
Mass Spectrometry-Based Method of Detecting and Distinguishing Type 1 and Type 2 Shiga-Like Toxins in Human Serum

Abstract
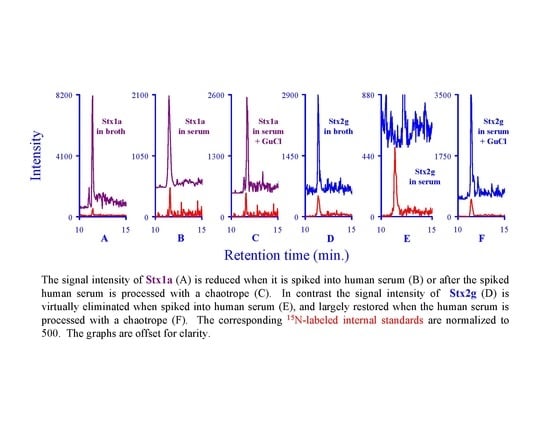
:
1. Introduction
2. Results
2.1. Generation of the Stx 15N-Labeled Internal Standard Protein (15N-Stx-ISP)
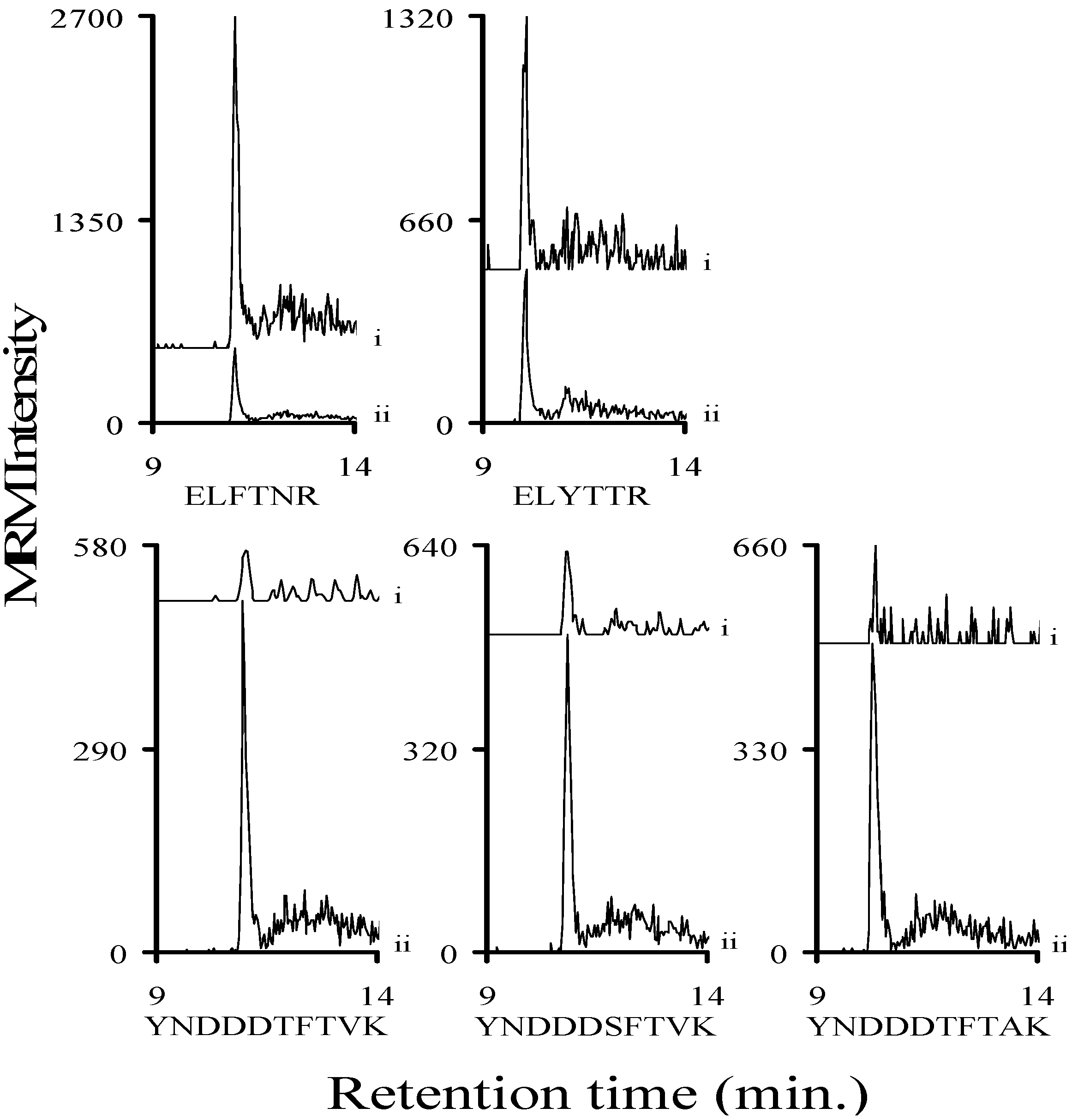
2.2. MRM Optimization of Stx1 Related Tryptic Peptides
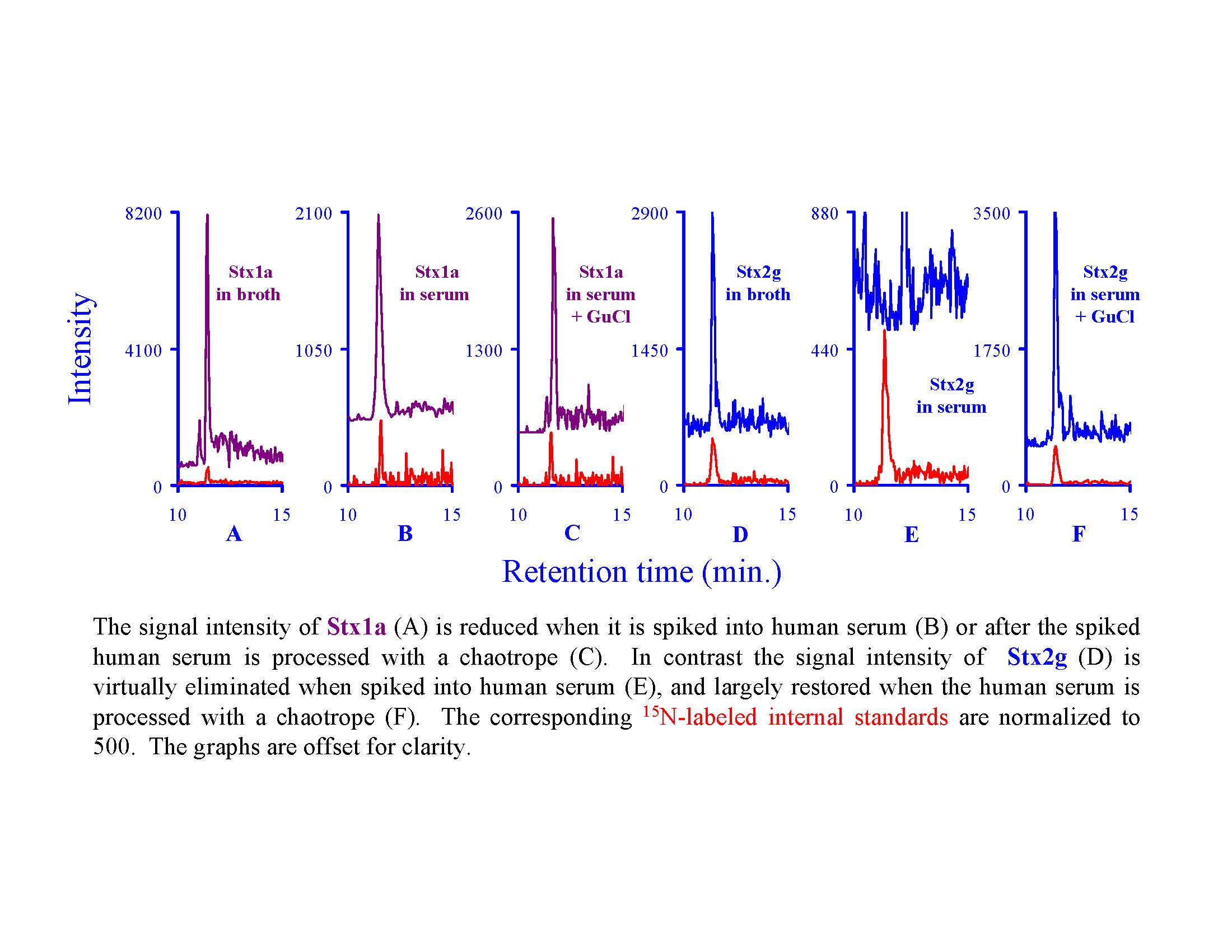
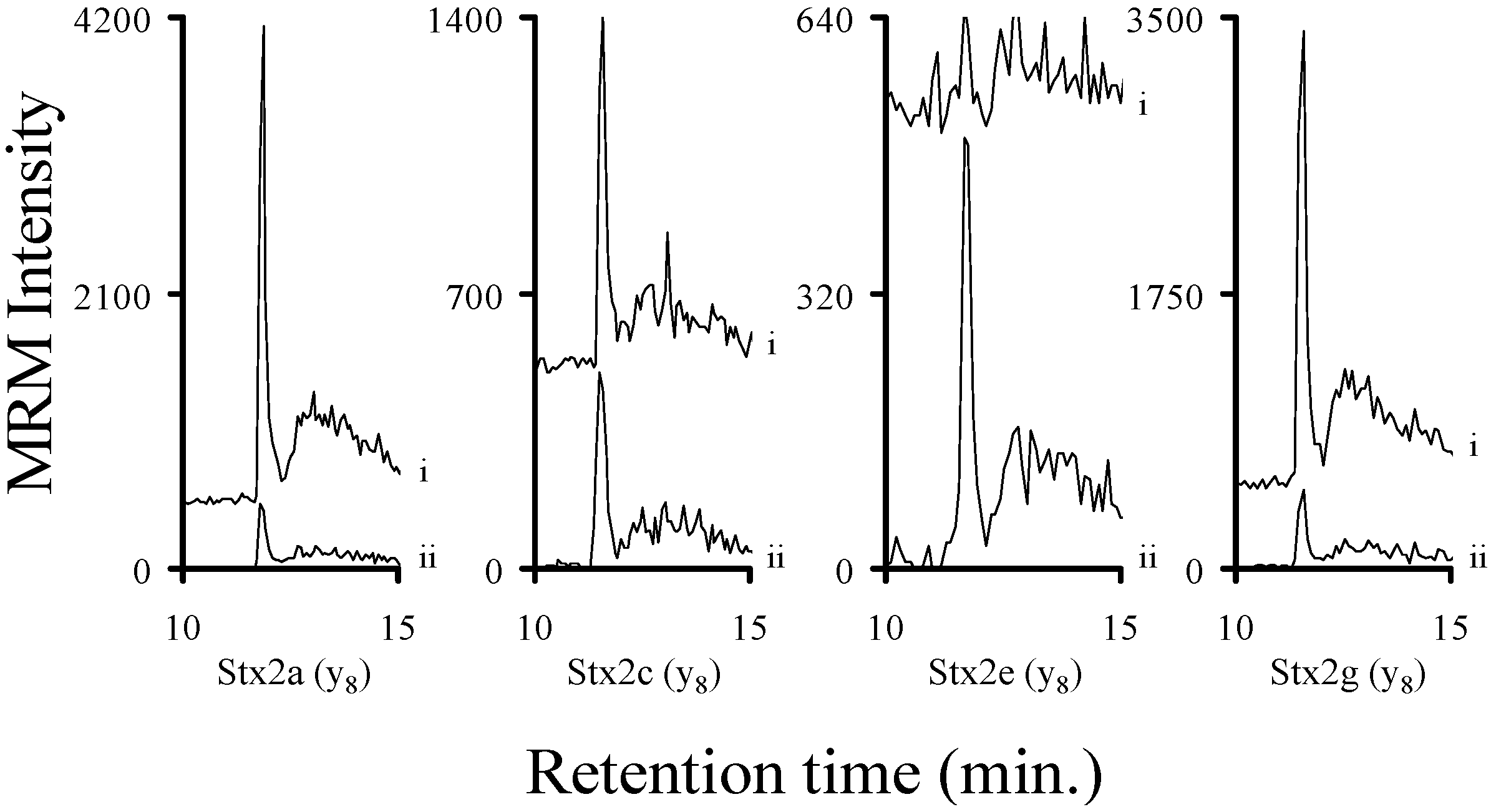
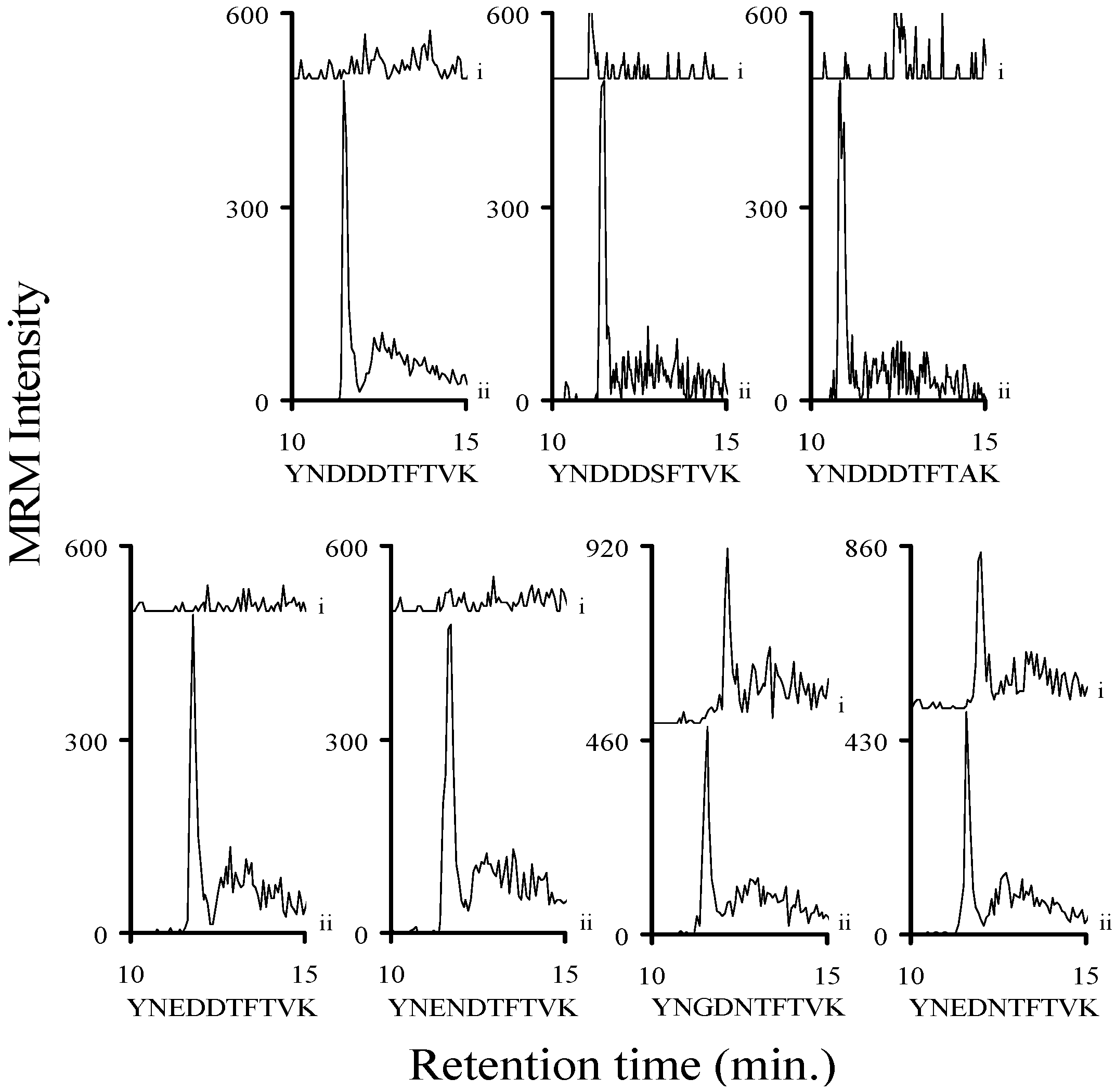
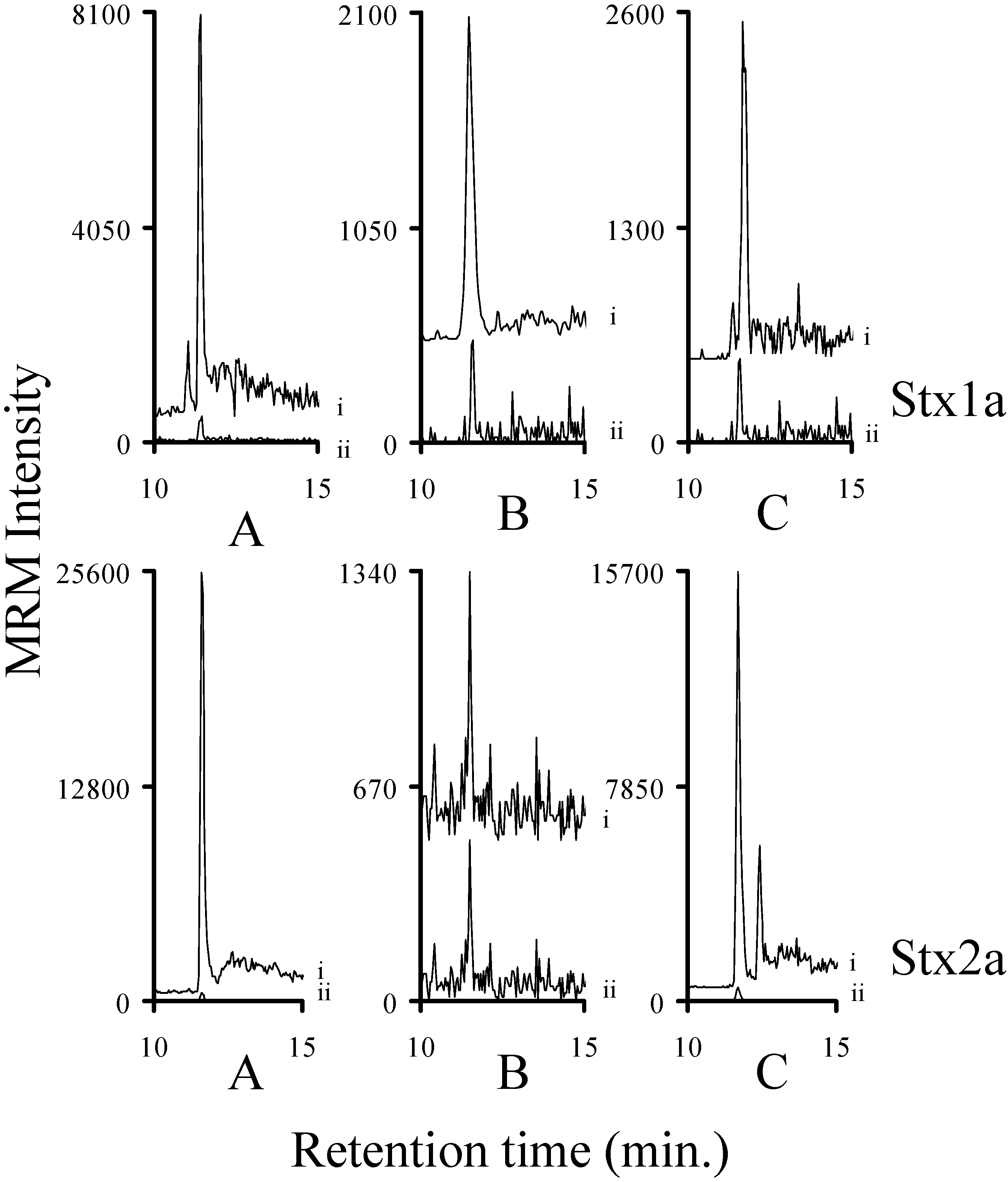
2.3. Using Characteristic Stx1 Tryptic Peptides to Distinguish between the Stx1 Subtypes
2.4. Quantitating Stx1 Toxins and Their Subtypes
2.5. Shortening the Reduction/Alkylation/Trypsin Time

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | 2 h | ON |
|---|---|---|
| Stx1a | 1.3 + 0.2 | 0.5 + 0.1 |
| Stx1e | 0.2 + 0.1 | 0.2 + 0.1 |
| Stx2g | 0.9 + 0.5 | 0.5 + 0.1 |
| Stx2e | 0.11 + 0.02 | 0.05 + 0.02 |
| Stx2a | 4.2 + 0.9 | 5.0 + 1.7 |
| Stx2c | 1.2 + 0.2 | 1.1 + 0.1 |
2.6. Detection of Stx1a, Stx1e, Stx2a, Stx2c, Stx2g, and Stx2e in Bacterial Broths


2.7. Assessing the Background in Human Serum

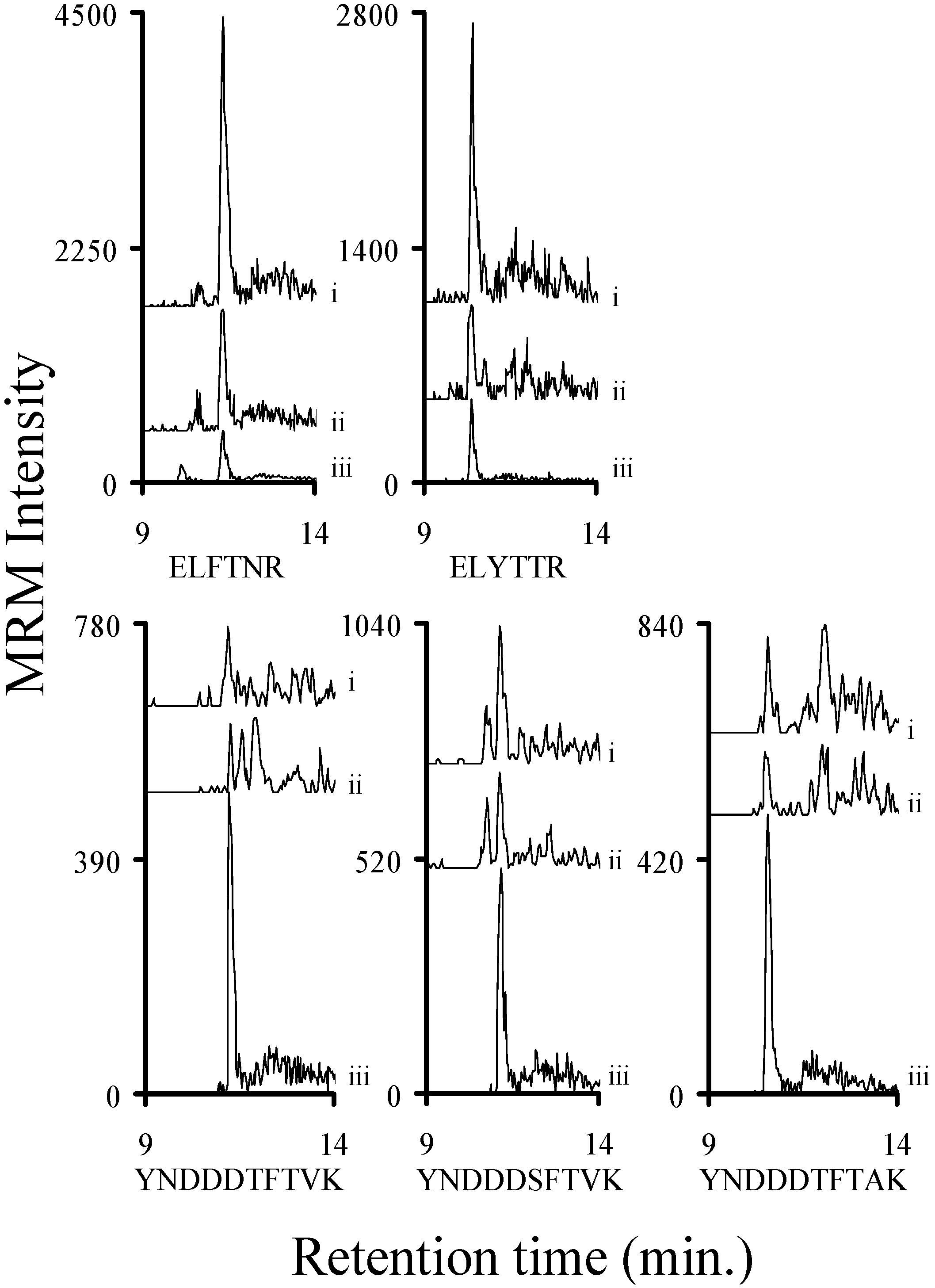
2.8. Determining a LOD for Relevant Stx1 Peptides in Buffer and Serum




| Sample Description | Proportion |
|---|---|
| Stx1a + HS | 1.00 |
| Filtered HS (100 K) + Stx1a | 0.99 |
| Stx1a + HS, Filtered (100 K) | 0.13 |
| Filtered HS (300 K) + Stx1a | 0.59 |
| Stx1a + HS, Filtered (300 K) | 0.15 |
| Stx2a + HS | 1 |
| Filtered HS (100 K) + Stx2a | 326 |
| Stx2a + HS, Filtered (100 K) | 14 |
| Filtered HS (300 K) + Stx2a | 115 |
| Stx2a + HS, Filtered (300 K) | 5 |
3. Discussion
3.1. The Stx 15N-Labeled Internal Standard Protein
3.2. Assessing the Background Interference with the Analyte Peptides
