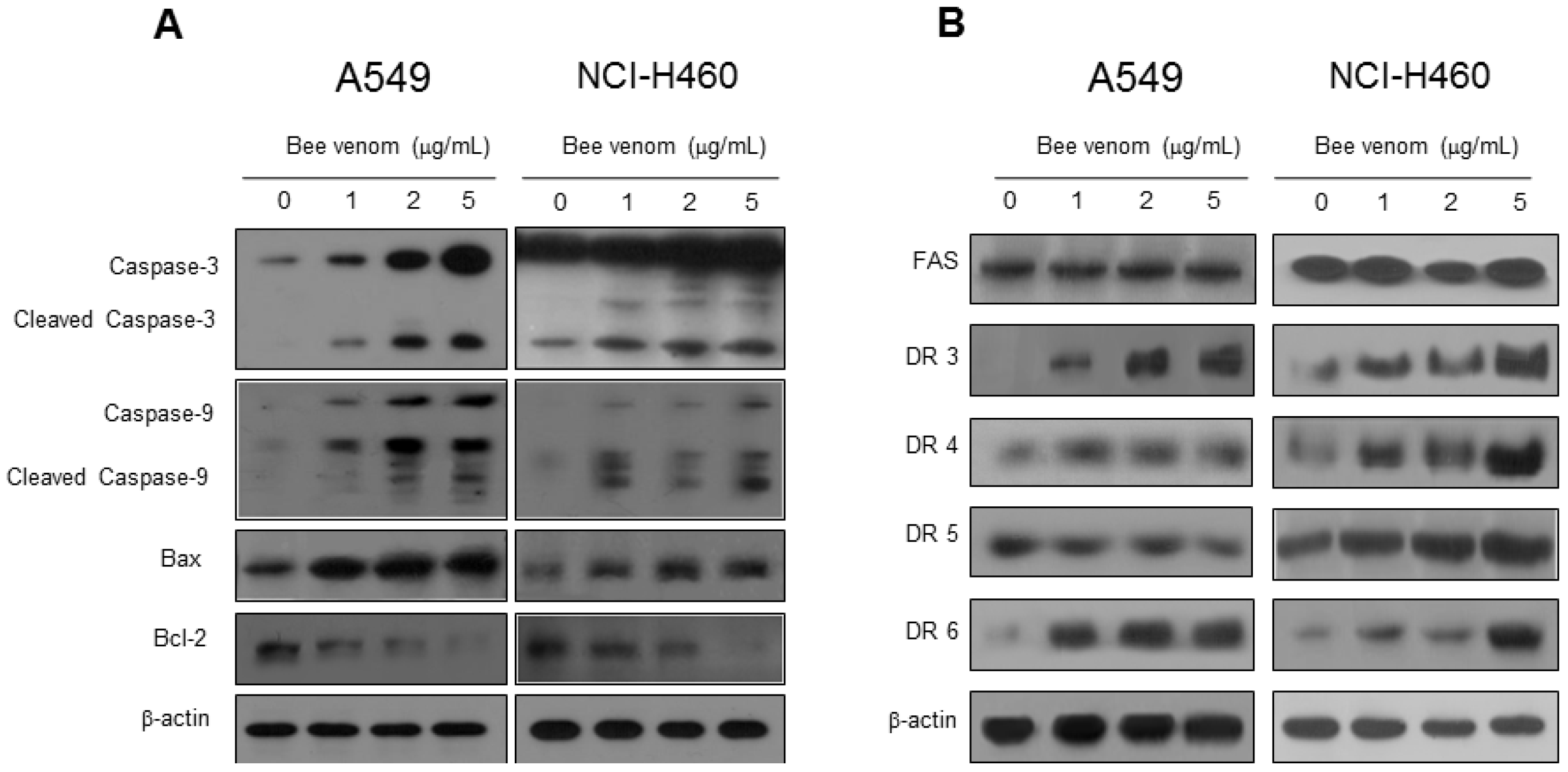
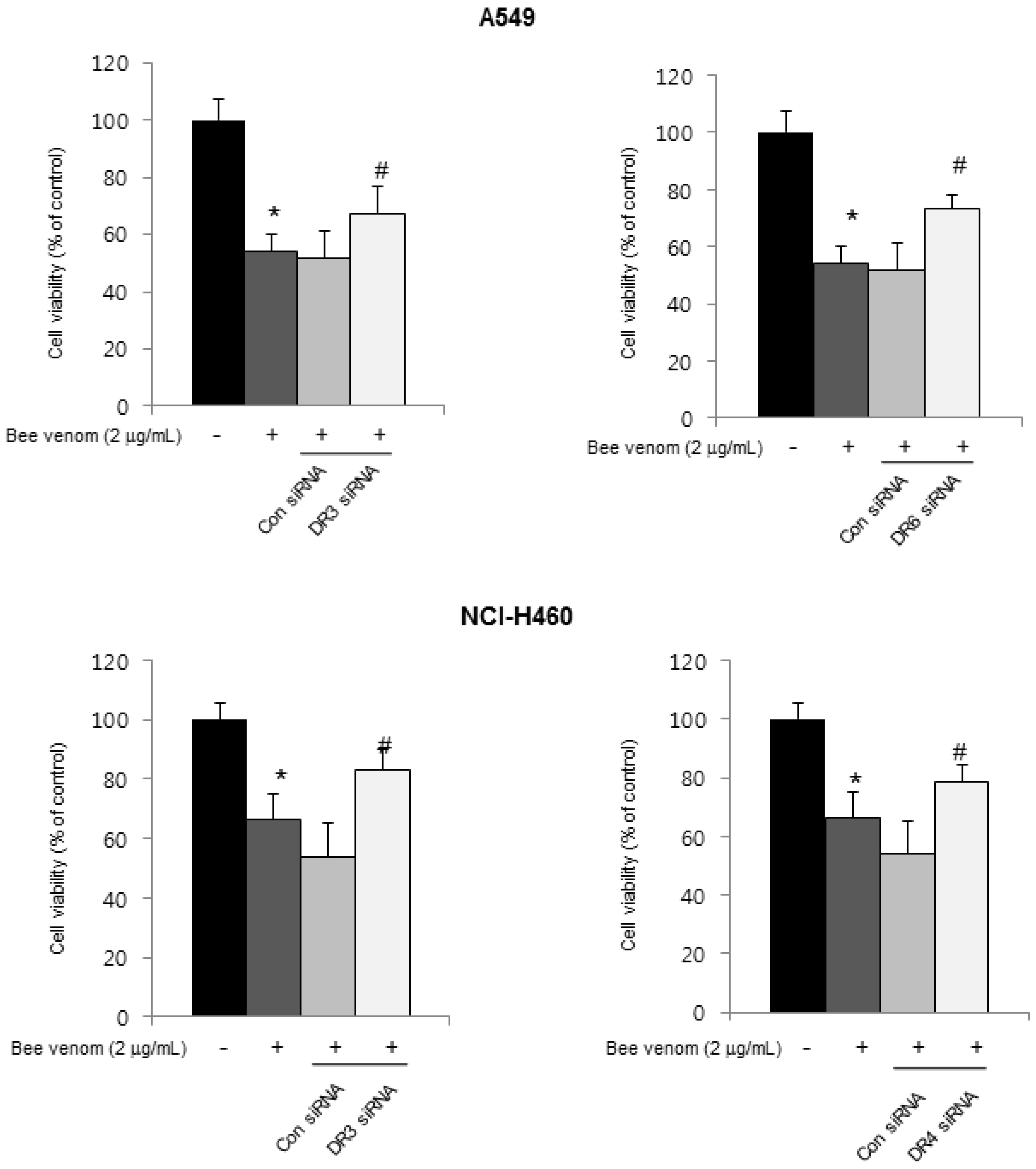
2.3. Expression of Apoptotic Regulatory Proteins and Death Receptor by BV


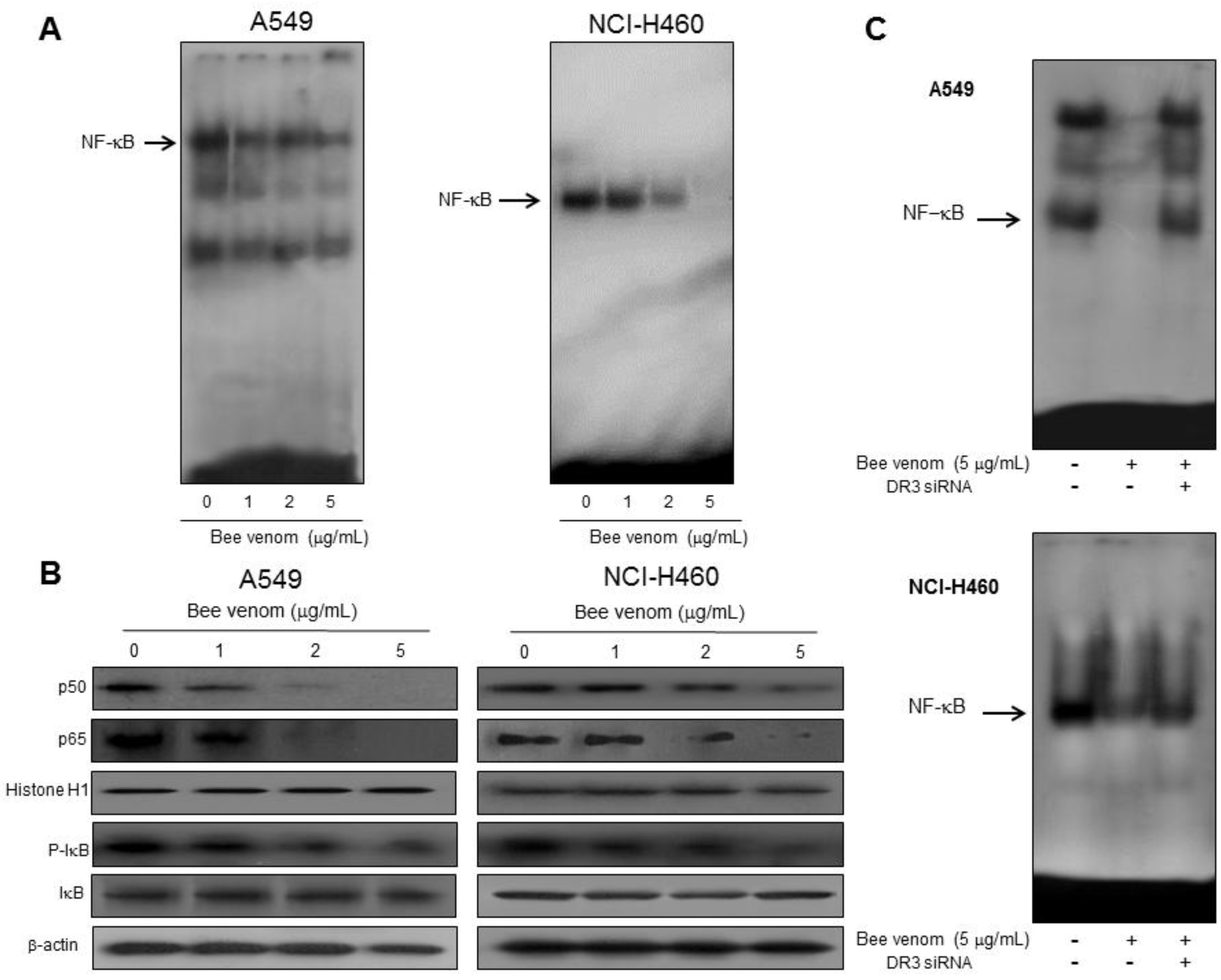
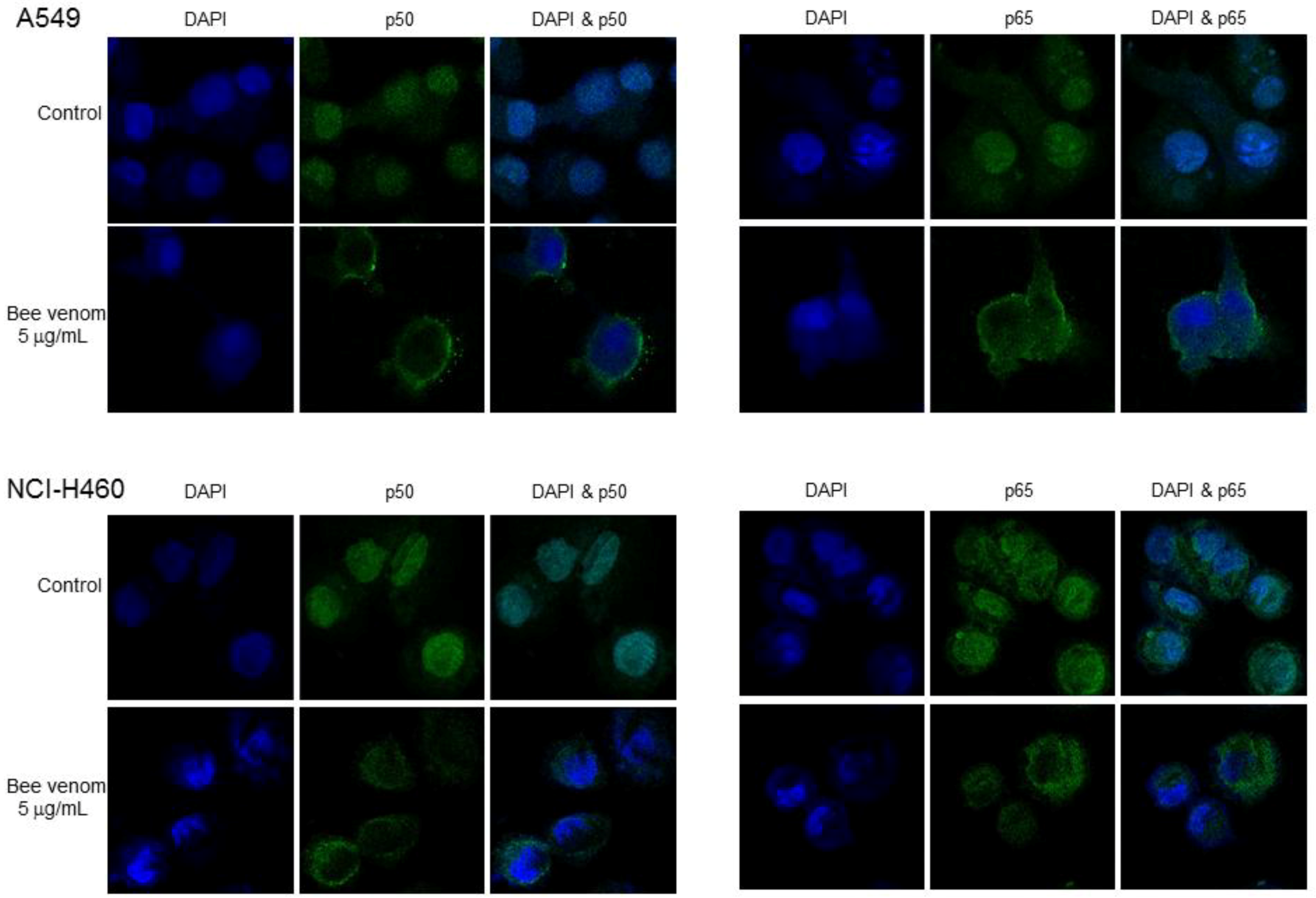
2.4. Involvement of NF-κB Signaling Pathway in Apoptotic Cell Death by BV

2.5. Combination of BV with TWEAK Further Induced DR3 Overexpression and Inactivation NF-κB

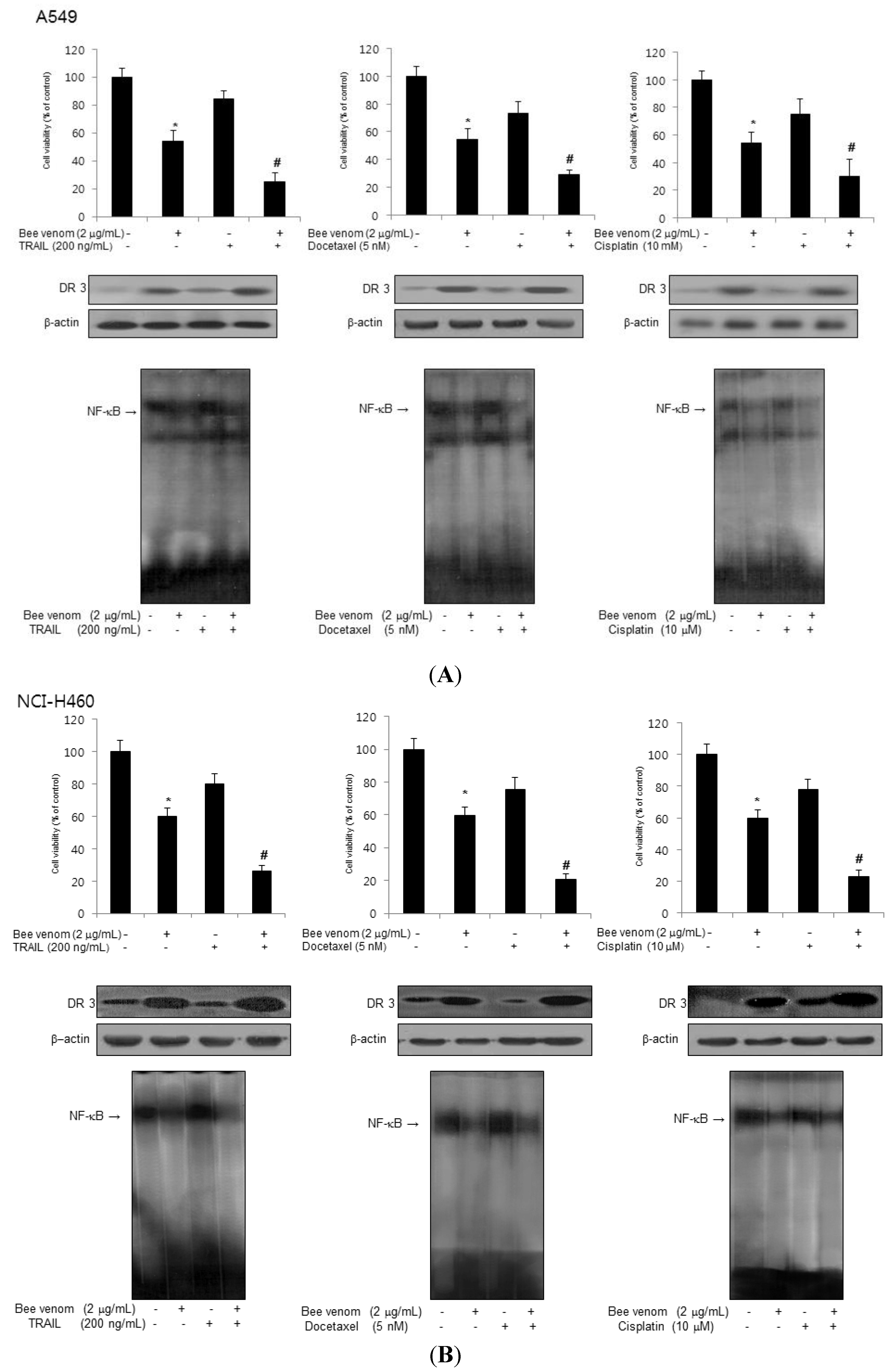
2.6. Combinations of BV with Chemotherapeutics on Inhibition of Lung Cancer Cell Growth, DR3 Overexpression and Inactivation of NF-κB

3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Apoptosis Evaluation
4.5. Western Blot Analysis
4.6. Transfection (siRNA)
4.7. Electromobility Shift Assay (EMSA)
4.8. Immunofluorescence Staining
4.9. Soft Agar Formation Assay
4.10. Data Analysis
5. Conclusions
Abbreviations
| BV | Bee venom |
| DRs | Death receptors |
| EMSA | Electromobility shift assay |
| FADD | Fas-associated death domain protein |
| FasL | Fas ligand; FKB: Favokawain B |
| LLC | Lewis lung carcinoma |
| MMP-2 | Matrix metalloproteinase-2 |
| NF-κB | Nuclear factor kappaB |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor-2 |
Acknowledgments
Author Contributions
Appendix


Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef]
- Goncalves, A.S.; Macedo, A.S.; Souto, E.B. Therapeutic nanosystems for oncology nanomedicine. Clin. Transl. Oncol. 2012, 14, 883–890. [Google Scholar] [CrossRef]
- Latz, C.; Huang, Q.; Kapadia, M.K.; Freitag, S.K. Metastasis to eyelid as initial presentation of non-small cell carcinoma. Ophthalmic Plast. Reconstr. Surg. 2009, 25, 406–408. [Google Scholar]
