
Identification and Quantification of Fumonisin A1, A2, and A3 in Corn by High-Resolution Liquid Chromatography-Orbitrap Mass Spectrometry

Abstract
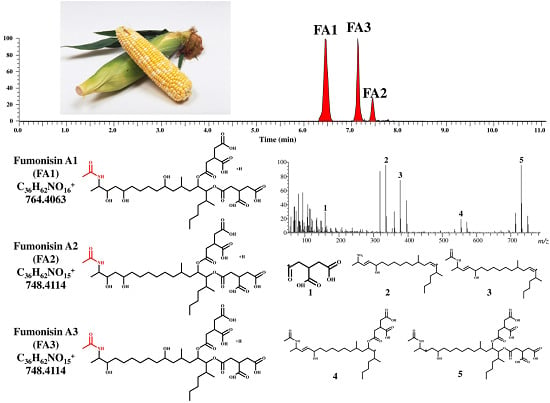
:
1. Introduction

2. Results and Discussion
2.1. Syntheses of FA2 and FA3 and Identification of Compounds II and III in MTC-9999E




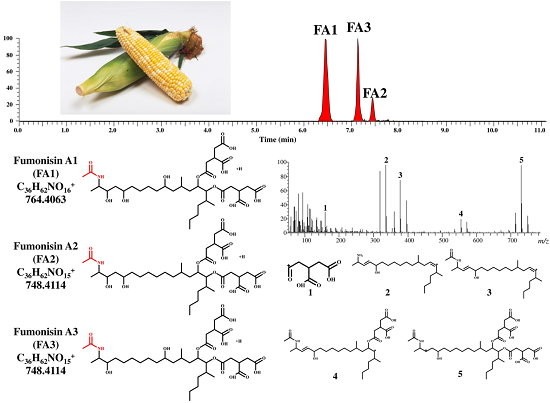{kind=link}
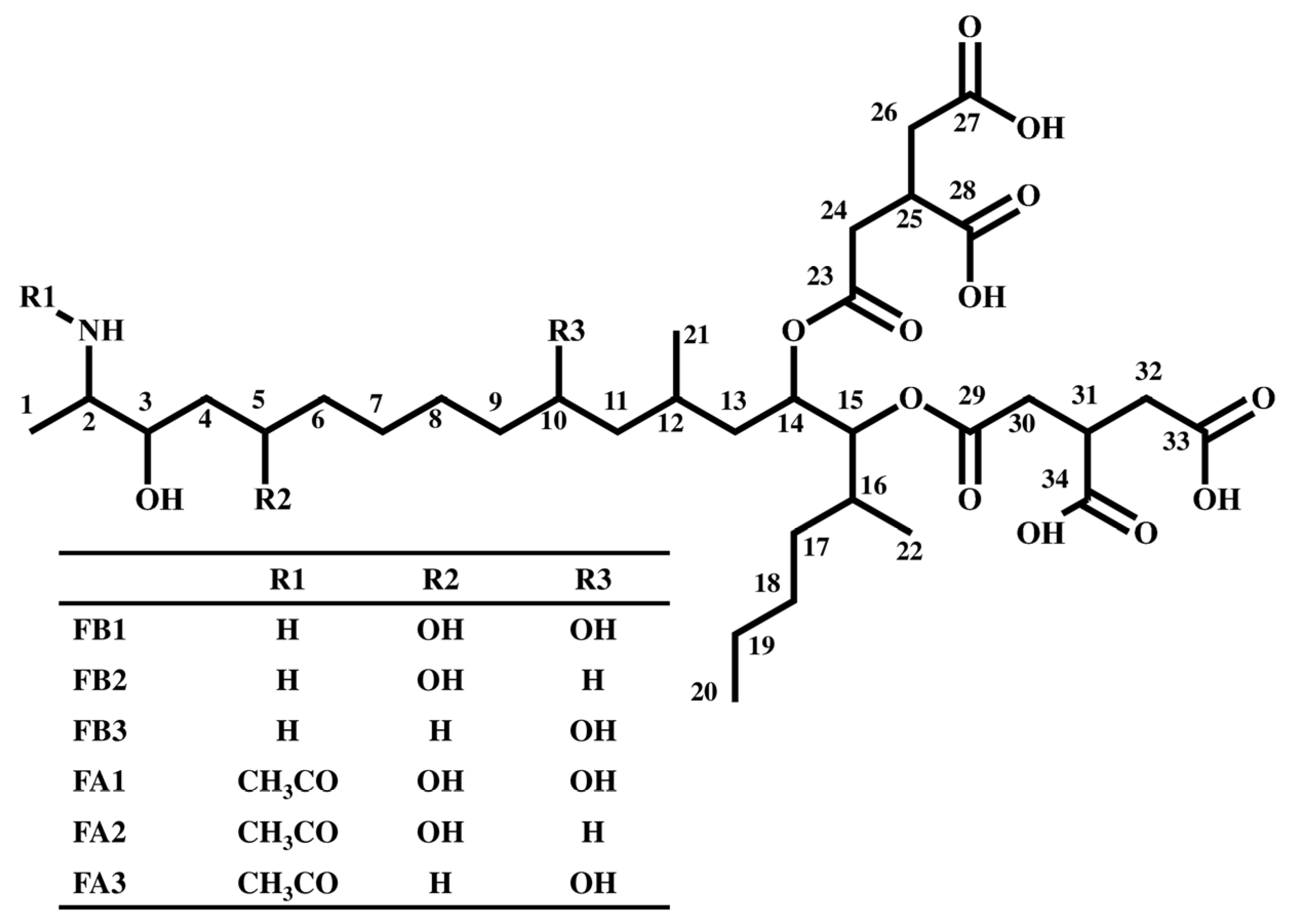{kind=link}
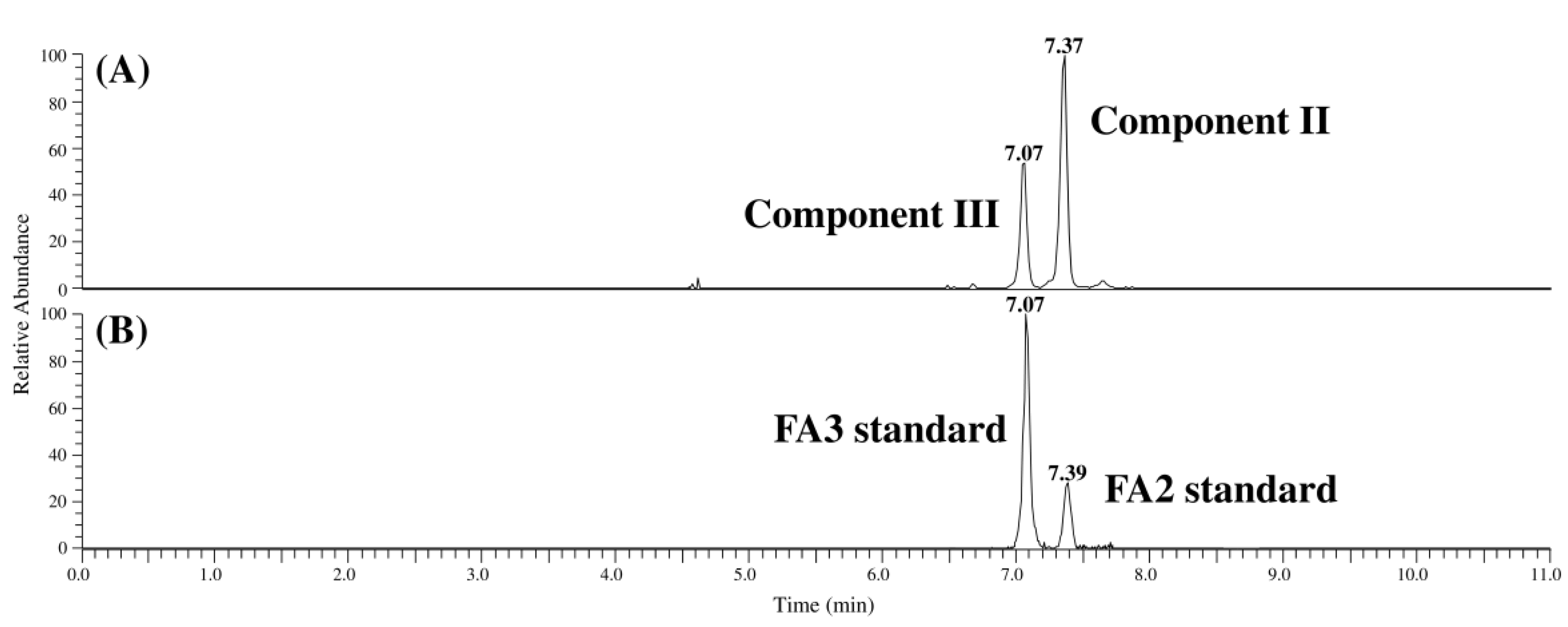{kind=link}
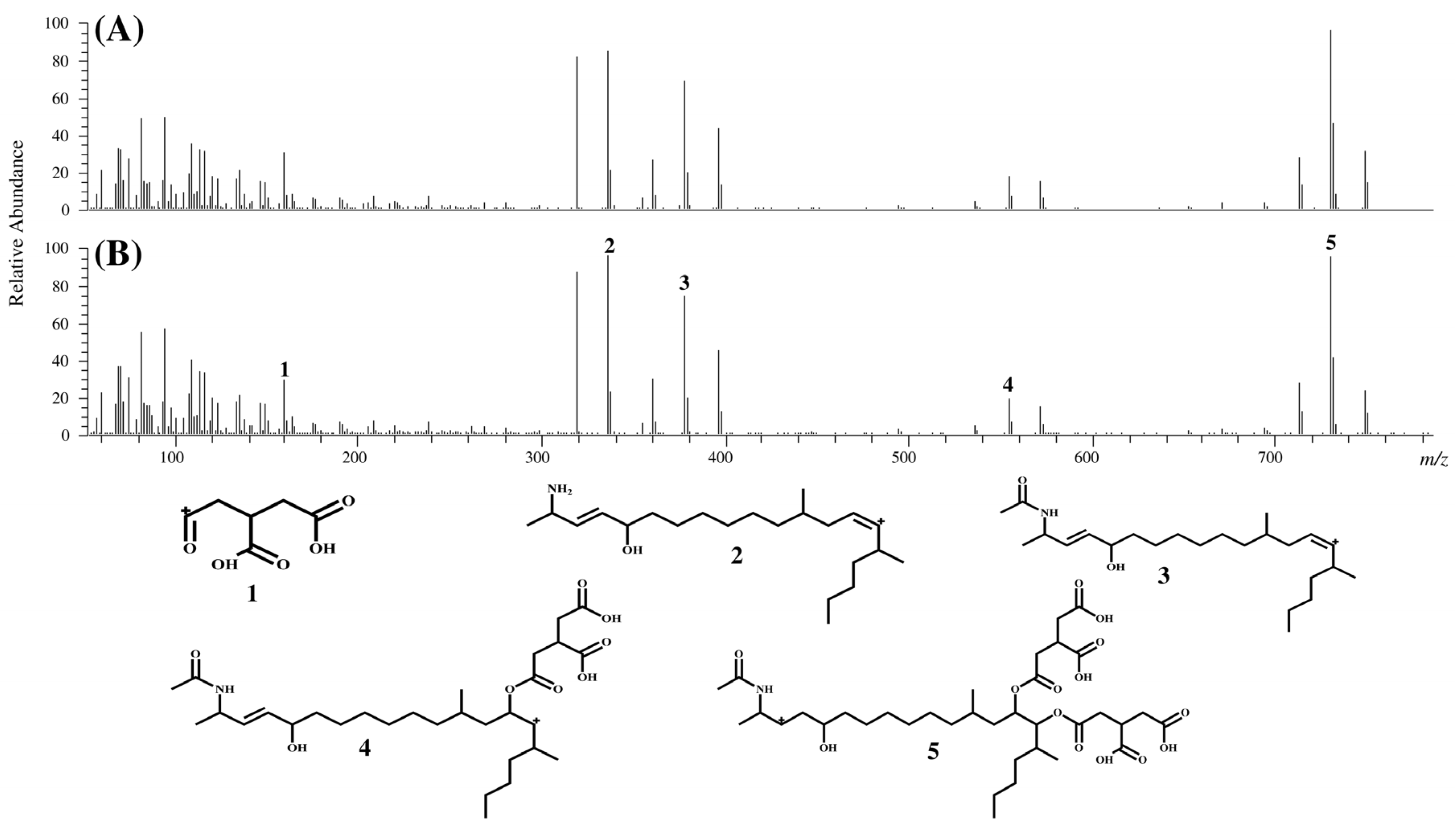{kind=link}
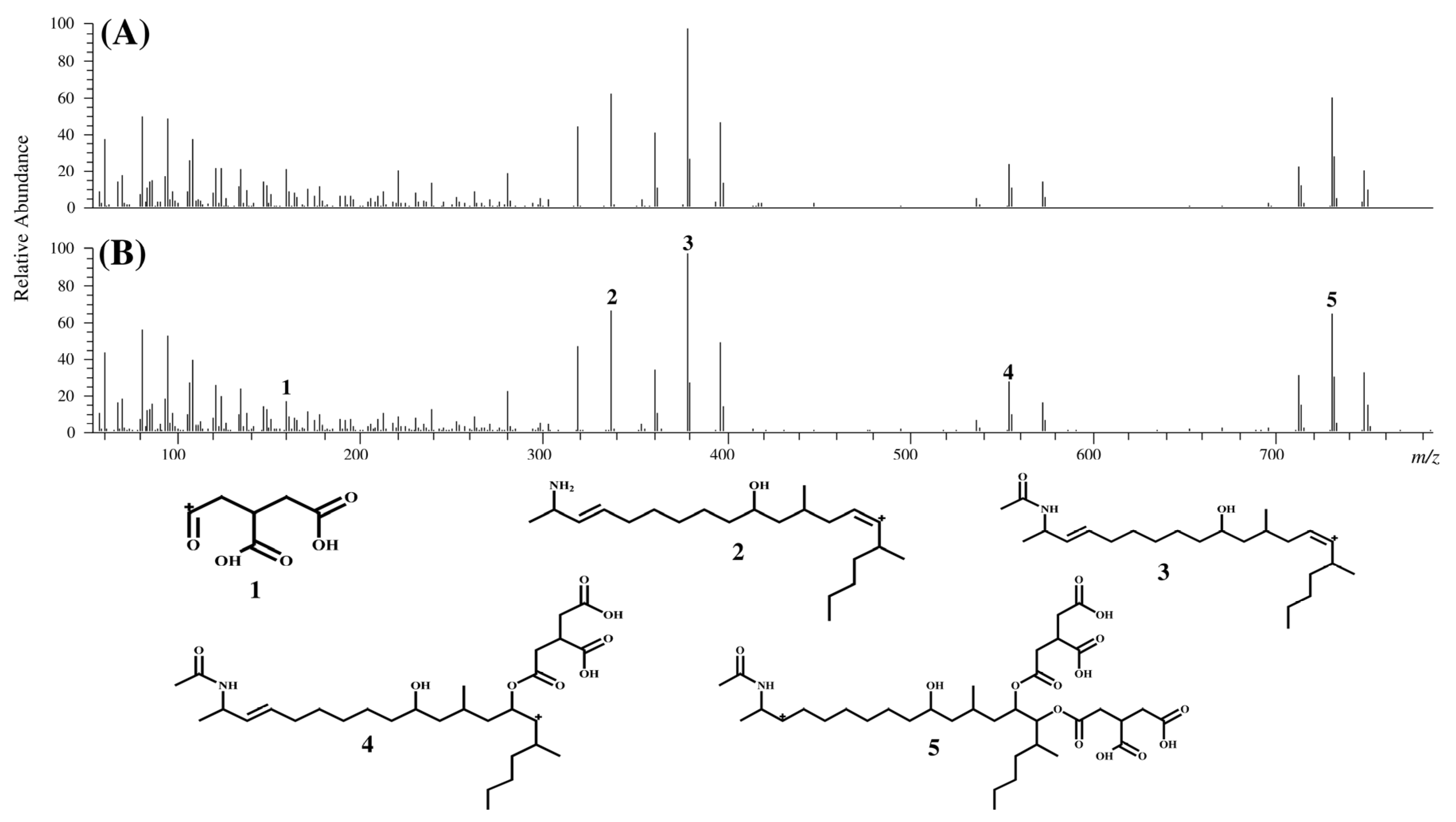{kind=link}
| ID | Measured mass (m/z) | Theoretical mass (m/z) | Calculated formula [M + H+] | Mass error (ppm) |
|---|---|---|---|---|
| 1 | 159.0285 | 159.0288 | C6H7O5 | −1.77 |
| 2 | 336.3266 | 336.3261 | C22H42NO | 1.60 |
| 3 | 378.3369 | 378.3367 | C24H44NO2 | 0.68 |
| 4 | 554.3661 | 554.3661 | C30H52NO8 | 0.05 |
| 5 | 730.4014 | 730.4017 | C36H60NO14 | −0.32 |
| ID | Measured mass (m/z) | Theoretical mass (m/z) | Calculated formula [M + H]+ | Mass error (ppm) |
|---|---|---|---|---|
| 1 | 159.0290 | 159.0288 | C6H7O5 | 1.21 |
| 2 | 336.3259 | 336.3261 | C22H42NO | −0.67 |
| 3 | 378.3366 | 378.3367 | C24H44NO2 | −0.12 |
| 4 | 554.3700 | 554.3687 | C30H52NO8 | 2.26 |
| 5 | 730.4009 | 730.4008 | C36H60NO14 | 0.06 |