3.3. Calibration Curves for the Quantification of Stx1 Toxins in a Sample
3.4. Quantifying Shiga-Like Toxins in Bacterial Media
3.5. Quantifying Shiga Like Toxins in Human Serum
4. Experimental Procedures
4.1. Generation of Shiga-Like Toxin Samples
4.2. Preparation of Serum Samples
4.3. Reduction, Alkylation, and Tryptic Cleavage of Shiga-Like Toxins
4.4. Mass Spectrometry
4.5. Nanospray LC-MS/MS
4.6. Preparation of the Gene Containing the Internal Standards [45]
- ATGGGCAGCAGCCATCATCATCATCATCACAGCAGCGGCCTGGTGCCGCGCGGCAGCCATATGAGAGCGGATTGTGCCAAGTACAATGACGACGACACCTTTACTGTCAAGATCAAGTATAACGGCGATAATACCTTTACTGTCAAGGCGGACTGTGCCGTCGGCAAGACTCCTGATTGTGTCACTGGTAAGTATAACGATGATGACTCCTTCACCGTGAAGTATAACGAGGATAATACGTTCACCGTTAAGGCCCGTGAGTTGTTTACCAACCGCTACAATGACGACGATACCTTTACTGCGAAGGGCCGCGAGTACTGGACCTCTCGCTATAACGAGAACGATACGTTCACAGTTAAGGCCCGCGAGTACTGGACCAACCGCTACAATGAGGACGATACCTTCACGGTCAAGTCCCGCGAATTATATACTACCCGCGTGGAGTATACTAAGATTGAGTTTTCCAAGAATGGCGAGGGCTTCTCAGAGGTGATTTTTCGCACTAACGCCTGCCACAATGGCGGTGGCTTTAGCGAGGTTATCTTTAGGACTACGGCCTGTCATAACGGAGGCGGTTTCTCTGAGGTCATTTTCCGTTCCTCGACCTGTGAGTCCGGCTCCGGCTTTGCTGAGGTGCAGTTTAACAACGACTAGTGACTCGAG
4.7. Peptides, Internal Standards, Purified Proteins, and Calibration Curves
5. Conclusions

Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tyler, J.S.; Livny, J.; Friedman, D.I. Lambdoid phages and Shiga toxin. In Phages: Their Role in Pathogenesis and Biotechnology; Waldor, M.K., Friedman, D.I., Adhya, S.L., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 131–164. [Google Scholar]
- Friedman, D.I.; Court, D.L. Bacteriophage lambda: Alive and well and still doing its thing. Curr. Opin. Microbiol. 2001, 4, 201–207. [Google Scholar] [CrossRef]