- Triller, N.; Korosec, P.; Kern, I.; Kosnik, M.; Debeljak, A. Multidrug resistance in small cell lung cancer: Expression of P-glycoprotein, multidrug resistance protein 1 and lung resistance protein in chemo-naive patients and in relapsed disease. Lung Cancer 2006, 54, 235–240. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Yan, Y.; Lemon, W.J.; LaRegina, M.; Morrison, C.; Lubet, R.; You, M. A chemically induced model for squamous cell carcinoma of the lung in mice: Histopathology and strain susceptibility. Cancer Res. 2004, 64, 1647–1654. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.H.; Son, D.J.; Oh, K.W.; Kim, K.H.; Song, H.S.; Kim, G.J.; Oh, G.T.; Yoon, D.Y.; Hong, J.T. Antiarthritic effect of bee venom: Inhibition of inflammation mediator generation by suppression of NF-κB through interaction with the p50 subunit. Arthritis Rheum. 2004, 50, 3504–3515. [Google Scholar] [CrossRef]
- Park, M.H.; Choi, M.S.; Kwak, D.H.; Oh, K.W.; Yoon do, Y.; Han, S.B.; Song, H.S.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom in prostate cancer cells through activation of caspase pathway via inactivation of NF-κB. Prostate 2011, 71, 801–812. [Google Scholar] [CrossRef]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef]
- Wang, C.; Chen, T.; Zhang, N.; Yang, M.; Li, B.; Lu, X.; Cao, X.; Ling, C. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IκBalpha kinase-NFκ. J. Biol. Chem. 2009, 284, 3804–3813. [Google Scholar]
- Ip, S.W.; Chu, Y.L.; Yu, C.S.; Chen, P.Y.; Ho, H.C.; Yang, J.S.; Huang, H.Y.; Chueh, F.S.; Lai, T.Y.; Chung, J.G. Bee venom induces apoptosis through intracellular Ca2+ -modulated intrinsic death pathway in human bladder cancer cells. Int. J. Urol. 2012, 19, 61–70. [Google Scholar] [CrossRef]