2.2. Validation of the Method
| Validation Item | FA1 | FA2 | FA3 | FB1 | FB2 | FB3 |
|---|---|---|---|---|---|---|
| Linearity (r) | 0.9996 | 0.9999 | 0.9993 | 0.9960 | 0.9946 | 0.9962 |
| Recovery (%) | 82.9 | 85.6 | 95.4 | 101.6 | 104.6 | 104.2 |
| Repeatability (%) | 2.7 | 9.5 | 6.5 | 5.3 | 3.7 | 7.1 |
| LOD (μg/kg) | 0.10 | 0.60 | 0.28 | 0.02 | 0.04 | 0.03 |
| LOQ (μg/kg) | 0.34 | 1.98 | 0.92 | 0.05 | 0.12 | 0.10 |
2.3. Quantification of FA1, FA2, FA3, FB1, FB2, and FB3 in Corn
| Sample | Concentration of Fumonisins (μg/kg) | |||||
|---|---|---|---|---|---|---|
| FA1 | FA2 | FA3 | FB1 | FB2 | FB3 | |
| MTC-9999E | 4177.7 | 4033.7 | 269.0 | 28633.2 | 8868.2 | 2033.2 |
| MTC-9990 | 256.0 | 221.9 | 30.2 | 1231.8 | 320.2 | 189.3 |
| FC-443 | 500.6 | 488.7 | 85.2 | 2661.0 | 714.8 | 358.0 |
| C-1 | 62.5 | 45.0 | 30.6 | 660.5 | 114.6 | 53.4 |
| C-2 | 10.6 | 6.6 | 2.6 | 309.4 | 37.2 | 19.8 |
| C-3 | 2.7 | 2.8 | 2.0 | 90.4 | 21.3 | 11.5 |
| C-4 | 42.4 | 23.4 | 8.7 | 461.8 | 86.4 | 52.1 |
| C-5 | 59.7 | 84.2 | 23.4 | 1181.8 | 276.2 | 183.9 |
| C-6 | 17.9 | 11.9 | 5.3 | 385.1 | 43.3 | 32.9 |
| C-7 | 8.0 | 5.9 | (<0.92) | 150.9 | 16.4 | 12.2 |
3. Experimental Section
3.1. Sample, Chemicals, and Reagents
| Sample | Acceptance Limit (mg/kg) | ||
|---|---|---|---|
| FB1 | FB2 | FB3 | |
| MTC-9999E | 28.3 ± 7.6 | 7.1 ± 1.9 | 1.7 ± 0.5 |