- Cantey, J.R. Shiga toxin—An expanding role in the pathogenesis of infectious diseases. J. Infect. Dis. 1985, 151, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.A.; Friedman, D.I.; Francis, G.J.; Tyler, J.S.; Young, V.B.; Haeger, J.; Abu-Ali, G.; Whittam, T.S. Pathogenesis of renal disease due to enterohemorrhagic Escherichia coli in germ-free mice. Infect. Immun. 2008, 76, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Tesh, V.L.; Donohue-Rolfe, A.; Jackson, M.P.; Olsnes, S.; Sandvig, K.; Lindberg, A.A.; Keusch, G.T. Shiga toxin: Biochemistry, genetics, mode of action, and role in pathogenesis. Curr. Top. Microbiol. Immunol. 1992, 180, 65–94. [Google Scholar] [PubMed]
- Fuller, C.A.; Pellino, C.A.; Flagler, M.J.; Strasser, J.E.; Weiss, A.A. Shiga toxin subtypes display dramatic differences in potency. Infect. Immun. 2011, 79, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, S.W.; Samuel, J.E.; Schmitt, C.K.; O’Brien, A.D. The specific activities of Shiga-like toxin type ii (SLT-II) and SLT-II-related toxins of enterohemorrhagic Escherichia coli differ when measured by Vero cell cytotoxicity but not by mouse lethality. Infect. Immun. 1994, 62, 623–631. [Google Scholar] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Smith, D.L.; Rooks, D.J.; Fogg, P.C.; Darby, A.C.; Thomson, N.R.; McCarthy, A.J.; Allison, H.E. Comparative genomics of Shiga toxin encoding bacteriophages. BMC genomics 2012, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- James, C.E.; Stanley, K.N.; Allison, H.E.; Flint, H.J.; Stewart, C.S.; Sharp, R.J.; Saunders, J.R.; McCarthy, A.J. Lytic and lysogenic infection of diverse Escherichia coli and Shigella strains with a verocytotoxigenic bacteriophage. Appl. Environ. Microbiol. 2001, 67, 4335–4337. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Paton, J.C. Enterobacter cloacae producing a Shiga-like toxin II-related cytotoxin associated with a case of hemolytic-uremic syndrome. J. Clin. Microbiol. 1996, 34, 463–465. [Google Scholar] [PubMed]