- Inoue, N.; Matsuda, F.; Goto, Y.; Manabe, N. Role of cell-death ligand-receptor system of granulosa cells in selective follicular atresia in porcine ovary. J. Reprod. Dev. 2011, 57, 169–175. [Google Scholar] [CrossRef]
- Zhu, H.; Ling, W.; Hu, B.; Su, Y.; Qiu, S.; Xiao, W.; Qi, Y. Adenovirus E1A reverses the resistance of normal primary human lung fibroblast cells to TRAIL through DR5 upregulation and caspase 8-dependent pathway. Cancer Biol. Ther. 2006, 5, 180–188. [Google Scholar] [CrossRef]
- Han, Z.; Pantazis, P.; Wyche, J.H.; Kouttab, N.; Kidd, V.J.; Hendrickson, E.A. A Fas-associated death domain protein-dependent mechanism mediates the apoptotic action of non-steroidal anti-inflammatory drugs in the human leukemic Jurkat cell line. J. Biol. Chem. 2001, 276, 38748–38754. [Google Scholar]
- Huang, Y.; He, Q.; Hillman, M.J.; Rong, R.; Sheikh, M.S. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Res. 2001, 61, 6918–6924. [Google Scholar]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Cramer, F.; Christensen, C.L.; Poulsen, T.T.; Badding, M.A.; Dean, D.A.; Poulsen, H.S. Insertion of a nuclear factor κ B DNA nuclear-targeting sequence potentiates suicide gene therapy efficacy in lung cancer cell lines. Cancer Gene Ther. 2012, 19, 675–683. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Ali, S.; Philip, P.A.; Wozniak, A.; Sarkar, F.H. Genistein enhances the effect of epidermal growth factor receptor tyrosine kinase inhibitors and inhibits nuclear factor κ B in nonsmall cell lung cancer cell lines. Cancer 2009, 115, 2165–2176. [Google Scholar] [CrossRef]