| MTC-9990 | 1.3 ± 0.3 | 0.2 ± 0.1 | ND |
| FC-443 | 3.6 ± 1.4 | 0.8 ± 0.3 | 0.3 ± 0.1 |
3.2. Sample Preparation
3.3. LC-Orbitrap MS Analysis
3.4. Syntheses of FA2 and FA3 and Identification of Their Structures by NMR Analysis
3.5. Validation of the Method
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Hussein, H.S.; Brasel, J.M. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology 2001, 167, 101–134. [Google Scholar]
- Norred, W.P. Fumonisins-mycotoxins produced by Fusarium moniliforme. J. Toxicol. Environ. Health 1993, 38, 309–328. [Google Scholar]
- Lazzaro, I.; Falavigna, C.; Dall’Asta, C.; Proctor, R.H.; Galaverna, G.; Battilani, P. Fumonisin, B, A and C profile and masking in Fusarium verticillioides strains on fumonisin-inducing and maize-based media. Int. J. Food Microbiol. 2012, 159, 93–100. [Google Scholar]
- Musser, S.M.; Eppley, R.M.; Mazzola, E.P.; Hadden, C.E.; Shockcor, J.P.; Crouch, R.C.; Martin, G.E. Identification of an N-acetyl keto derivative of fumonisin B1 in corn cultures of Fusarium proliferatum. J. Nat. Prod. 1995, 58, 1392–1397. [Google Scholar]
- Musser, S.M.; Plattner, R.D. Fumonisin composition in cultures of Fusarium moniliforme, Fusarium proliferatum, and Fusarium nygami. J. Agric. Food Chem. 1997, 45, 1169–1173. [Google Scholar]