- Schmidt, H.; Montag, M.; Bockemuhl, J.; Heesemann, J.; Karch, H. Shiga-like toxin II-related cytotoxins in Citrobacter freundii strains from humans and beef samples. Infect. Immun. 1993, 61, 534–543. [Google Scholar] [PubMed]
- Probert, W.S.; McQuaid, C.; Schrader, K. Isolation and identification of an Enterobacter cloacae strain producing a novel subtype of Shiga toxin type 1. J. Clin. Microbiol. 2014, 52, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Paton, J.C.; Paton, A.W. Instability of a Shiga toxin type 2 gene in Enterobacter cloacae. J. Clin. Microbiol. 1997, 35, 1917. [Google Scholar] [PubMed]
- Kimura, T.; Tani, S.; Yi, Y.M.; Takeda, T. Serum amyloid P component is the Shiga toxin 2-neutralizing factor in human blood. J. Biol. Chem. 2001, 276, 41576–41579. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; van der Helm, K.; Mulvey, G.L.; Armstrong, G.D. Serum amyloid P component binding to Shiga toxin 2 requires both a subunit and B pentamer. Infect. Immun. 2003, 71, 6075–6078. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Mulvey, G.L.; Marcato, P.; Griener, T.P.; Kahan, M.C.; Tennent, G.A.; Sabin, C.A.; Chart, H.; Pepys, M.B. Human serum amyloid P component protects against Escherichia coli O157:H7 Shiga toxin 2 in vivo: Therapeutic implications for hemolytic-uremic syndrome. J. Infect. Dis. 2006, 193, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, A.; Luzzi, I.; Seganti, L.; Marchetti, M.; Karmali, M.A.; Clarke, I.; Boyd, B. Frequency and nature of verocytotoxin 2 (VT2) neutralizing activity (NA) in human and animal sera. In Recent Advances in Verocytotoxin-Producing Escherichia coli Infections; Karmali, M.A., Goglio, A., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 353–356. [Google Scholar]