- Liao, H.F.; Kuo, C.D.; Yang, Y.C.; Lin, C.P.; Tai, H.C.; Chen, Y.Y.; Chen, Y.J. Resveratrol enhances radiosensitivity of human non-small cell lung cancer NCI-H838 cells accompanied by inhibition of nuclear factor-κ B activation. J. Radiat. Res. 2005, 46, 387–393. [Google Scholar] [CrossRef]
- Lind, D.S.; Hochwald, S.N.; Malaty, J.; Rekkas, S.; Hebig, P.; Mishra, G.; Moldawer, L.L.; Copeland, E.M., 3rd; Mackay, S. Nuclear factor-κB is upregulated in colorectal cancer. Surgery 2001, 130, 363–369. [Google Scholar] [CrossRef]
- Ban, J.O.; Lee, H.S.; Jeong, H.S.; Song, S.; Hwang, B.Y.; Moon, D.C.; Yoon do, Y.; Han, S.B.; Hong, J.T. Thiacremonone augments chemotherapeutic agent-induced growth inhibition in human colon cancer cells through inactivation of nuclear factorκ-B. Mol. Cancer Res. 2009, 7, 870–879. [Google Scholar] [CrossRef]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Ban, J.O.; Yuk, D.Y.; Woo, K.S.; Kim, T.M.; Lee, U.S.; Jeong, H.S.; Kim, D.J.; Chung, Y.B.; Hwang, B.Y.; Oh, K.W.; et al. Inhibition of cell growth and induction of apoptosis via inactivation of NF-κB by a sulfurcompound isolated from garlic in human colon cancer cells. J. Pharmacol. Sci. 2007, 104, 374–383. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, S.Y.; Cho, J.S.; Son, S.M.; Choi, S.S.; Yun, Y.P.; Yoo, H.S.; Yoon do, Y.; Oh, K.W.; Han, S.B.; et al. Combination of ginsenoside rg3 with docetaxel enhances the susceptibility of prostate cancer cells via inhibition of nf-κb. Eur. J. Pharmacol. 2010, 631, 1–9. [Google Scholar] [CrossRef]