- Rheeder, J.P.; Marasas, W.F.O.; Vismer, H.F. Production of fumonisin analogs by Fusarium species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar]
- Lazzaro, I.; Falavigna, C.; Galaverna, G.; Dall’Asta, C.; Battilani, P. Cornmeal and starch influence the dynamic of fumonisin B, A and C production and masking in Fusarium verticillioides and F. proliferatum. Int. J. Food Microbiol. 2014, 166, 21–27. [Google Scholar]
- Sewram, V.; Mshicileli, N.; Shephard, G.S.; Vismer, H.F.; Rheeder, J.P.; Lee, Y.-W.; Leslie, J.F.; Marasas, W.F.O. Production of fumonisin B and C analogs by several Fusarium species. J. Agric. Food Chem. 2005, 53, 4861–4866. [Google Scholar]
- Nakagawa, H.; Sakamoto, S.; Sago, Y.; Nagashima, H. Detection of type A trichothecene di-glucosides produced in corn by high-resolution liquid chromatography-orbitrap mass spectrometry. Toxins 2013, 5, 590–604. [Google Scholar]
- Zachariasova, M.; Vaclavikova, M.; Lacina, O.; Vaclavik, L.; Hajslova, J. Deoxynivalenol oligoglycosides: New “masked” Fusarium toxins occurring in malt, beer, and breadstuff. J. Agric. Food Chem. 2012, 60, 9280–9291. [Google Scholar]
- De Girolamo, A.; Lattanzio, V.M.T.; Schena, R.; Visconti, A.; Pascale, M. Use of liquid chromatography-high-resolution mass spectrometry for isolation and characterization of hydrolyzed fumonisins and relevant analysis in maize-based products. J. Mass Spectrom. 2014, 49, 297–305. [Google Scholar]