- He, X.; Qi, W.; Quinones, B.; McMahon, S.; Cooley, M.; Mandrell, R.E. Sensitive detection of Shiga toxin 2 and some of its variants in environmental samples by a novel immuno-PCR assay. Appl. Environ. Microbiol. 2011, 77, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.C.; Jinneman, K.; Scheutz, F.; Monday, S.R. Specificity of PCR and serological assays in the detection of Escherichia coli Shiga toxin subtypes. Appl. Environ. Microbiol. 2011, 77, 6699–6702. [Google Scholar] [CrossRef] [PubMed]
- Willford, J.; Mills, K.; Goodridge, L.D. Evaluation of three commercially available enzyme-linked immunosorbent assay kits for detection of Shiga toxin. J. Food Prot. 2009, 72, 741–747. [Google Scholar] [PubMed]
- Skinner, C.; Patfield, S.; Hernlem, B.J.; He, X. New Stx2e Monoclonal Antibodies for Immunological Detection and Distinction of Stx2 Subtypes. PloS ONE 2015, 10, e0132419. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.I.; Kumar, B.; Kamboj, D.V. Multiplex detection of protein toxins using MALDI-TOF-TOF tandem mass spectrometry: Application in unambiguous toxin detection from bioaerosol. Anal. Chem. 2012, 84, 10500–10507. [Google Scholar] [CrossRef] [PubMed]
- Conrady, D.G.; Flagler, M.J.; Friedmann, D.R.; van der Wielen, B.D.; Kovall, R.A.; Weiss, A.A.; Herr, A.B. Molecular basis of differential B-pentamer stability of Shiga toxins 1 and 2. PloS ONE 2010, 5, e15153. [Google Scholar] [CrossRef] [PubMed]
- Fagerquist, C.K.; Sultan, O. Top-down proteomic identification of furin-cleaved alpha-subunit of Shiga toxin 2 from Escherichia coli O157:H7 using MALDI-TOF-TOF-MS/MS. J. Biomed. Biotechnol. 2010, 2010, 123460. [Google Scholar] [CrossRef] [PubMed]