- Lee, S.Y.; Cho, J.S.; Yuk, D.Y.; Moon, D.C.; Jung, J.K.; Yoo, H.S.; Lee, Y.M.; Han, S.B.; Oh, K.W.; Hong, J.T. Obovatol enhances docetaxel-induced prostate and colon cancer cell death through inactivation of nuclear transcription factor-κb. J. Pharmacol. Sci. 2009, 111, 124–136. [Google Scholar] [CrossRef]
- Orsolic, N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012, 31, 173–194. [Google Scholar] [CrossRef]
- Ip, S.W.; Liao, S.S.; Lin, S.Y.; Lin, J.P.; Yang, J.S.; Lin, M.L.; Chen, G.W.; Lu, H.F.; Lin, M.W.; Han, S.M.; et al. The role of mitochondria in bee venom-induced apoptosis in human breast cancer MCF7 cells. In Vivo 2008, 22, 237–245. [Google Scholar]
- Moon, D.O.; Park, S.Y.; Heo, M.S.; Kim, K.C.; Park, C.; Ko, W.S.; Choi, Y.H.; Kim, G.Y. Key regulators in bee venom-induced apoptosis are Bcl-2 and caspase-3 in human leukemic U937 cells through downregulation of ERK and Akt. Int. Immunopharmacol. 2006, 6, 1796–1807. [Google Scholar] [CrossRef]
- Valentin, E.; Lambeau, G. What can venom phospholipases A(2) tell us about the functional diversity of mammalian secreted phospholipases A(2)? Biochimie 2000, 82, 815–831. [Google Scholar] [CrossRef]
- Holle, L.; Song, W.; Holle, E.; Wei, Y.; Li, J.; Wagner, T.E.; Yu, X. In vitro- and in vivo-targeted tumor lysis by an MMP2 cleavable melittin-LAP fusion protein. Int. J. Oncol. 2009, 35, 829–835. [Google Scholar]
- Huh, J.E.; Baek, Y.H.; Lee, M.H.; Choi, D.Y.; Park, D.S.; Lee, J.D. Bee venom inhibits tumor angiogenesis and metastasis by inhibiting tyrosine phosphorylation of VEGFR-2 in LLC-tumor-bearing mice. Cancer Lett. 2010, 292, 98–110. [Google Scholar] [CrossRef]
- Shanker, A.; Brooks, A.D.; Tristan, C.A.; Wine, J.W.; Elliott, P.J.; Yagita, H.; Takeda, K.; Smyth, M.J.; Murphy, W.J.; Sayers, T.J. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J. Natl. Cancer Inst. 2008, 100, 649–662. [Google Scholar] [CrossRef]