- Fang, L.; Xiong, A.; Wang, R.; Ji, S.; Yang, L.; Wang, Z. A strategy for screening and identifying mycotoxins in herbal medicine using ultra-performance liquid chromatography with tandem quadrupole time-of-flight mass spectrometry. J. Sep. Sci. 2013, 36, 3115–3122. [Google Scholar]
- Bartók, T.; Tölgyesi, L.; Mesterházy, Á.; Bartók, M.; Szécsi, Á. Identification of the first fumonisin mycotoxins with three acyl groups by ESI-ITMS and ESI-TOFMS following RP-HPLC separation: palmitoyl, linoleoyl and oleoyl EFB1 fumonisin isomers from a solid culture of Fusarium verticillioides. Food Addit. Contam. 2010, 27, 1714–1723. [Google Scholar]
- Bartók, T.; Tölgyesi, L.; Szécsi, Á.; Mesterházy, Á.; Bartók, M.; Gyimes, E.; Véha, A. Detection of previously unknown fumonisin P analogue mycotoxins in a Fusarium verticillioides culture by high-performance liquid chromatography–electrospray ionization time-of-flight and ion trap mass spectrometry. J. Chromatogr. Sci. 2014, 52, 508–513. [Google Scholar]
- Tamura, M.; Mochizuki, N.; Nagatomi, Y.; Toriba, A.; Hayakawa, K. Characterization of fumonisin A-series by high-resolution liquid chromatography-orbitrap mass spectrometry. Toxins 2014, 6, 2580–2593. [Google Scholar]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar]