- Fagerquist, C.K.; Sultan, O. Induction and identification of disulfide-intact and disulfide-reduced beta-subunit of Shiga toxin 2 from Escherichia coli O157:H7 using MALDI-TOF-TOF-MS/MS and top-down proteomics. Analyst 2011, 136, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Kitova, E.N.; Daneshfar, R.; Marcato, P.; Mulvey, G.L.; Armstrong, G.; Klassen, J.S. Stability of the homopentameric B subunits of Shiga toxins 1 and 2 in solution and the gas phase as revealed by nanoelectrospray fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Kitova, E.N.; Kitov, P.I.; Bundle, D.R.; Klassen, J.S. The observation of multivalent complexes of Shiga-like toxin with globotriaoside and the determination of their stoichiometry by nanoelectrospray fourier-transform ion cyclotron resonance mass spectrometry. Glycobiology 2001, 11, 605–611. [Google Scholar] [PubMed]
- Kitova, E.N.; Kitov, P.I.; Paszkiewicz, E.; Kim, J.; Mulvey, G.L.; Armstrong, G.D.; Bundle, D.R.; Klassen, J.S. Affinities of Shiga toxins 1 and 2 for univalent and oligovalent PK-trisaccharide analogs measured by electrospray ionization mass spectrometry. Glycobiology 2007, 17, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Kitova, E.N.; Mulvey, G.L.; Dingle, T.; Sinelnikov, I.; Wee, S.; Griener, T.P.; Armstrong, G.D.; Klassen, J.S. Assembly and stability of the Shiga toxins investigated by electrospray ionization mass spectrometry. Biochemistry 2009, 48, 5365–5374. [Google Scholar] [CrossRef] [PubMed]
- Kondo, F.; Kobayashi, S.; Matsumoto, M.; Yamada, S.; Saito, M.; Suzuki, Y.; Ishikawa, N.; Nakanishi, T.; Shimizu, A. Analysis of Vero toxins 1 and 2 by high-performance liquid chromatography/electrospray ionization mass spectrometry. J. Mass Spectrom. 1997, 32, 1140–1142. [Google Scholar] [CrossRef]