- Moibi, J.A.; Mak, A.L.; Sun, B.; Moore, R.B. Urothelial cancer cell response to combination therapy of gemcitabine and TRAIL. Int. J. Oncol. 2011, 39, 61–71. [Google Scholar]
- Tang, Y.; Li, X.; Liu, Z.; Simoneau, A.R.; Xie, J.; Zi, X.; Flavokawain, B. A kava chalcone, induces apoptosis via up-regulation of death-receptor 5 and Bim expression in androgen receptor negative, hormonal refractory prostate cancer cell lines and reduces tumor growth. J. Int. Cancer 2010, 127, 1758–1768. [Google Scholar] [CrossRef]
- Festuccia, C.; Dondi, D.; Piccolella, M.; Locatelli, A.; Gravina, G.L.; Tombolini, V.; Ozarelix, M.M. A fourth generation GnRH antagonist, induces apoptosis in hormone refractory androgen receptor negative prostate cancer cells modulating expression and activity of death receptors. Prostate 2010, 70, 1340–1349. [Google Scholar]
- Bodmer, J.L.; Burns, K.; Schneider, P.; Hofmann, K.; Steiner, V.; Thome, M.; Bornand, T.; Hahne, M.; Schroter, M.; Becker, K.; et al. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo-1/CD95). Immunity 1997, 6, 79–88. [Google Scholar] [CrossRef]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar] [CrossRef]
- Zhu, G.; Yin, F.; Deng, X. Effect of NF-κB on inhibition of non-small cell lung cancer cell cyclooxygenase-2 by brucine. China J. Chin. Mater. Med. 2012, 37, 1269–1273. [Google Scholar]
- Jones, D.R.; Broad, R.M.; Comeau, L.D.; Parsons, S.J.; Mayo, M.W. Inhibition of nuclear factor κB chemosensitizes non-small cell lung cancer through cytochrome c release and caspase activation. J. Thorac. Cardiovasc. Surg. 2002, 123, 310–317. [Google Scholar] [CrossRef]