- Tamura, M.; Matsumoto, K.; Watanabe, J.; Iida, J.; Nagatomi, Y.; Mochizuki, N. Minimization of carryover for high-throughput LC-MS/MS analysis of 14 mycotoxins in corn grits. J. Sep. Sci. 2014, 37, 1552–1560. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamura, M.; Mochizuki, N.; Nagatomi, Y.; Harayama, K.; Toriba, A.; Hayakawa, K. Identification and Quantification of Fumonisin A1, A2, and A3 in Corn by High-Resolution Liquid Chromatography-Orbitrap Mass Spectrometry. Toxins 2015, 7, 582-592. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7020582
Tamura M, Mochizuki N, Nagatomi Y, Harayama K, Toriba A, Hayakawa K. Identification and Quantification of Fumonisin A1, A2, and A3 in Corn by High-Resolution Liquid Chromatography-Orbitrap Mass Spectrometry. Toxins. 2015; 7(2):582-592. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7020582
Chicago/Turabian StyleTamura, Masayoshi, Naoki Mochizuki, Yasushi Nagatomi, Koichi Harayama, Akira Toriba, and Kazuichi Hayakawa. 2015. "Identification and Quantification of Fumonisin A1, A2, and A3 in Corn by High-Resolution Liquid Chromatography-Orbitrap Mass Spectrometry" Toxins 7, no. 2: 582-592. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7020582