- Kondo, F.; Saito, H.; Hayashi, R.; Onda, H.; Kobayashi, S.; Matsumoto, M.; Suzuki, M.; Ito, Y.; Oka, H.; Nakanishi, T.; et al. Identification of Shiga toxins in Shiga toxin-producing Escherichia coli using immunoprecipitation and high-performance liquid chromatography-electrospray ionization mass spectrometry. Analyst 2003, 128, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Meisen, I.; Friedrich, A.W.; Karch, H.; Witting, U.; Peter-Katalinic, J.; Muthing, J. Application of combined high-performance thin-layer chromatography immunostaining and nanoelectrospray ionization quadrupole time-of-flight tandem mass spectrometry to the structural characterization of high- and low-affinity binding ligands of Shiga toxin 1. Rapid Commun. Mass Spectrom. 2005, 19, 3659–3665. [Google Scholar] [PubMed]
- Silva, C.J.; Erickson-Beltran, M.L.; Skinner, C.B.; Dynin, I.; Hui, C.; Patfield, S.A.; Carter, J.M.; He, X. Safe and effective means of detecting and quantitating Shiga-like toxins in attomole amounts. Anal. Chem. 2014, 86, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Aebersold, R.; Chen, R.; Rush, J.; Goodlett, D.R.; McIntosh, M.W.; Zhang, J.; Brentnall, T.A. Mass spectrometry based targeted protein quantification: Methods and applications. J. Proteome Res. 2009, 8, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- ExPASy. Available online: http://www.expasy.org/tools/ (accessed on 16 November 2015).
- Silva, C.J.; Dynin, I.; Erickson, M.L.; Requena, J.R.; Balachandran, A.; Hui, C.; Onisko, B.C.; Carter, J.M. Oxidation of Methionine 216 in Sheep and Elk Prion Protein Is Highly Dependent upon the Amino Acid at Position 218 but Is not Important for Prion Propagation. Biochemistry 2013, 52, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, P.; Fohlman, J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 