- Chien, S.T.; Lin, S.S.; Wang, C.K.; Lee, Y.B.; Chen, K.S.; Fong, Y.; Shih, Y.W. Acacetin inhibits the invasion and migration of human non-small cell lung cancer A549 cells by suppressing the p38alpha MAPK signaling pathway. Mol. Cell. Biochem. 2011, 350, 135–148. [Google Scholar] [CrossRef]
- Herr, I.; Ucur, E.; Herzer, K.; Okouoyo, S.; Ridder, R.; Krammer, P.H.; von Knebel Doeberitz, M.; Debatin, K.M. Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res. 2003, 63, 3112–3120. [Google Scholar]
- Murtaza, I.; Adhami, V.M.; Hafeez, B.B.; Saleem, M.; Mukhtar, H. Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3-mediated inhibition of NF-κB. Int. J. Cancer 2009, 125, 2465–2473. [Google Scholar] [CrossRef]
- Kollipara, P.S.; Jeong, H.S.; Han, S.B.; Hong, J.T. Anti-proliferative effect of (E)-2,4-bis(p-hydroxyphenyl)-2-butenal by suppression of NF-κB and up regulation of DR5 via up-regulation of p38 MAPK in NSCLC. Br. J. Pharmacol. 2013, 168, 1471–1484. [Google Scholar] [CrossRef]
- Cooper, J.T.; Stroka, D.M.; Brostjan, C.; Palmetshofer, A.; Bach, F.H.; Ferran, C. A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J. Biol Chem. 1996, 271, 18068–18073. [Google Scholar] [CrossRef]
- Daigeler, A.; Brenzel, C.; Bulut, D.; Geisler, A.; Hilgert, C.; Lehnhardt, M.; Steinau, H.U.; Flier, A.; Steinstraesser, L.; Klein-Hitpass, L.; et al. TRAIL and Taurolidine induce apoptosis and decrease proliferation in human fibrosarcoma. J. Exp. Clin. Cancer Res. 2008, 27. [Google Scholar] [CrossRef]