1984, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Skinner, C.; McMahon, S.; Rasooly, R.; Carter, J.M.; He, X. Purification and characterization of Shiga toxin 2f, an immunologically unrelated subtype of Shiga toxin 2. PLoS ONE 2013, 8, e59760. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Patfield, S.; Hnasko, R.; Rasooly, R.; Mandrell, R.E. A polyclonal antibody based immunoassay detects seven subtypes of Shiga toxin 2 produced by Escherichia coli in human and environmental samples. PLoS ONE 2013, 8, e76368. [Google Scholar] [CrossRef] [PubMed]
- Onisko, B.; Dynin, I.; Requena, J.R.; Silva, C.J.; Erickson, M.; Carter, J.M. Mass spectrometric detection of attomole amounts of the prion protein by nanoLC/MS/MS. J. Am. Soc. Mass Spectrom. 2007, 18, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1989; p. 1626. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, C.J.; Erickson-Beltran, M.L.; Skinner, C.B.; Patfield, S.A.; He, X. Mass Spectrometry-Based Method of Detecting and Distinguishing Type 1 and Type 2 Shiga-Like Toxins in Human Serum. Toxins 2015, 7, 5236-5253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7124875
Silva CJ, Erickson-Beltran ML, Skinner CB, Patfield SA, He X. Mass Spectrometry-Based Method of Detecting and Distinguishing Type 1 and Type 2 Shiga-Like Toxins in Human Serum. Toxins. 2015; 7(12):5236-5253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7124875
Chicago/Turabian StyleSilva, Christopher J., Melissa L. Erickson-Beltran, Craig B. Skinner, Stephanie A. Patfield, and Xiaohua He. 2015. "Mass Spectrometry-Based Method of Detecting and Distinguishing Type 1 and Type 2 Shiga-Like Toxins in Human Serum" Toxins 7, no. 12: 5236-5253. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7124875