- Kumar, A.; Malik, F.; Bhushan, S.; Shah, B.A.; Taneja, S.C.; Pal, H.C.; Wani, Z.A.; Mondhe, D.M.; Kaur, J.; Singh, J. A novel parthenin analog exhibits anti-cancer activity: Activation of apoptotic signaling events through robust NO formation in human leukemia HL-60 cells. Chem. Biol. Interact. 2011, 193, 204–215. [Google Scholar] [CrossRef]
- Lee, S.H.; Son, S.M.; Son, D.J.; Kim, S.M.; Kim, T.J.; Song, S.; Moon, D.C.; Lee, H.W.; Ryu, J.C.; Yoon, D.Y.; et al. Epothilones induce human colon cancer SW620 cell apoptosis via the tubulin polymerization independent activation of the nuclear factor-κB/IκB kinase signal pathway. Mol. Cancer Ther. 2007, 6, 2786–2797. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choi, K.E.; Hwang, C.J.; Gu, S.M.; Park, M.H.; Kim, J.H.; Park, J.H.; Ahn, Y.J.; Kim, J.Y.; Song, M.J.; Song, H.S.; et al. Cancer Cell Growth Inhibitory Effect of Bee Venom via Increase of Death Receptor 3 Expression and Inactivation of NF-kappa B in NSCLC Cells. Toxins 2014, 6, 2210-2228. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6082210
Choi KE, Hwang CJ, Gu SM, Park MH, Kim JH, Park JH, Ahn YJ, Kim JY, Song MJ, Song HS, et al. Cancer Cell Growth Inhibitory Effect of Bee Venom via Increase of Death Receptor 3 Expression and Inactivation of NF-kappa B in NSCLC Cells. Toxins. 2014; 6(8):2210-2228. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6082210
Chicago/Turabian StyleChoi, Kyung Eun, Chul Ju Hwang, Sun Mi Gu, Mi Hee Park, Joo Hwan Kim, Joo Ho Park, Young Jin Ahn, Ji Young Kim, Min Jong Song, Ho Sueb Song, and et al. 2014. "Cancer Cell Growth Inhibitory Effect of Bee Venom via Increase of Death Receptor 3 Expression and Inactivation of NF-kappa B in NSCLC Cells" Toxins 6, no. 8: 2210-2228. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6082210